Elastic Properties and Electronic Properties of MxNy (M = Ti, Zr) from First Principles Calculations

College of Science, Hohai University, Nanjing 210098, China
*
Author to whom correspondence should be addressed.
Materials 2018, 11(9), 1640; https://doi.org/10.3390/ma11091640
Submission received: 10 August 2018
/
Revised: 28 August 2018
/
Accepted: 29 August 2018
/
Published: 7 September 2018
(This article belongs to the Section Manufacturing Processes and Systems)
Abstract
:The elastic properties and electronic properties of MxNy (M = Ti, Zr) TiN, Ti2N, Zr3N4, ZrN with different structures have been investigated using density functional theory. Through the calculation of the elastic constants, it was found that all of these structures meet the mechanical stability except for ZrN with space group P63mc. Their mechanical properties are studied by a comparison of various parameters. The stiffness of TiN is larger than that of ZrN with space group Fmm. Ti2N’s stiffness with space group I41/amdz is larger than Ti2N with space group P42/mnm. Zr3N4’s stiffness with space group Pnam is largest in three structures of Zr3N4. TiN, Ti2N and ZrN are non-central force, Zr3N4 is central force. TiN and ZrN with space group Fmm are brittle, and TiN is brittler than ZrN with space group Fmm. The two kinds of Ti2N are brittle and Ti2N with space group I41/amdz is larger. Three structures of Zr3N4 are tough and Zr3N4 with space group I3d is the toughest. Meanwhile, the electronic properties of TiN, Ti2N, Zr3N4 and ZrN were calculated, possible superconducting properties of the studied materials were predicted.
1. Introduction
In modern society and significant scientific research realms, superhard materials have been investigated by many people [1,2,3,4,5]. Titanium nitride has been widely used as a superhard material in the aerospace industry [6], oil-producing industry [7] and other fields due to its high strength, good heat-resistance and excellent corrosion resistance. Zirconium nitride has aroused interest because it manifests immense potentialities in cutting tools [8] and it is a good superconductor [9] owing to its high superconducting critical temperature. So, it is of high research value to investigate the properties of titanium nitride and zirconium nitride. Studying their properties can help us make better use of them as well as giving full play to their value.
Therefore, on account of the importance in fundamental science and technological applications, there has been plenty of research about the structural and physical properties of these two kinds of materials in the past few years. Saha et al. used first-principles calculations based on density functional theory to study ZrN’s electronic structure, vibrational spectra and thermal properties [10]. Guo et al. researched cubic zirconium nitride (c-Zr3N4) with the same structure as Th3P4. They found the structural and electronic properties by the local density functional pseudopotential method [11]. Wang et al. adopted an efficacious strain-stress way to compute the elastic stiffness constants of TiN, and got some related properties by first-principles calculations within the generalized gradient approximation [12]. Ivashchenko et al. studied the electromagnetic properties of Ti2N with two different structures under pressure [13]. Kim et al. obtained the color of TiN and ZrN by first-principles calculations [14]. Some others also have studied TiN, Ti2N, ZrN or Zr3N4’s properties in a specific aspect [15,16,17,18,19,20]. When we use the Inorganic Crystal Structure Database, we can find that these crystals have many different lattice parameters and space groups. The elastic properties and electronic structures of these substances have never been compared by unified research so far. So, we studied them and made comparative analyses.
The first-principles calculation with the pseudopotential method based on density functional theory (DFT) has been rapidly developed into a standard tool for material modeling simulation in the fields of physics, mechanics and material science [21,22,23]. Using density functional theory (DFT) and generalized gradient approximation (GGA) can calculate the various properties of the crystals. In this paper, we used the first-principles calculation with pseudopotential density functional theory (PDFT), the generalized gradient approximation (GGA) and the quasi-harmonic Debye model to build different structures of ZrN, Zr3N4, TiN and Ti2N. After that, the stabilities of these structures were predicted by simulation. By studying the elastic properties and electronic properties of ZrN, Zr3N4, TiN and Ti2N, the change rules were analyzed and summarized.
2. Computational Method
With the purpose of calculating the exact properties of the materials, all calculations were computed by using the CASTEP [24] software package of Materials Studio software created by Accelrys (San Diego, CA, USA) according to the density functional theory (DFT). The atoms calculated are Zr (4s24p64d25s2), Ti (3s23p63d24s2) and N (2s22p3). We used the combination of the Perdew-Burke-Ernzerhof (PBE) system and the generalized gradient function (GGA) to calculate the exchange-correlation energy [25]. Two kinds of Ti2N, two kinds of ZrN and three kinds of Zr3N4 were studied as shown in Figure 1. At the same time, Ti and Zr are subgroup elements and TiN’s space group is the same as ZrN, so TiN was also studied as a contrast. We set the suitable cutoff energy and K point as shown in Table 1. Based on these parameters, we can ensure that the total energy of these substances converges from a proper set. Calculated structural data under zero pressure are given in Table 2. We have studied the elastic properties using the optimized stable structure.
3. Results and Discussion
3.1. Result of the Elastic Properties
TiN, ZrN with space group Fmm and Zr3N4 with space group I3d belong to the cubic system, and their elastic tensors have three independent components , and . Its equations are derived from reference [26]. All Ti2N and ZrN with space group P63mc belong to the tetragonal system, whose elastic tensors have six independent components , , , , and . Zr3N4 with space group Pna21 and Zr3N4 with space group Pnam belongs to the orthonormal system, whose elastic tensors have nine independent components , , , , , , , and . Its equations are derived from reference [27]. On account of the fact that the Voigt band is acquired by the average polycrystalline moduli, it is the upper band of the real modulus. Meanwhile, the Reuss band is acquired by hypothetical stress, so it describes the lower limit. The arithmetic mean of the two bands is called the Voigt-Reuss-Hill approximation. B represents bulk modulus, and G expresses shear modulus. By using the subscript which indicates the Voigt band, Reuss band and Hill average by V, R and H, respectively, they are calculated by the following equations.
For cubic system:
The criterion of mechanical stability is
For the tetragonal system:
The criterion of mechanical stability is
For the orthonormal system
The criterion of mechanical stability is
BV, BR, GV, GR of different substances were obtained by the calculation of the Equations (1)–(14). Under the Voigt-Reuss-Hill approximation, it can be found that the modulus of polycrystal is the mean value under the Voigt bound and Reuss bound,
Young’s modulus (E) and poisson’s ratio () were obtained by these equations:
The associated Equations (15)–(18) can calculate the relevant physical quantities. From the relevant literature, we know that G indicates the anti-plastic deformability of materials and B indicates the resistance to fracture of materials [28]. Pugh proposed the estimation of the ductility of a given material by the ratio of G/B between the shear modulus G and the bulk modulus B [29]. According to his theory, the small G/B values represent the toughness of the corresponding materials, while the larger G/B values represent brittleness. G/B = 0.57 is the critical value. The results we got were written in Table 3, Table 4, Table 5 and Table 6.
First of all, according to the results, ZrN with space group P63mc fails to meet the conditions of mechanical stability. So, it is necessary to compare ZrN and TiN in the same space group. The data show that the Young’s modulus of TiN is larger than that of ZrN. It indicates that the stiffness of TiN is larger than that of ZrN with space group Fmm. Poisson’s ratios can show the binding force of the atom [30]. Poisson’s ratios of TiN and ZrN are both less than 0.25, so they are non-central force. G/B of TiN is 0.665, which shows that TiN is brittle. With regard to ZrN, G/B is 0.615. Therefore, ZrN with space group Fmm is brittle, and there is a reduction from TiN to ZrN. For Ti2N, we found that Ti2N’s stiffness with space group I41/amdz is larger than that with space group p42/mnm. Poisson’s ratios are the same, and they are non-central force. G/B of Ti2Nwith space group I41/amdz is 0.69. At the same time, G/B of Ti2Nwith space group P42/mnm is 0.665. The two kinds of Ti2N are brittle and Ti2N with space group I41/amdz is larger. As for Zr3N4, Zr3N4 with space group Pnam has the largest stiffness; the second largest is Zr3N4 with space group Pna21. Poisson’s ratios are the opposite, but all of them are central force. Meanwhile, three structures of Zr3N4 are tough and Zr3N4 with space group I3d is the toughest. The remaining two substances are not very different, which proves that the symmetry of the structure has little effect on this property.
