1. Introduction
Although significant advancements have already been made in the development of biomaterials, regenerative technologies still demand polymer materials that can be processed without harming the environment, are biocompatible, biologically active, and capable of safe bioresorption. The biopolymers chitin (CN) and chitosan (CS) completely meet the above requirements; in addition to these properties, they have some other advantages, such as antibacterial and fungicidal activity, anesthetic, wound healing, and hemostatic properties [
1].
Two important physicochemical parameters of CS are its degree of deacetylation and its molecular mass. These characteristics have a significant influence on the properties of CS-based materials and, therefore, on their biomedical applications [
2]. Use of high molecular mass chitosan is restricted in a number of areas since it has low solubility in water and diluted solutions of acids; besides, its solutions demonstrate high viscosities even at low concentrations. Low molecular mass chitosan samples have biomedical applications due to their high solubility in water and low viscosity of aqueous solutions at physiological pH values [
3].
The degree of deacetylation has an effect on biocompatibility, biodegradability, solubility in acidic solutions, hydrophilicity, swelling in water, and, therefore, on the biological activity of CS [
4]. Since the matrices intended for various tissue regeneration techniques require different degradation rates, the possibility of controlling the chitosan bioresorption process makes this polymer applicable for the regeneration of almost all types of tissues.
The cationic nature of CS favors the formation of polyelectrolyte complexes with a wide range of anionic glycosaminoglycans, including heparin, heparin sulfate, and chondroitin sulfate. Glycosaminoglycans, especially heparin and heparin sulfate, are able to bind growth factors and cytokines, as well as initiate or induce their synthesis. Thus, the constructions containing chitosan-glycosaminoglycan complexes can be used for retaining and accumulating the necessary factors that are released by colonizing cells and for accumulating growth factors from the surrounding biological fluids. The important role of these complexes in regeneration technologies for bone and cartilage tissues has been demonstrated [
5].
The presence of functional groups in CS provides an opportunity to obtain conjugates between chitosan and biologically active molecules, such as laminin peptides, RGD-peptides/proteins, γ-poly(glutamic acid), etc. Thus, the matrix can be used for immobilization of biologically active molecules that participate in the adhesion, proliferation, differentiation of cells, and other processes that are important for tissue regeneration [
6].
Studies of physicochemical and mechanical properties of CS made it possible to develop numerous techniques for processing, modifying, and preparing different types of chitosan-based matrices intended for regenerative medicine. A wide selection of matrices (fibers, films, sponges, and hydrogels) provides a means to simulate surface properties and mechanical characteristics of a natural tissue.
Despite all the advantages described above, chitosan has several limitations, such as the absence of biological response, which is partially caused by mechanical properties of CS (inappropriate for many applications) and strong swelling of CS in liquid media. Besides, difficulties in the use of pure CS as a basis for cell cultivation matrices may be related to electrostatic interactions between positively charged amino groups of CS and the cell surface. This interaction disturbs the activity of trans-membrane cell receptors. This disturbance, in turn, has an adverse effect on cell-surface adhesion and cell proliferation [
7]. A decrease of surface charge helps to reduce this effect.
With this, one method for optimizing properties of CS-containing matrices is introducing mineral or organic nano-sized fillers. Successful use of the composites strongly depends on high specific area of nanoparticles, which provides a high surface-area-to-volume ratio and, therefore, stronger interaction between the polymer and the nanofiller. Besides, during stable interphase interaction, excellent properties of nanoparticles are transferred to the composite [
8]. Preparation of nanocomposites allows varying properties of natural polymers, including chitosan (e.g., their porosity, surface morphology, and mechanical strength), over a wide range, and imparts new functional properties to native macromolecules [
9].
A variety of biomedical technologies require completely bioresorbable materials (i.e., containing bioresorbable matrix and filler). One of the biocompatible, bioactive, and bioresorbable polymers is chitin, which can be successfully used as a filler. The main structural units of chitin are nanofibrils; they are highly oriented macromolecular aggregates with transverse sizes of 15–20 nm and longitudinal sizes of 400–500 nm [
10]. This structure provides a high mechanical strength of natural chitin-containing tissues. The crystalline structure of chitin is polymorph and consists of two crystalline modifications with an antiparallel (α-chitin) or a parallel (β-chitin) arrangement of macromolecules.