3.2. Result of the Electronic Properties
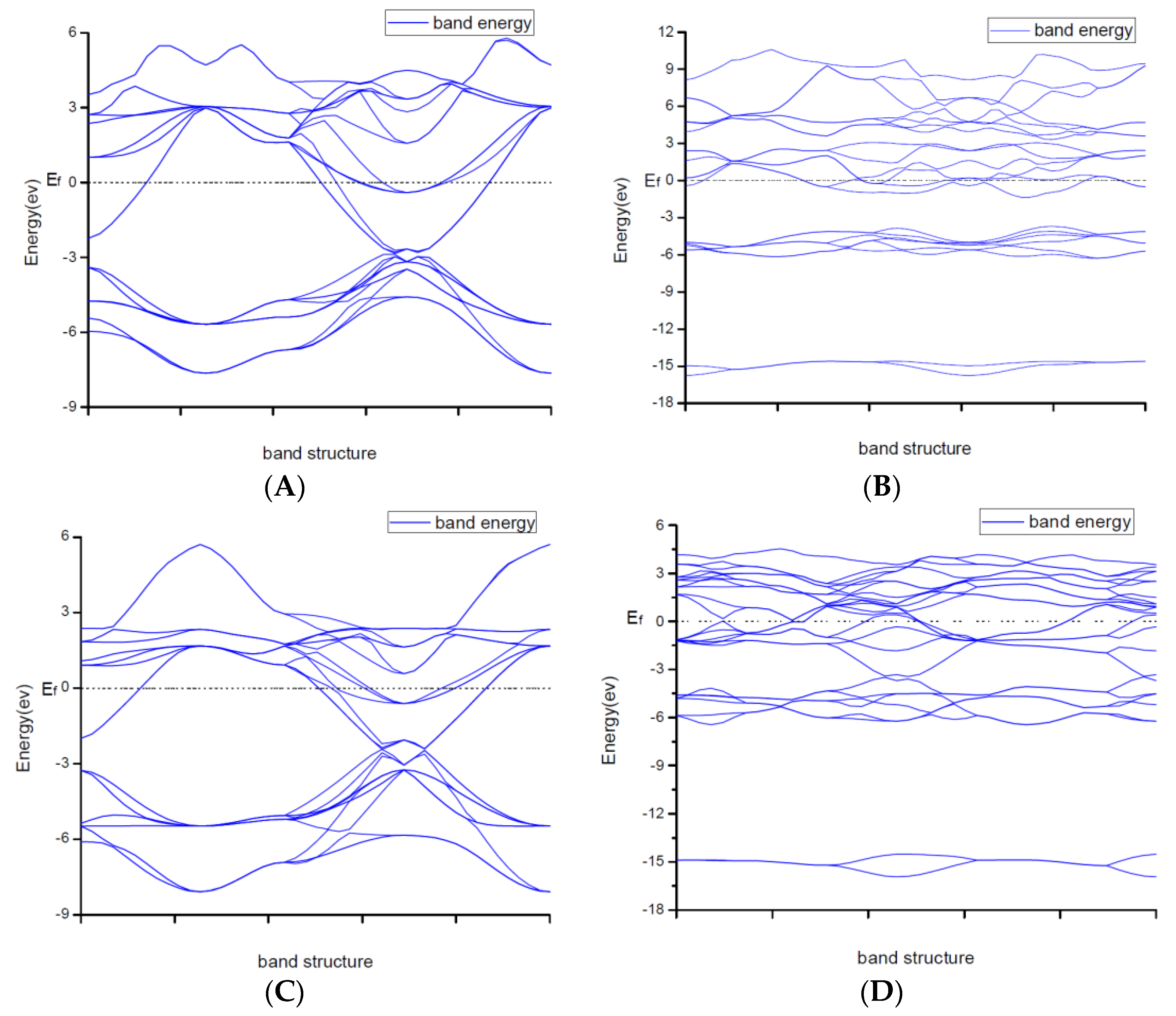
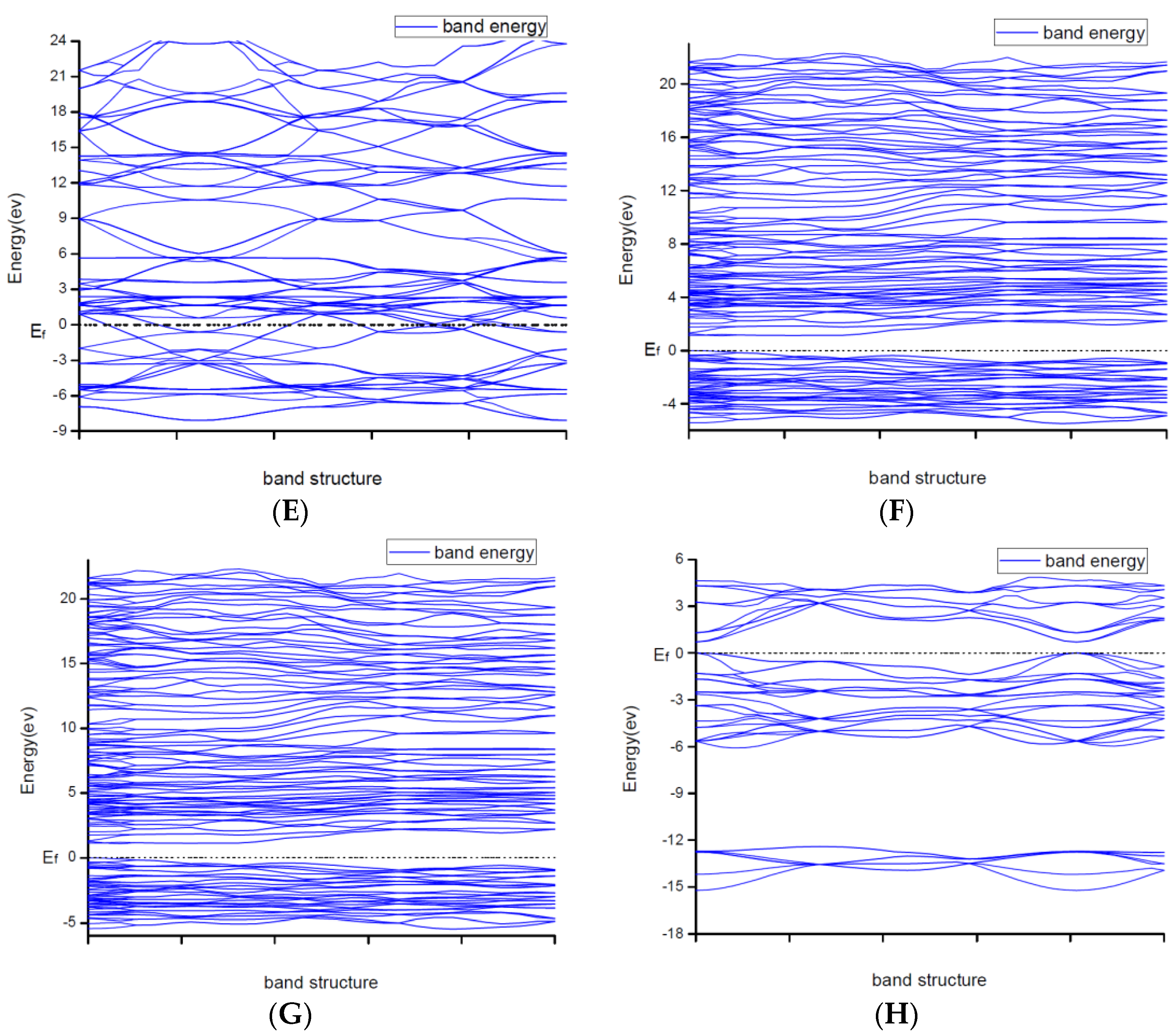
Due to explaining the macroscopic properties, we investigated the electronic band structures, density of states and difference of the charge density of all structures under zero pressure. Figure 4 shows the calculated band structures along the high symmetry directions in the Brillouin zone. It is worth mentioning that DFT cannot give reliable results for the energy gap because DFT does not consider the correlation effect of electrons in 3d orbit and 4f orbitin some strongly correlated systems. As a result, band gaps became smaller. So, the judgement on whether the system is metallic or insulating and on the value of the gap was semi-quantitative.
From Figure 2, the conduction band minimum and valence band maximum of TiN, ZrN with space group Fmm and Ti2N with two kinds of structures are located at the G-point with a very small band gap. The conduction band minimum and valence band maximum of ZrN with space group P63mc is located at the M-point. The conduction band minimum and valence band maximum of Zr3N4 with space group I3d is located at the G-point. From the results, their band gaps are less than 1 eV. So, for these substances, electrons can easily gain energy at room temperature and jump to the transfer band to conduct electricity. The conduction band minimum and valence band maximum of Zr3N4 with space groups Pna21 and Pnam are located at the Z-point with band gap between1eVand 3eV. These two substances are between conductors and insulators. Therefore, they are electrically conductive as long as the appropriate energy is given or the gaps between their energy are changed. It indicates that the electrical conductivity of ZrN with space group Fmm is better than that of ZrN with space group P63mc. For TiN and ZrN with the same space group, TiNhas smaller electron effective mass and larger atomic non-localization than ZrN due to the bigger width of band structure. For three different structures of Zr3N4, symmetry and asymmetry have little influence on the electronic properties. The electrical conductivities of Zr3N4 with space groups Pna21 and Pnam are better than that of Zr3N4 with space group I3d. Ti2N with space group I41/amdz has a smaller electron effective mass and a degree of non-localization with stronger atomic orbital extension than Ti2N with space group P42/mnm.
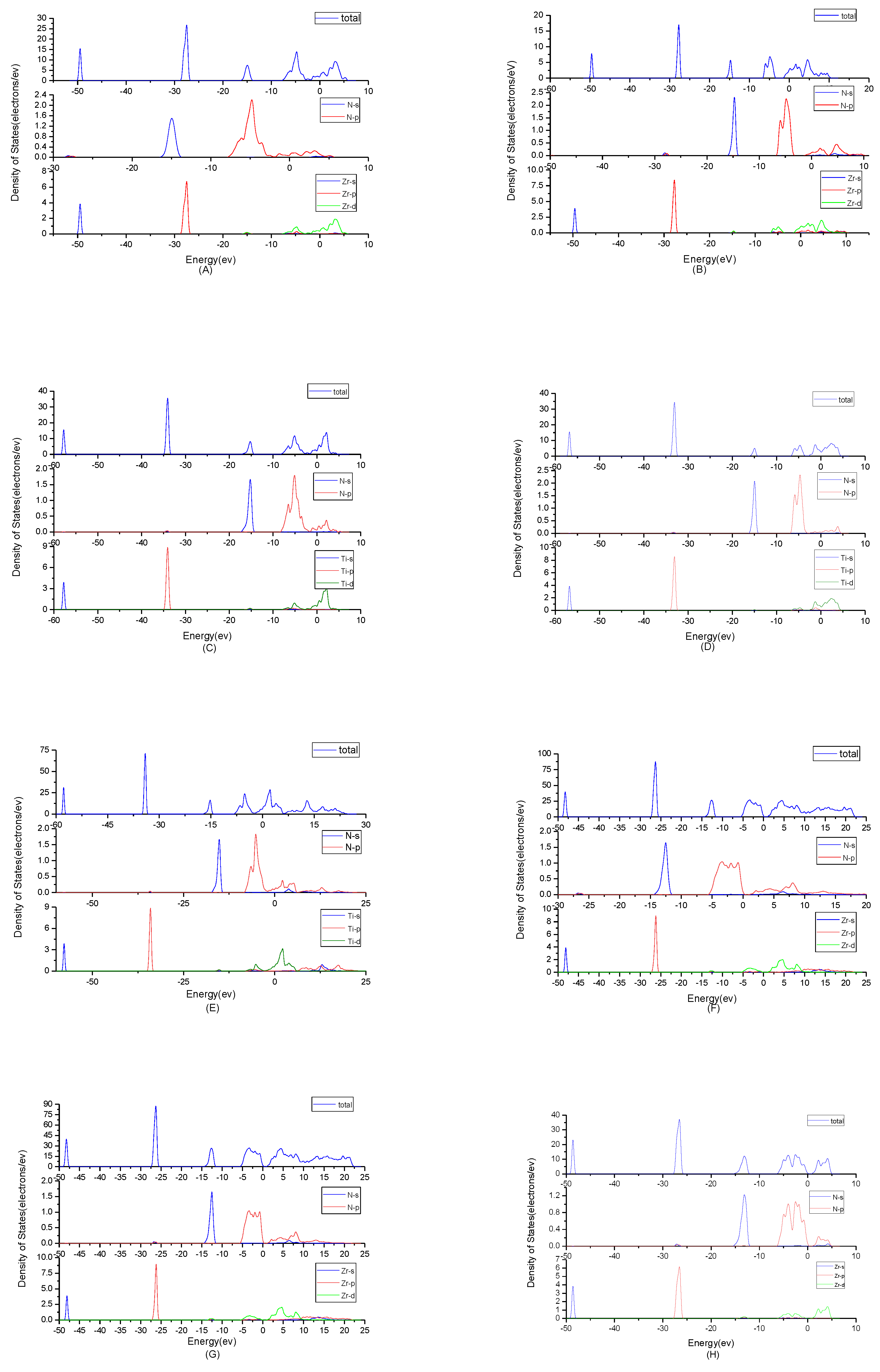
To find origins of band structures, the total density of states and partial density of states were calculated as shown in Figure 3. PBE cannot accurately show the weak interaction between molecules, because it will make the values of d track and f track be slightly lower than the results of dispersion correction and there will besome differences in the hybridization at the top of the conduction band. But the corrected value is very small and will not affect the conclusion of this work. At the same time, the focus of this study is near the Fermi level, where the situation will not change substantially due to the effect of dispersion. By observing the density of states of ZrN, the results showed that the contributions of the atoms to the energy band are similar. N2p and Zr4d have obvious peaks at −5 eV, and they have a state density resonance, so N2p and Zr4d are bonded. Zr4d, Zr4P and Zr5s have a state density contribution to N2p, so they also form covalent bonds. Meanwhile, ZrN with space group P63mc has a greater span of density of state and stronger domain. Accordingly, it has stronger bonds. For TiN, N2p have obvious peaks at Ti3d; they are bonded. In addition, Ti4p and Ti4s have a part in the contribution of state density to N2p. Ti’s s orbit and p orbit are the main sources of the valence band. N’s s orbits, p orbits and Ti’s have a small contribution. The d orbit of Ti and the p orbit of N are the main sources of the guide band. As for Ti2N, Ti4p, Ti4s and Ti3d are bonded respectively to N2s and N2p in the two structures. The s, p orbit of Ti (sTi and pTi) and N (sN and pN) are the main sources of the valence band. dTi orbit has a small contribution. For Ti2N with space group P42/mnm, the d orbit of Ti (dTi) and the p orbit of N (pN) are the main sources of the guide band. For another structure of Ti2N, the sTi, dTi and pTi orbits are the main sources of the guide band. The sN orbits and pN orbits have some contributions. Finally, according to the density of states of Zr3N4, the differences between Zr3N4 with space group Pna21 and Zr3N4 with space group Pnam are minimal; this situation shows that the symmetry of the space group has little effect on it. For Zr3N4 with space group I3d, sN orbits, pZr orbits and dZr orbits form covalent bonds. The 2pN forms covalent bonds with 5sZr, 4pZrand 4dZr. For the other two structures, 2pN forms covalent bonds with 5sZr, 4dZr and 4pZr and 2sN forms covalent bonds with 4dZr and 4pZr. At the same time, Zr3N4 with space group Pna21 and Zr3N4 with space group Pnam have a greater span of density of state and stronger domain. Therefore, they have stronger bonds.
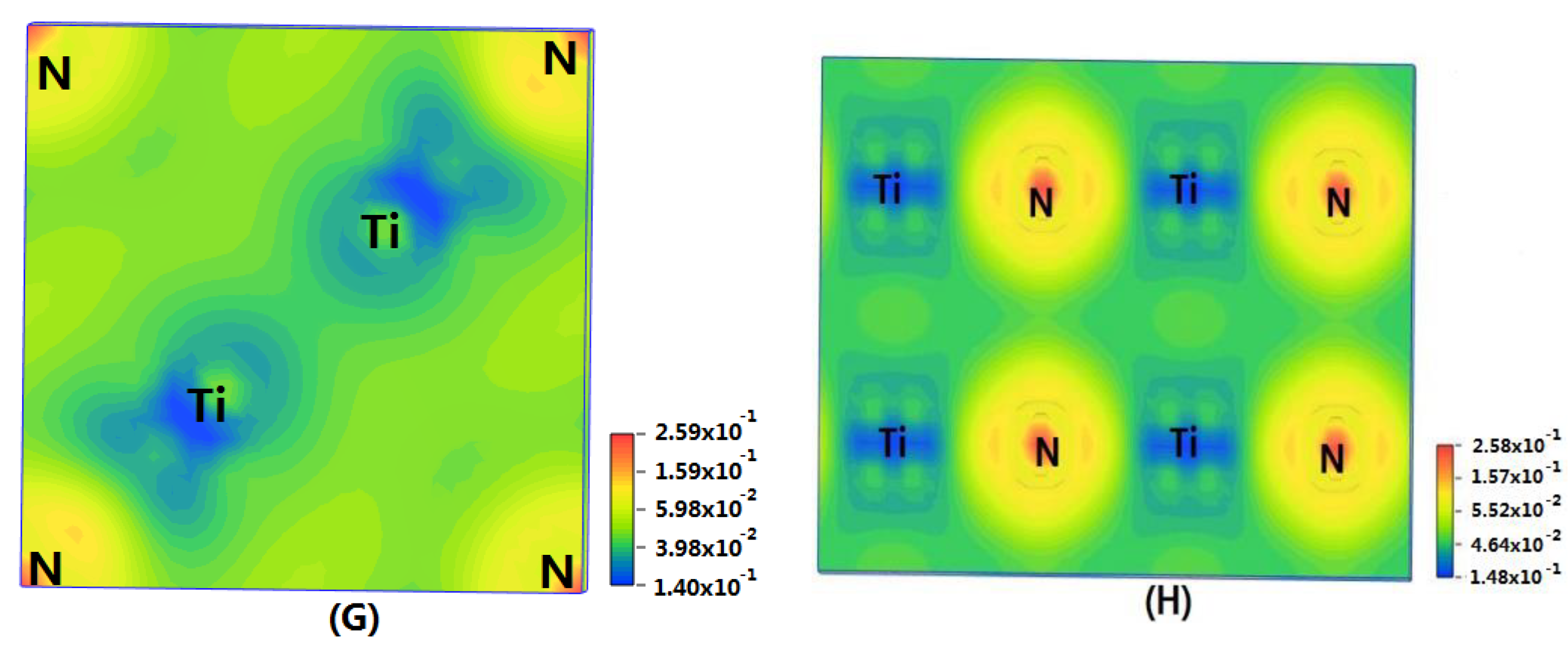