A number of biomedical studies have demonstrated the possibility of modifying chitosan and other polymers by introducing a filler consisting of chitin nanofibrils (chitin nanowhiskers, CNWs) [
11]. In vitro studies show that CNWs (which possess a high surface area) effectively interact with enzymes, proteins, and cell components of blood, immune cells, fibroblasts, and other cells involved in tissue regeneration. Thus, when CNWs are used in regenerative technologies, modulation of granulation processes takes place, angiogenesis and regular deposition of collagen fibers in the damaged areas are stimulated, and re-epithelialization proceeds more effectively [
12]. At the same time, the antiseptic action of CNWs plays an important role in the healing process; besides, their presence reduces the risks of scar tissue hypertrophy and contraction [
13].
However, any reinforcement agent not only changes the mechanical behavior of the basic material, but it might also influence its biological performance, such as its biocompatibility, biodegradability, or bioactivity. In the best case, the contribution of a reinforcing compound is beneficial and leads to an improvement of the biological performance of a composite. To evaluate the influence of the structure, chemical composition, and content of a reinforcing agent on biological properties of a composite, a generic approach should be used. An ideal system for such investigations should be readily producible, form homogeneous composite structures, and allow for an easy modification of a reinforcing agent. It should be possible to use a wide range of physicochemical, mechanical, and biological methods to characterize the prepared materials. Furthermore, the interaction between a biomaterial’s surface and biological matter is a crucial aspect in the design of biomaterials and a critical factor for their performance. Protein adsorption is important since the initial protein layer mediates cell colonization and directs the overall biocompatibility. The initial studies of a biomaterial’s surface should include the determination of general surface properties (chemical composition, mechanical properties, degree of hygroscopicity, surface charge) and the investigation of its interactions with different kinds of cells.
The first aim of the present study was to obtain film matrices that possess the mechanical properties necessary for sterilization, cell cultivation, and other manipulations in liquid media. For this purpose, we studied the effect of chitin nanofibril additives on mechanical properties, moisture sorption, and surface charge of chitosan films. Furthermore, analysis of proliferation characteristics of the cells cultivated on modified substrates allowed us to find the optimal composition of the nanocomposites. The investigation of these relationships will help to optimize biocompatibility and bioactivity of the advanced matrices for tissue engineering.
2. Materials and Methods
2.1. Preparation of Composite Films
Chitosan (Biolog Heppe GmbH, Landsberg, Germany) and chitin nanofibrils (Mavi Sud srl, Aprilia, Italy) were used for preparing composite films. The molecular mass (M
m) of chitosan was 1.64 × 10
5 g/mol and the degree of deacetylation (DD) was 92%. The diameter of chitin nanofibrils was 20 ± 4 nm and the length was 700 ± 100 nm [
10,
14]. For evaluating the effect of the film matrix on the properties of the composite films, chitosan with a similar M
m of 1.4 × 10
5 g/mol but a lower DD of 80% was used (Sigma Aldrich, St. Louis, MO, USA). Glacial acetic acid (Vecton ZAO, Moscow, Russia) was used as a solvent.
Composite films were prepared from aqueous acetic acid solutions of chitosan containing chitin nanofibrils. In an amount necessary to produce a solution with a chitosan concentration of 4 wt %, chitosan was added to the aqueous dispersion of chitin nanofibrils. The content of chitin nanofibrils with respect to chitosan was 0, 0.5, 1, 5, 10, 20, 30 wt %. Before the addition of chitosan, a suspension containing chitin nanofibrils was dispersed to uniformly distribute particles throughout the volume. This was done using an ultrasonic generator IL10-0.63 for 15 min at a frequency of 23 kHz. The resulting solution was stirred for 30 min at 1000 rpm, after which acetic acid was added in such an amount that its concentration in the solution was 2%. The resulting solution was mixed for 60 min at 1000 rpm. After that, the chitosan solution with chitin nanofibrils was filtered and deaerated at a low pressure of 0.8 atm for 24 h.
The chitosan and composite films were obtained by casting the solution through a slit draw die onto a glass substrate followed by drying at RT (room temperature) for 1 day. The thickness of the films was 80 ± 15 μm. The prepared films were kept in a mixture of 10% aqueous solution of NaOH and C2H5OH for 15 min, then washed with distilled water, and air-dried.