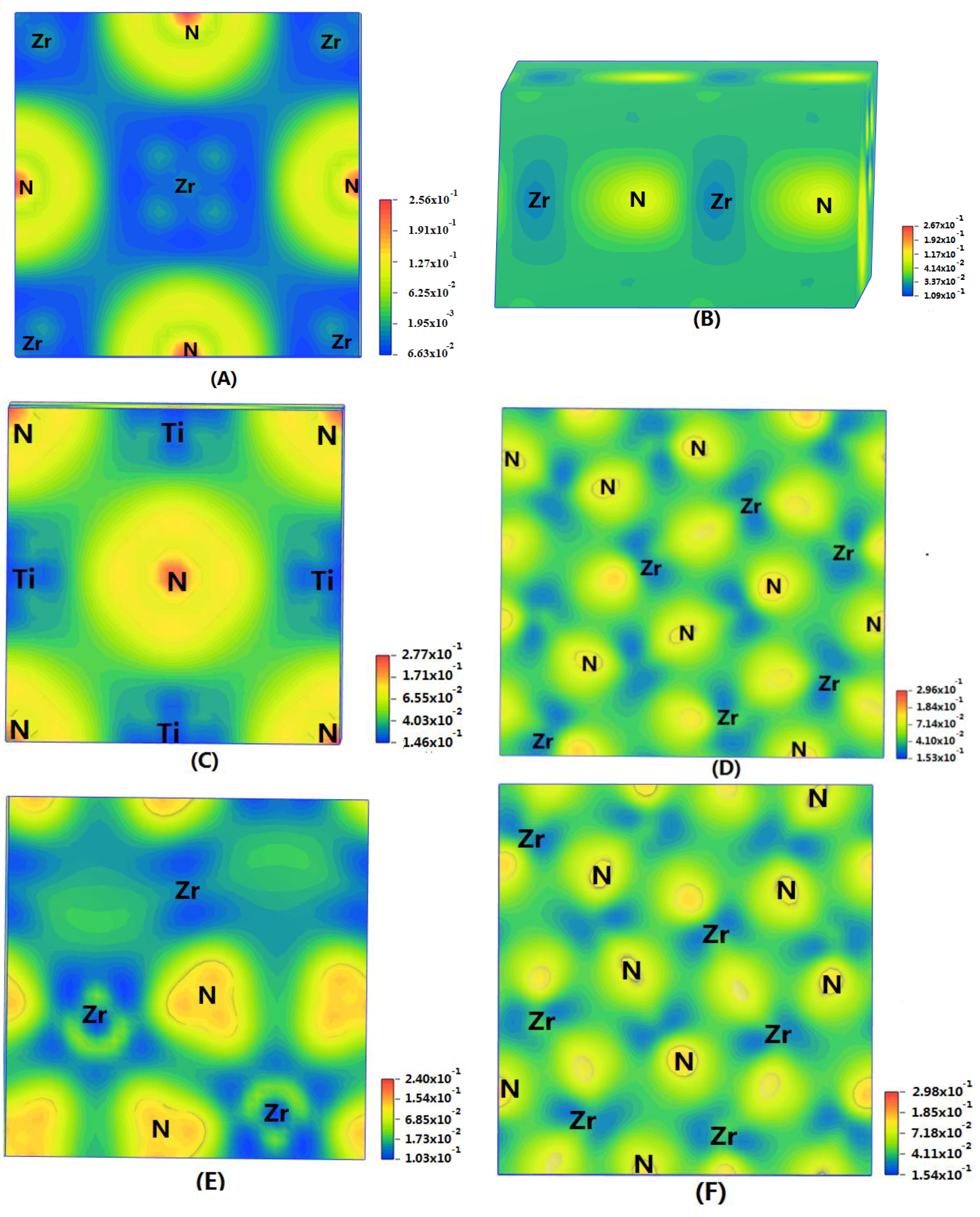
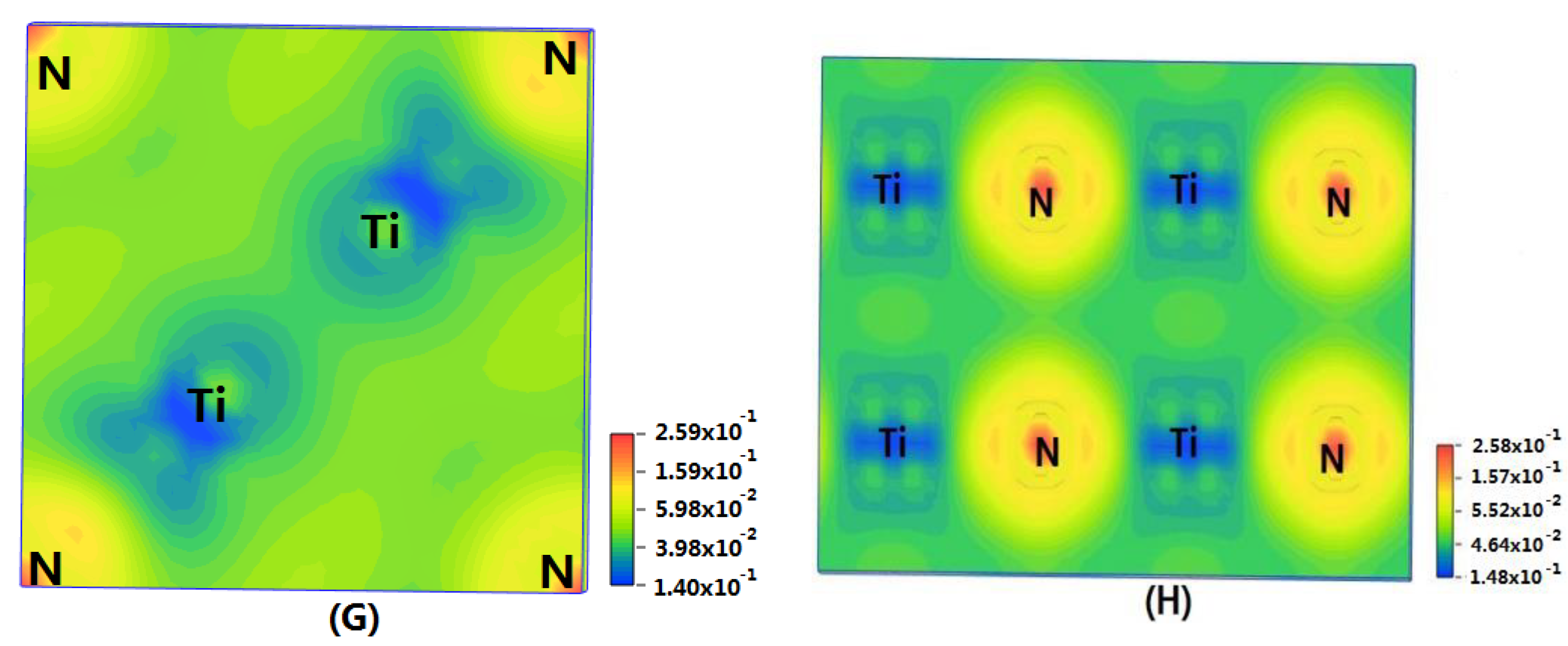
In order to show the bonding between atoms of different crystals more intuitively, difference charge density maps are shown in Figure 4. According to plot A and plot C, for TiN and ZrNwith space group Fmm, the charge densities between Ti and N are larger than that of Zr and N, which means that the effects between Ti–N are stronger. Meanwhile, ZrN with space group Fmm has stronger Zr–N bonds than ZrN with space group P63mc. So, the interatomic interaction of ZrN with space group Fmm is greater than that of ZrN with space group P63mc. For three different structures of Zr3N4, symmetry and asymmetry have little influence on the electronic properties. There are larger charge densities between Zr and N in Zr3N4 with space group Pna21 and Zr3N4 with space group Pnam than the other structure. So, there are stronger interactions between their atoms. The last two graphs (Figure 4G,H) represent the charge density of TiN with two different structures. Ti2N with space group P42/mnm has a larger charge density than Ti2N with space group I41/amdz. Therefore, the interatomic effect of Ti2N with space group P42/mnm is stronger.
3.3. Superconducting Properties
After obtaining the elastic properties and electronic properties of the studied materials, the possible superconducting properties were discussed based on the results. According to the simplified theory of superconductivity, if a material can become a superconductor, three conditions must be satisfied. First of all, atoms that make up crystals are lighter. Second, the coefficient of elasticity of the crystal is as large as possible, and crystals are relatively tough. Third, the effective Fermi level of materials should be low. Zr and Ti are two of the 28 superconducting elements, as their nitrogen compounds satisfy the first condition. From the results of the elastic properties, TiN is tougher than ZrN when used as superconducting material. Ti2N with space group I41/amdz is better than Ti2N with space group P42/mnm. The three structures of Zr3N4 all have potential as superconducting materials; Zr3N4 with space group I3d is the best of them. From the third point of view, metal systems are more likely to superconduct. So, TiN, ZrN and Zr3N4 with space group I3d are better choices to satisfy the third condition. Combining three characteristics, Zr3N4 with space group I3d is most likely to have superconductivity in all materials. TiN is more likely to be used as a superconducting material than ZrN with same space group. Ti2N with space group I41/amdz is more suitable to be used as superconducting material than Ti2N with space group P42/mnm.
4. Conclusions
In short, we have investigated the elastic properties and electronic properties of the four materials TiN, Ti2N, ZrN and Zr3N4 with different structures by using the first principles method.
In order to study the elastic properties, we have calculated elastic stiffness constants Cij, bulk modulus B, shear modulus G, Young modulus E, Poisson’s ratio and G/B. Research on Young’s modulus indicated that the stiffness of TiN is larger than that of ZrN with space group Fmm. Ti2N’s stiffness with space group I41/amdz is larger than that with space group p42/mnm. Zr3N4 with space group Pnam’s stiffness is largest; the second is Zr3N4 with space group Pna21. For Poisson’s ratio, TiN, Ti2N and ZrN are non-central force, Zr3N4 are central force. Meanwhile, G/B shows that TiN and ZrN with space group Fmm are brittle, and TiN is larger than ZrN. The two kinds of Ti2N are brittle and Ti2N with space group I41/amdz is larger. Three structures of Zr3N4 are tough and Zr3N4 with space group I3d is the toughest. It is worth mentioning that ZrN with space group P63mc fails to meet the conditions of mechanical stability.
Based on the analysis of band structures, density of states and the difference in the charge density of these substances, their electronic properties are clear at a glance. The electrical conductivity of ZrN with space group Fmm is better than that of ZrN with space group P63mc. The interatomic interaction of ZrN with space group Fmm is greater than that of ZrN with space group P63mc. Contributions of the atom of ZrN to the energy band are similar. However, ZrN with space group P63mc has a greater span of density of state and stronger domain. Accordingly, it has stronger bonds. For TiN and ZrN with the same space group, TiN has a smaller electron effective mass and larger atomic non-localization than ZrN due to the bigger width of band structure. The charge density between Ti and N is larger than that of Zr and N, which means that the effect between Ti–N is stronger. For three different structures of Zr3N4, symmetry and asymmetry have little influence on the electronic properties. Because the electrical conductivities of Zr3N4 with space groups Pna21 and Pnam are better than that of Zr3N4 with space group I3d, the interactions between their atoms are stronger. They have a greater span of density of state and stronger domain. Therefore, they have stronger bonds. Ti2N with space group I41/amdz has smaller electron effective mass and the degree of non-localization with stronger atomic orbital extension than Ti2N with space group P42/mnm. The interatomic effect of Ti2N with space group P42/mnm is stronger. For Ti2N with space group P42/mnm, the d orbit of Ti and the p orbit of N are the main sources of the guide band. For another structure of Ti2N, the s, d and p orbit of Ti are the main sources of the guide band. N’s s orbits and p orbits have some contribution. They have different orbital bonding methods.
From the elastic and electronic results, Zr3N4 with space group I3d is most likely to have superconductivity in all materials. TiN is more likely to be used as superconducting material than ZrN with same space group. Ti2N with space group I41/amdz is more suitable to be used as superconducting material than Ti2N with space group P42/mnm.
Author Contributions
Supervision, X.Y.; Writing–original draft Preparation, Y.J.
Funding
This research was funded by the Fundamental Research Funds for the Central Universities grant number: [2015B03814], the National Natural Science Foundation of China grant number: [11402072], and Jiangsu Province Natural Science Foundation grant number: [BK20140840].
Conflicts of Interest
The authors declare no conflict of interest.
References
- Veprek, S.J. The search for novel, superhard materials. Vac. Sci. Technol. A 1999, 17, 2401. [Google Scholar] [CrossRef]
- Léger, J.M.; Haines, J. The search for superhard materials. Endeavour 1997, 21, 121–124. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Gregoryanz, E. Synthesis of superhard materials. Mater. Today 2005, 8, 44–51. [Google Scholar] [CrossRef]
- Kaner, R.B.; Gilman, J.J.; Tolbert, S.H. Designing superhard materials. Mater. Sci. 2005, 308, 1268. [Google Scholar]
- Richter, A.; Smith, R.; Dubrovinskaia, N.; Mcgee, E. Mechanical properties of superhard materials synthesised at various pressure-temperature conditions investigated by nanoindentation. High Press. Res. 2006, 26, 99–109. [Google Scholar] [CrossRef]
- Zalnezhad, E.; Sarhan, A.D.; Hamdi, M. Optimizing the PVD TiN thin film coating’s parameters on aerospace AL7075-T6 alloy for higher coating hardness and adhesion with better tribological properties of the coating surface. Int. J. Adv. Manuf. Technol. 2013, 64, 281–290. [Google Scholar] [CrossRef]