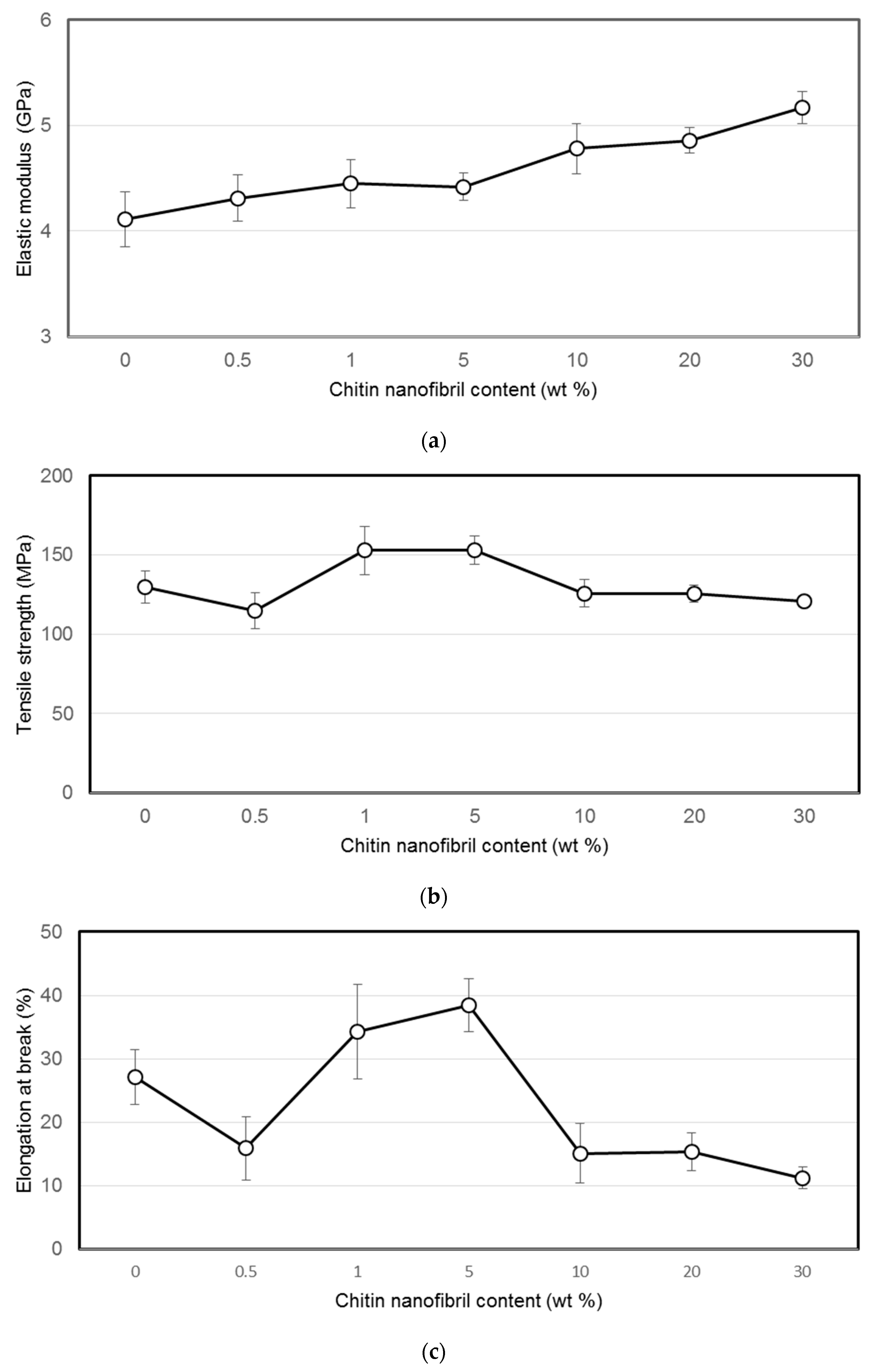
2.2. Mechanical Testing of Materials
The measurements of the mechanical properties of the films were carried out with the electrodynamic system ElectroPuls E1000 (Instron, Norwood, MA, USA) in the mode of stretching the sample in the form of strips 2 mm wide and 20 mm long. Tensile tests were carried out at a speed of 5 mm/min at room temperature. Prior to the mechanical testing, films were stored at a relative humidity (RH) of 66% for 24 h. Ten measurements were taken at each point.