- Gardy, J.; Hassanpour, A.; Lai, X.; Ahmed, M.H. Synthesis of Ti(SO4)O solid acid nano-catalyst and its application for biodiesel production from used cooking oil. Appl. Catal. A Gen. 2016, 527, 81–95. [Google Scholar] [CrossRef]
- Siow, P.C.; Ghani, J.A.; Ghazali, M.J.; Jaafar, T.R.; Selamat, M.A.; Haron, C.H.C. Characterization of TiCN and TiCN/ZrN coatings for cutting tool application. Ceram. Inter. 2013, 39, 1293–1298. [Google Scholar] [CrossRef]
- Weht, R.; Filippetti, A.; Pickett, W.E. Electron doping in the honeycomb bilayer superconductors (Zr, Hf) NCl. Europhys. Lett. 2007, 48, 320–325. [Google Scholar] [CrossRef]
- Saha, B.; Acharya, J.; Sands, T.D.; Waghmare, U.V. Electronic structure, phonons, and thermal properties of ScN, ZrN, and HfN: A first-principles study. J Appl. Phys. 2010, 107, 960. [Google Scholar] [CrossRef]
- Guo, Q.X.; Kwan, W.K.; Cheng, X.L.; Zhang, H. First-principles study of the structural and electronic properties of the cubic Zr3N4, under high pressure. Phys. Status Solidi 2010, 247, 67–71. [Google Scholar] [CrossRef]
- Wang, A.J.; Shang, S.L.; Du, Y.; Kong, Y.; Zhang, L.J.; Chen, L.; Liu, Z.K. Structural and elastic properties of cubic and hexagonal TiN and AlN from first-principles calculations. Comput. Mater. Sci. 2010, 48, 705–709. [Google Scholar] [CrossRef]
- Ivashchenko, V.I.; Turchi, P.E.A.; Shevchenko, V.I.; Olifan, E.I. First-principles study of phase stability of Ti2N under pressure. Phys. Rev. B 2012, 86, 2602–2607. [Google Scholar] [CrossRef]
- Kim, J.; Jhi, S.H.; Ryeol, L.K. Color of TiN and ZrN from first-principles calculations. J. Appl. Phys. 2011, 110, 4889. [Google Scholar] [CrossRef]
- Brik, M.G.; Ma, C.G. First-principles studies of the electronic and elastic properties of metal nitrides XN (X = Sc, Ti, V, Cr, Zr, Nb). Comput. Mater. Sci. 2012, 51, 380–388. [Google Scholar] [CrossRef]
- Mattesini, M.; Ahuja, R.; Johansson, B. Cubic Hf3N4, and Zr3N4: A class of hard materials. Phys. Rev. B 2003, 68, 184108. [Google Scholar] [CrossRef]
- Kroll, P. Hafnium nitride with thorium phosphide structure: Physical properties and an assessment of the Hf-N, Zr-N, and Ti-N phase diagrams at high pressures and temperatures. Phys. Rev. Lett. 2003, 90, 125501. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.; Yin, G.; Li, J.; Zheng, Y.; Chen, L.; Jia, Y. Optical properties of cubic Ti3N4, Zr3N4, and Hf3N4. Appl. Phys. Lett. 2006, 89, 151908. [Google Scholar] [CrossRef]
- Xu, Y.X.; Chen, L.; Pei, F.; Chang, K.K.; Du, Y. Effect of the modulation ratio on the interface structure of TiAlN/TiN and TiAlN/ZrN multi-layers: First-principles and experimental investigations. Acta Mater. 2017, 130, 281–288. [Google Scholar] [CrossRef]
- Kobayashi, K. First-principles study of the electronic properties of transition metal nitride surfaces. Surf. Sci. 2001, 493, 665–670. [Google Scholar] [CrossRef]
- Gonze, X.; Beuken, J.M.; Caracas, R.; Detraux, F.; Fuchs, M.; Rignanese, G.M.; Torrent, M. First-principles computation of material properties: The ABINIT software project. Comput. Mater. Sci. 2002, 25, 478–492. [Google Scholar] [CrossRef]
- Shang, S.L.; Wang, Y.; Kim, D.; Liu, Z.K. First-principles thermodynamics from phonon and Debye model: Application to Ni and Ni3Al. Comput. Mater. Sci. 2010, 47, 1040–1048. [Google Scholar] [CrossRef]
- Wang, B.T.; Shi, H.; Li, W.D.; Zhang, P. First-principles study of ground-state properties and high pressure behavior of ThO2. J. Nucl. Mater. 2010, 399, 181–188. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, A.J. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.; Léger, J.M.; Bocquillon, G. Synthesis and design of superhard materials. Annu. Rev. Mater. Res. 2001, 31, 1–23. [Google Scholar] [CrossRef]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Ghebouli, B.; Fatmi, M.; Ghebouli, M.A.; Choutri, H.; Louail, L.; Chihi, T.; Bin-Omran, S. Theoretical study of the structural, elastic, electronic and optical properties of XCaF3, (X = K and Rb). Solid State Sci. 2015, 43, 9–14. [Google Scholar] [CrossRef]
- Chen, X.Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Greaves, G.N.; Greer, A.L.; Lakes, R.S.; Rouxel, T. Poisson’s ratio and modern materials. Nat. Mater. 2011, 10, 823–837. [Google Scholar] [CrossRef] [PubMed]
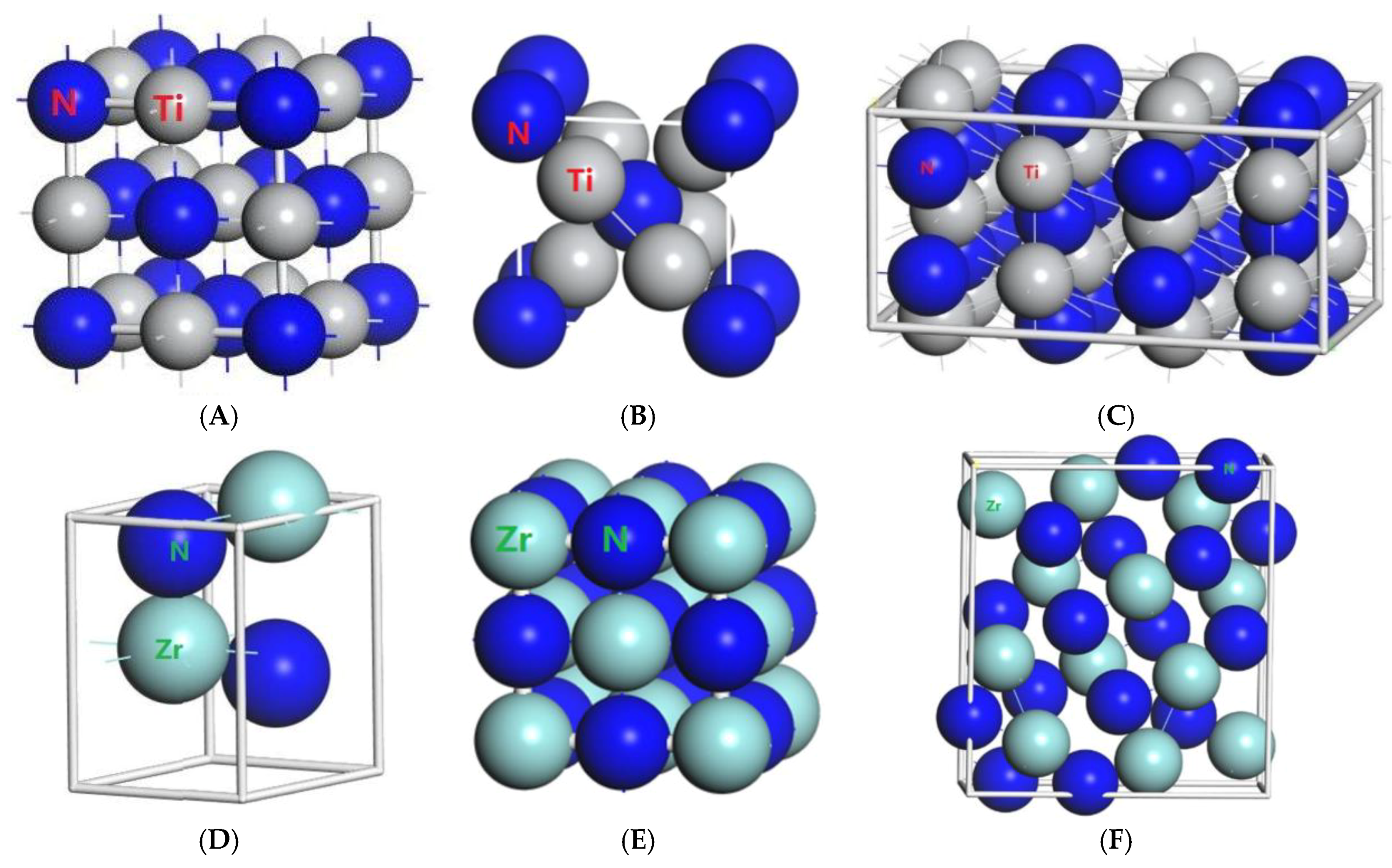
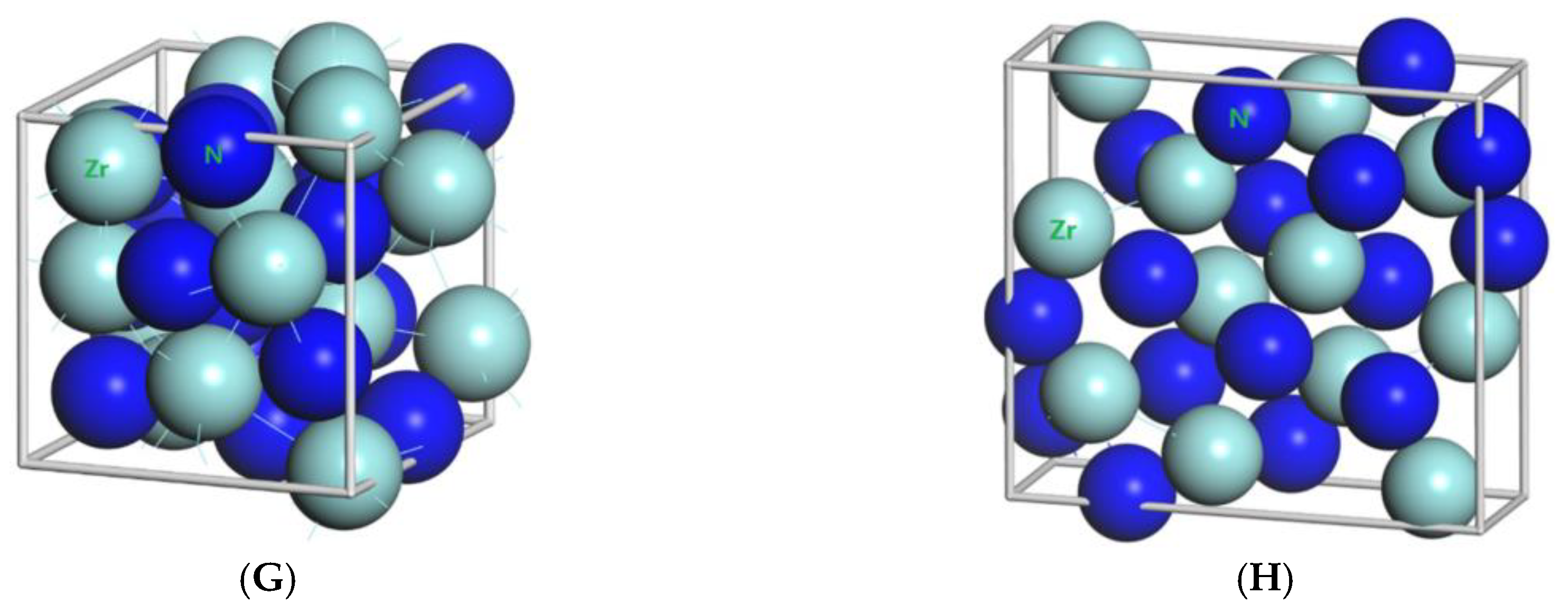
Figure 1.
Primitive unit cells of (A) TiN with space group of Fmm; (B) Ti2N with space group of P42/mnm; (C) Ti2N with space group of I41/amdz; (D) ZrN with space group of P63mc; (E) ZrN with space group of Fmm; (F) Zr3N4 with space group of Pna21; (G) Zr3N4 with space group of I3d; (H) Zr3N4 with space group of Pnam.
Figure 1.
Primitive unit cells of (A) TiN with space group of Fmm; (B) Ti2N with space group of P42/mnm; (C) Ti2N with space group of I41/amdz; (D) ZrN with space group of P63mc; (E) ZrN with space group of Fmm; (F) Zr3N4 with space group of Pna21; (G) Zr3N4 with space group of I3d; (H) Zr3N4 with space group of Pnam.
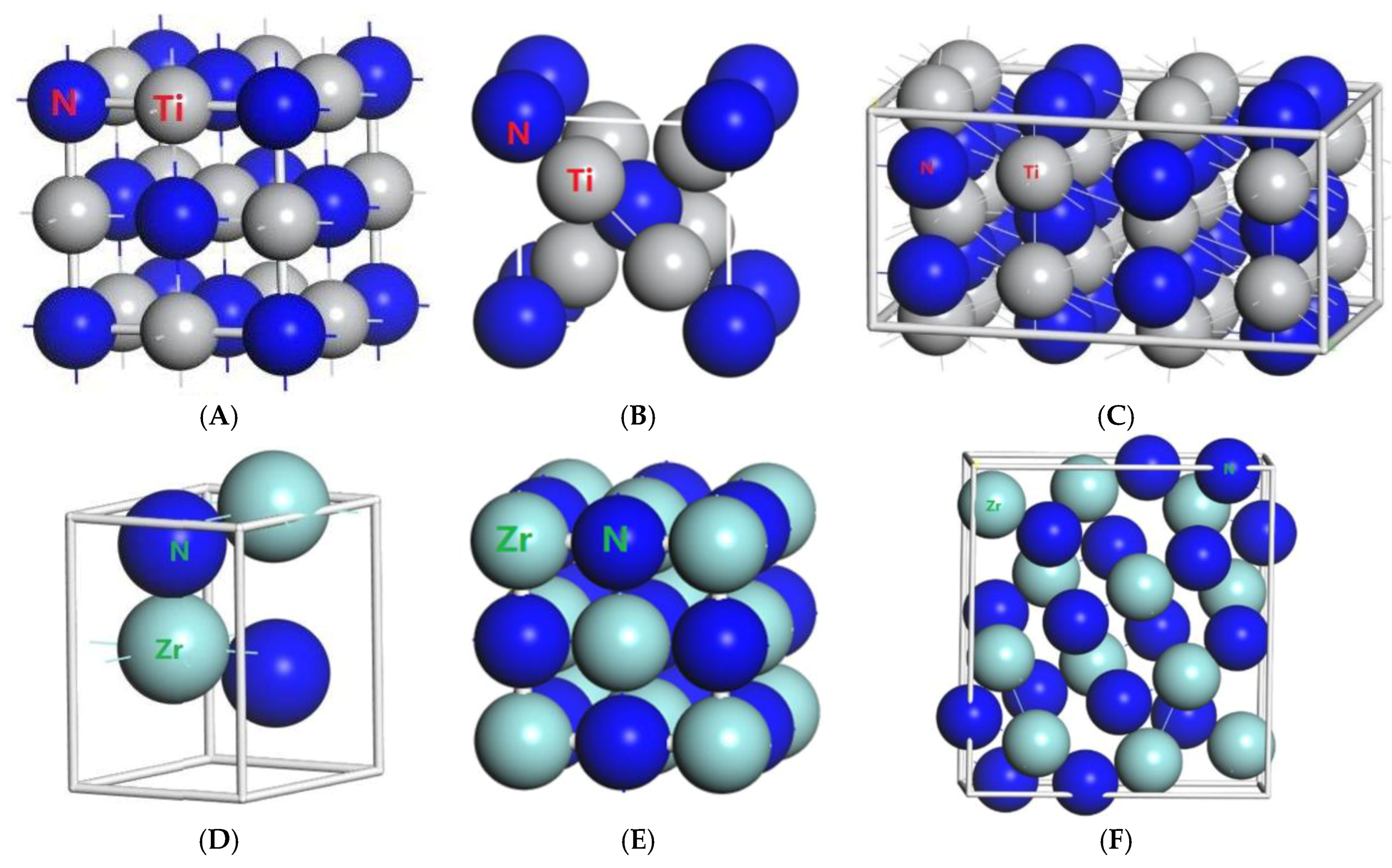
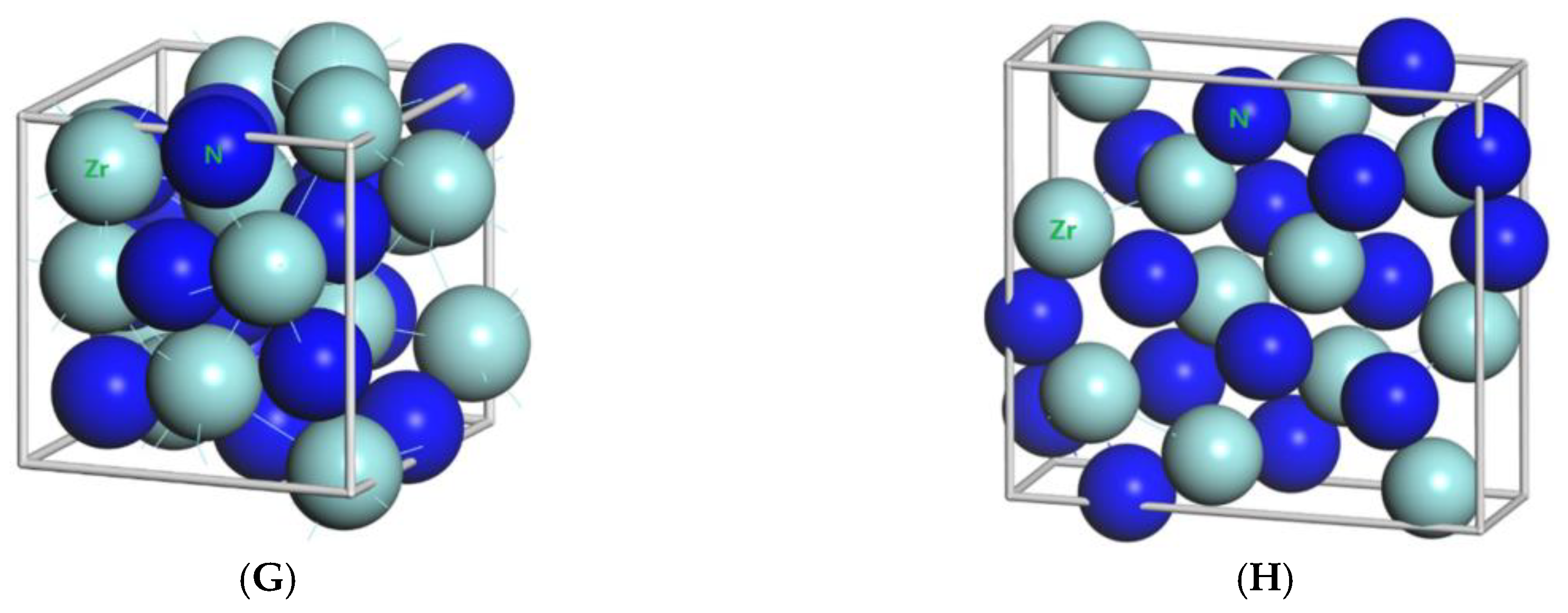
Figure 2.
Band structures of (A) ZrN with space group Fmm; (B) ZrN with space group P63mc; (C) TiN; (D) Ti2N with space group P42/mnm; (E) Ti2N with space group I41/amdz; (F) Zr3N4 with space group Pna21; (G) Zr3N4 with space group Pnam; (H) Zr3N4 with space group I3d.
Figure 2.
Band structures of (A) ZrN with space group Fmm; (B) ZrN with space group P63mc; (C) TiN; (D) Ti2N with space group P42/mnm; (E) Ti2N with space group I41/amdz; (F) Zr3N4 with space group Pna21; (G) Zr3N4 with space group Pnam; (H) Zr3N4 with space group I3d.


Figure 3.
Total density of states plots and partial density of states plots of (A) ZrN with space group Fmm; (B) ZrN with space group P63mc; (C) TiN, (D) Ti2N with space group P42/mnm; (E) Ti2N with space group I41/amdz; (F) Zr3N4 with space group Pna21; (G) Zr3N4 with space group Pnam; (H) Zr3N4 with space group I3d.
Figure 3.
Total density of states plots and partial density of states plots of (A) ZrN with space group Fmm; (B) ZrN with space group P63mc; (C) TiN, (D) Ti2N with space group P42/mnm; (E) Ti2N with space group I41/amdz; (F) Zr3N4 with space group Pna21; (G) Zr3N4 with space group Pnam; (H) Zr3N4 with space group I3d.