2.3. Moisture Sorption Study
Water vapour sorption was measured by the isopiestic method; the RH in the desiccators was created with saturated salt solutions. The desiccators were thermostated at 25 °C. The samples were kept at each partial water vapour pressure until sorption equilibrium was established, where the equilibrium sorption values were established for 3–7 weeks. Before measurement, the samples were dried at 80 °C to a constant weight [
15]; the control over the moisture content was carried out by the weight method.
2.4. Water Contact Angle
The water contact angle on chitosan and chitosan-based composite films was determined by the sessile drop method using the drop shape analyzer DSA30 (Krüss GmbH, Hamburg, Germany). Distilled water was used as the test liquid. Five measurements were taken for each film sample.
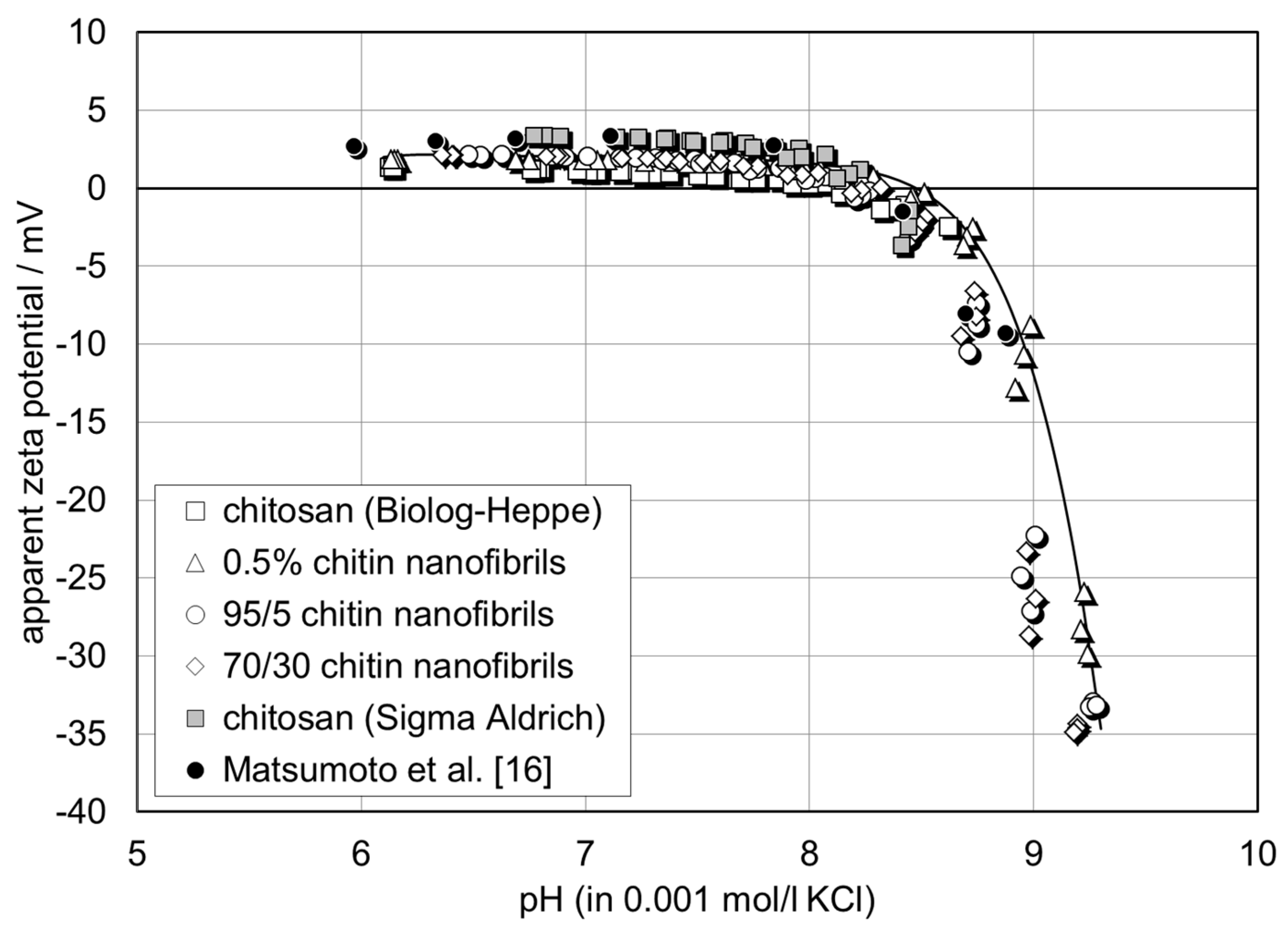
2.5. Zeta Potential Analysis
The surface charge of the chitin-chitosan nanocomposite films was assessed by the zeta potential at the film-water interface. The surface zeta potential was determined from the measurement of the streaming potential using the instrument SurPASS™ 3 (Anton Paar, Graz, Austria). The streaming potential was generated by applying a pressure gradient across a capillary channel, which provokes flow of an aqueous solution and is related to the surface zeta potential by
where d
Ustr/dΔ
p is the streaming potential coupling coefficient, η and ε are the viscosity and dielectric coefficient of water, ε
0 is the vacuum permittivity, and κ
B is the electric conductivity of the bulk solution.