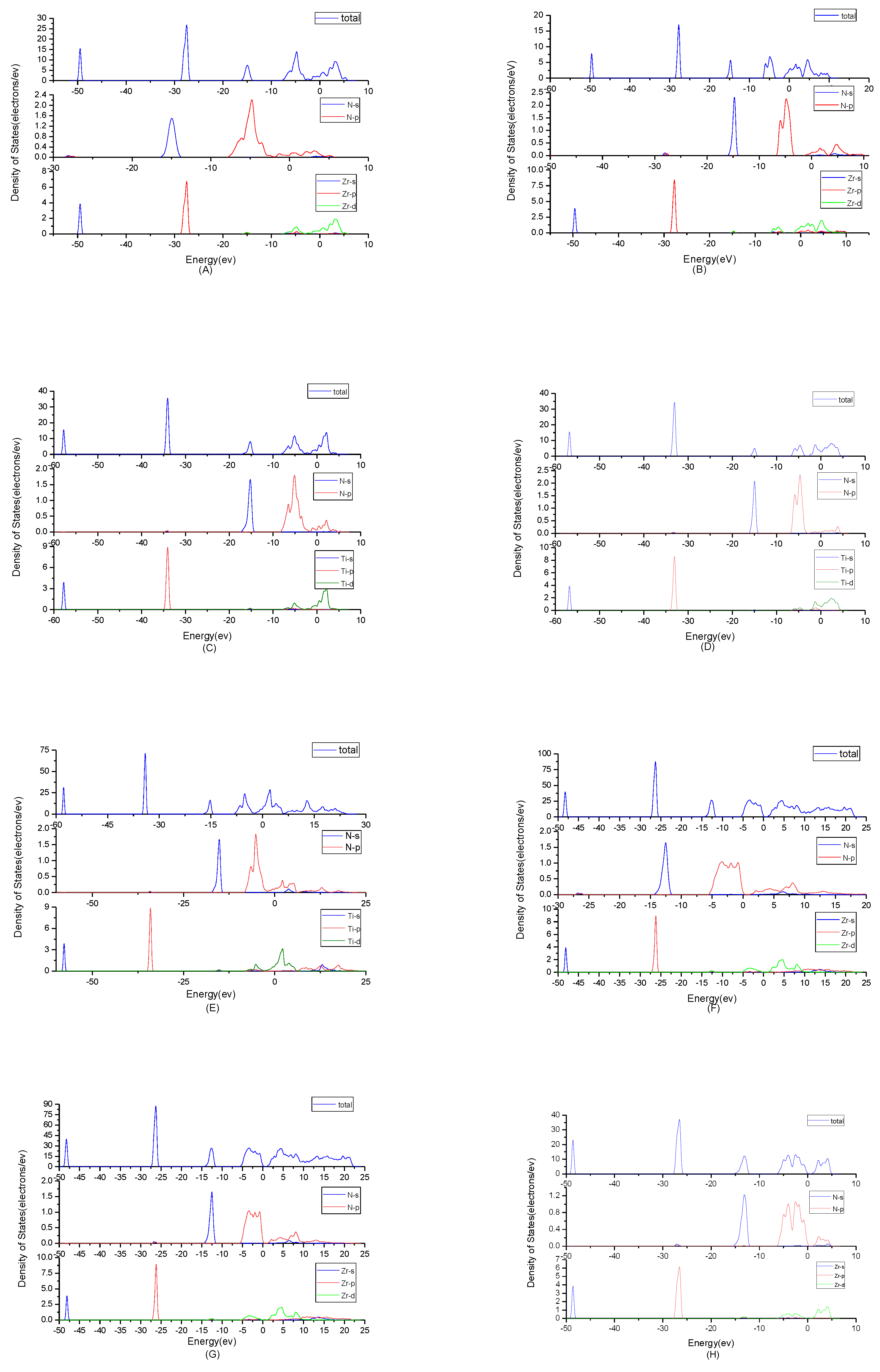
Figure 4.
Difference charge density maps of (A) ZrN with space group Fmm; (B) ZrN with space group P63mc; (C) TiN; (D) Zr3N4 with space group Pna21; (E) Zr3N4 with space group I3d; (F) Zr3N4 with space group Pnam; (G)Ti2N with space group P42/mnm; (H)Ti2N with space group I41/amdz.
Figure 4.
Difference charge density maps of (A) ZrN with space group Fmm; (B) ZrN with space group P63mc; (C) TiN; (D) Zr3N4 with space group Pna21; (E) Zr3N4 with space group I3d; (F) Zr3N4 with space group Pnam; (G)Ti2N with space group P42/mnm; (H)Ti2N with space group I41/amdz.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Lattice parameters a, b, c, cell volume (V), the cutoff energy (eV) and K point of Ti2N, ZrN and Zr3N4.
Table 1.
Lattice parameters a, b, c, cell volume (V), the cutoff energy (eV) and K point of Ti2N, ZrN and Zr3N4.
| Compound | Space Group | a (Å) | b (Å) | c (Å) | V (Å3) | Cutoff Energy (eV) | K Point |
|---|---|---|---|---|---|---|---|
| TiN | Fmm | 4.25 | - | - | 76.77 | 750 | 20 × 20 × 20 |
| Ti2N | I41/amz | 4.149 | 4.149 | 8.786 | 74.20 | 600 | 8 × 8 × 13 |
| P42/mm | 4.945 | 4.945 | 3.034 | 151.26 | 680 | 9 × 9 × 4 | |
| ZrN | Fmm | 4.573 | - | - | 95.76 | 800 | 20 × 20 × 20 |
| P63mc | 3.128 | 3.128 | 5.013 | 42.49 | 720 | 22 × 22 × 6 | |
| Zr3N4 | I3d | 6.74 | - | - | 306.18 | 500 | 15 × 15 × 15 |
| Pna21 | 9.729 | 10.818 | 3.281 | 345.32 | 480 | 15 × 15 × 15 | |
| Pnam | 9.788 | 10.854 | 3.300 | 350.59 | 490 | 6 × 17 × 5 |
Table 2.
Data calculated in this work (some experimental data and some the results of others) of TiN, Ti2N, ZrN and Zr3N4 under zero pressure. (TW = this work, Exp = exsperiment, Cal. = other calculated, Space group = S.G).
Table 2.
Data calculated in this work (some experimental data and some the results of others) of TiN, Ti2N, ZrN and Zr3N4 under zero pressure. (TW = this work, Exp = exsperiment, Cal. = other calculated, Space group = S.G).
| Compound | Data Type | S.G | a (Å) | b (Å) | c (Å) |
|---|---|---|---|---|---|
| TiN | TW | Fmm | 4.246 | - | - |
| Exp. | Fmm | 4.250 | - | - | |
| Cal. | Fmm | 4.256 [12] | - | - | |
| Ti2N | TW | I41/amdz | 6.003 | 6.003 | 8.502 |
| TW | P42/mnm | 4.952 | 4.952 | 3.034 | |
| Cal. | P42/mnm | 4.928 [13] | 4.928 [13] | 3.021 [13] | |
| ZrN | TW | Fmm | 4.591 | - | - |
| Cal. | Fmm | 4.59 | - | - | |
| TW | P63mc | 3.564 [10] | 3.564 [10] | 5.538 [10] | |
| Zr3N4 | TW | I3d | 6.783 | - | - |
| Pna21 | 9.823 | 10.843 | 3.291 | ||
| Pnam | 9.814 | 10.840 | 3.294 | ||
| Exp. | I3d | 6.740 [11] | - | - |
Table 3.
Elastic stiffness constants Cij (GPa) of ZrN with space group Fmm and TiN.
| Compound | S.G | C11 | C12 | C44 |
|---|---|---|---|---|
| TiN | Fmm | 563.93 | 133.28 | 165.91 |
| ZrN | Fmm | 536.17 | 105.19 | 121.21 |
Table 4.
Elastic stiffness constants Cij (GPa) of Ti2N and ZrN with space group P63mc.
| Compound | S.G | C11 | C12 | C13 | C33 | C44 | C66 |
|---|---|---|---|---|---|---|---|
| Ti2N | I41/amdz | 512.26 | 188.64 | 120.05 | 568.49 | 166.22 | 231.47 |
| P42/mnm | 341.57 | 147.80 | 103.12 | 429.89 | 141.11 | 146.57 | |
| ZrN | P63mc | 219.22 | 162.29 | 165.69 | 120.14 | 51.24 | 28.46 |
Table 5.
Elastic stiffness constants Cij (GPa) of Zr3N4.
| S.G | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 |
|---|---|---|---|---|---|---|---|---|---|
| I3d | 332.4 | 119.7 | - | - | - | - | 64.91 | - | - |
| Pna21 | 211.4 | 126.1 | 125.0 | 383.9 | 116.2 | 425.5 | 56.44 | 95.7 | 132.8 |
| Pnam | 215.2 | 126.9 | 126.8 | 389.3 | 109.1 | 428.5 | 55.7 | 96.6 | 134.9 |
Table 6.
Bulk modulus B (GPa), Shear modulus G (GPa), Young’s modulus E (GPa), Poisson’s ratio and G/B of ZrN, Zr3N4, TiN and Ti2N.
Table 6.
Bulk modulus B (GPa), Shear modulus G (GPa), Young’s modulus E (GPa), Poisson’s ratio and G/B of ZrN, Zr3N4, TiN and Ti2N.
| Compound | S.G | B | G | E | ν | G/B |
|---|---|---|---|---|---|---|
| ZrN | Fmm | 248.85 | 152.92 | 380.77 | 0.245 | 0.615 |
| P63mc | 196.59 | −569.37 | - | - | - | |
| TiN | Fmm | 276.83 | 184.18 | 452.24 | 0.2277 | 0.665 |
| Ti2N | I41/amdz | 272.27 | 187.96 | 458.40 | 0.220 | 0.690 |
| P42/mnm | 202.12 | 134.43 | 330.11 | 0.228 | 0.665 | |
| Zr3N4 | I3d | 190.6 | 79.18 | 208.7 | 0.318 | 0.415 |
| Pna21 | 186.5 | 93.69 | 240.8 | 0.285 | 0.502 | |
| Pnam | 187.6 | 94.74 | 243.3 | 0.284 | 0.505 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ji, Y.; Yuan, X. Elastic Properties and Electronic Properties of MxNy (M = Ti, Zr) from First Principles Calculations. Materials 2018, 11, 1640. https://doi.org/10.3390/ma11091640
AMA Style
Ji Y, Yuan X. Elastic Properties and Electronic Properties of MxNy (M = Ti, Zr) from First Principles Calculations. Materials. 2018; 11(9):1640. https://doi.org/10.3390/ma11091640
Chicago/Turabian StyleJi, Yangqi, and Xiaoli Yuan. 2018. "Elastic Properties and Electronic Properties of MxNy (M = Ti, Zr) from First Principles Calculations" Materials 11, no. 9: 1640. https://doi.org/10.3390/ma11091640
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.