The chitin–chitosan nanocomposite films were stored in 0.001 mol/l KCl for 24 h prior to the zeta potential analysis. For each film composition, two samples with a size of 20 mm × 10 mm were then mounted on sample holders with the same cross-section using double-sided adhesive tape. The sample holders were inserted in the measuring cell (adjustable gap cell) and the distance between the film surfaces was adjusted to 109 ± 4 µm by monitoring the temporal change of the applied pressure difference. The streaming potential was recorded in the pressure range of 200–500 mbar and the streaming potential coupling coefficient dU
str/dΔp in Equation (1) was taken as the slope of the linear dependence of streaming potential on pressure. The isoelectric point (iep; the iep coincides with the pH of the aqueous solution where ζ = 0 mV) was determined by a pH scan of the zeta potential in the range of pH 6–9.5. At each pH, the zeta potential was recorded four times. A 0.001 mol/l KCl solution was used as the background electrolyte and the pH was adjusted automatically with 0.05 mol/l KOH. Prior to the streaming potential, the Ohm resistance
R inside the capillary channel was measured under stagnant (no-flow) conditions. The electric conductance, i.e., the inverse Ohm resistance, is an indicator for the swelling propensity of the chitin–chitosan nanocomposite films. To compensate for the effect of a variation in the electrolyte conductivity of the bulk solution, either due to a shift in the solution temperature or due to an increase in the ionic strength by the addition of 0.05 mol/l KOH, the cell constant of the capillary channel calculated according to
was evaluated and correlated with the swelling propensity instead of either the Ohm resistance or the conductance.
L,
A,
W, and
H are the length, cross-section, width, and height of the rectangular flow channel between chitin–chitosan nanocomposite films.
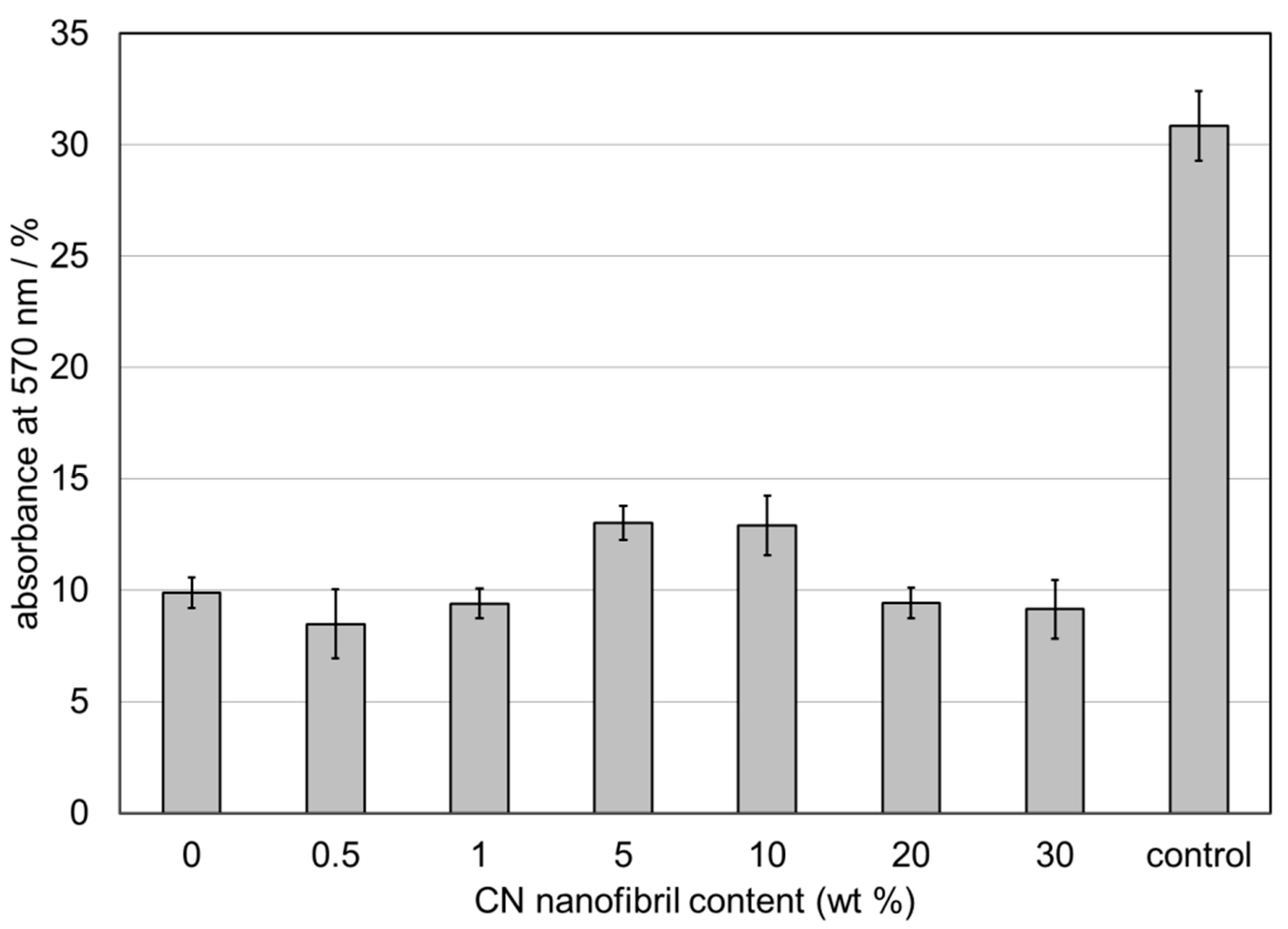
2.6. Fibroblast Attachment, Viability, Proliferation, and Morphology
Human dermal fibroblasts obtained from fresh skin biopsy were from the Institute of Cytology, RAS (St. Petersburg, Russia) and were used up to the 10th passage. The cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 1% penicillin, 1% streptomycin, 1% fungizone, 2 mM L-glutamine, and 10% fetal bovine serum (FBS). The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. For the experiments, fibroblasts from pre-confluent cultures were harvested with 0.25% trypsin/EDTA. Trypsin was neutralized with FBS and the cells were re-suspended in DMEM (all Thermo Fisher Scientific, Waltham, MA, USA).
Studies of the viability and proliferation of the cells in the samples were performed using the tetrazolium bromide (MTT) test. Film matrices were placed in 24-well plates, and culture medium was added. Sterilized silicone rings were placed on top to keep the scaffolds submersed, and 25 × 103 fibroblasts were seeded on top of the scaffolds. The cells were resuspended in culture medium and were then incubated in a humidified atmosphere of 5% CO2. Following 4 days of incubation, the culture medium was removed and each well was treated with 10 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Thermo Fisher Scientific, Waltham, MA, USA), 5 mg/mL in culture medium, and was incubated for 4 hours at 37°C in a humidified atmosphere of 5% CO2. The yellow MTT is reduced to blue-purple formazan in the presence of the mitochondrial dehydrogenase. This enzyme is present in intact living cells, hence the blue-purple color produced should be proportional to the number of viable cells present. The MTT solution was then replaced with 100 µL/well of dimethylsulfoxide (DMSO, Paneco Ltd., Moscow, Russia) to dissolve the formazan salts, followed by 10 minutes of slow agitation, yielding a blue-purple solution. The absorbance of this solution was measured at 570 nm using a SPECTROstar® Nano microplate reader for absorbance measurements (BMG LABTECH, Ortenberg, Germany).
Studies of the cellular morphology and attachment were observed using an inverted light microscope Primo Vert (Zeiss, Oberkochen, Germany) and images were captured with a digital camera.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







