3.1. Result of the Elastic Properties
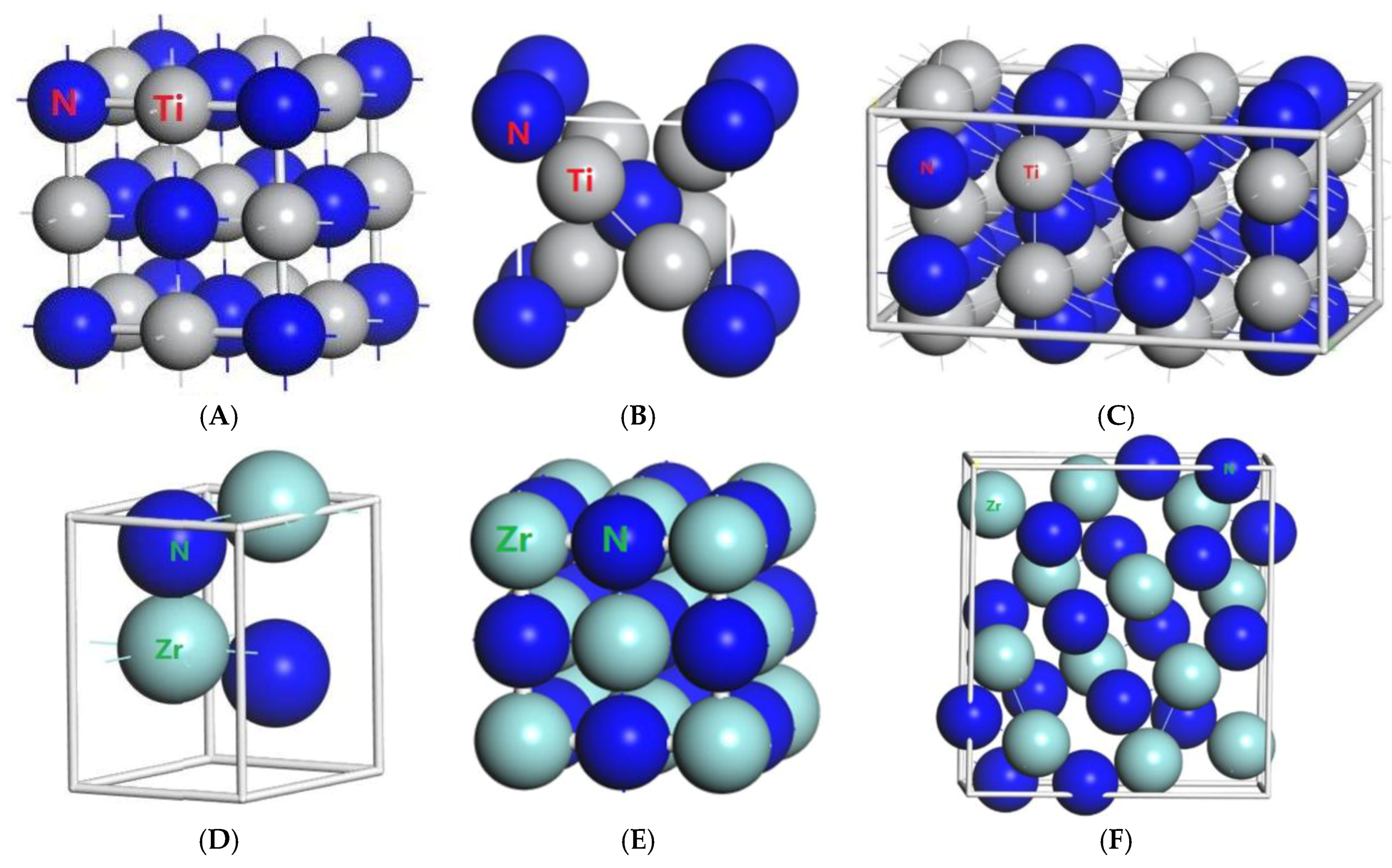
TiN, ZrN with space group Fm
m and Zr
3N
4 with space group I
3d belong to the cubic system, and their elastic tensors
have three independent components
,
and
. Its equations are derived from reference [
26]. All Ti
2N and ZrN with space group P6
3mc belong to the tetragonal system, whose elastic tensors
have six independent components
,
,
,
,
and
. Zr
3N
4 with space group Pna2
1 and Zr
3N
4 with space group Pnam belongs to the orthonormal system, whose elastic tensors
have nine independent components
,
,
,
,
,
,
,
and
. Its equations are derived from reference [
27]. On account of the fact that the Voigt band is acquired by the average polycrystalline moduli, it is the upper band of the real modulus. Meanwhile, the Reuss band is acquired by hypothetical stress, so it describes the lower limit. The arithmetic mean of the two bands is called the Voigt-Reuss-Hill approximation.
B represents bulk modulus, and
G expresses shear modulus. By using the subscript which indicates the Voigt band, Reuss band and Hill average by V, R and H, respectively, they are calculated by the following equations.
The criterion of mechanical stability is
For the tetragonal system:
The criterion of mechanical stability is
For the orthonormal system
The criterion of mechanical stability is
BV,
BR,
GV,
GR of different substances were obtained by the calculation of the Equations (1)–(14). Under the Voigt-Reuss-Hill approximation, it can be found that the modulus of polycrystal is the mean value under the Voigt bound and Reuss bound,
Young’s modulus (
E) and poisson’s ratio (
) were obtained by these equations:
The associated Equations (15)–(18) can calculate the relevant physical quantities. From the relevant literature, we know that
G indicates the anti-plastic deformability of materials and
B indicates the resistance to fracture of materials [
28]. Pugh proposed the estimation of the ductility of a given material by the ratio of
G/
B between the shear modulus
G and the bulk modulus
B [
29]. According to his theory, the small
G/
B values represent the toughness of the corresponding materials, while the larger
G/
B values represent brittleness.
G/
B = 0.57 is the critical value. The results we got were written in
Table 3,
Table 4,
Table 5 and
Table 6.
First of all, according to the results, ZrN with space group P6
3mc fails to meet the conditions of mechanical stability. So, it is necessary to compare ZrN and TiN in the same space group. The data show that the Young’s modulus of TiN is larger than that of ZrN. It indicates that the stiffness of TiN is larger than that of ZrN with space group Fm
m. Poisson’s ratios can show the binding force of the atom [
30]. Poisson’s ratios of TiN and ZrN are both less than 0.25, so they are non-central force.
G/
B of TiN is 0.665, which shows that TiN is brittle. With regard to ZrN,
G/
B is 0.615. Therefore, ZrN with space group Fm
m is brittle, and there is a reduction from TiN to ZrN. For Ti
2N, we found that Ti
2N’s stiffness with space group I4
1/amdz is larger than that with space group p4
2/mnm. Poisson’s ratios are the same, and they are non-central force.
G/
B of Ti
2Nwith space group I4
1/amdz is 0.69. At the same time,
G/
B of Ti
2Nwith space group P4
2/mnm is 0.665. The two kinds of Ti
2N are brittle and Ti
2N with space group I4
1/amdz is larger. As for Zr
3N
4, Zr
3N
4 with space group Pnam has the largest stiffness; the second largest is Zr
3N
4 with space group Pna2
1. Poisson’s ratios are the opposite, but all of them are central force. Meanwhile, three structures of Zr
3N
4 are tough and Zr
3N
4 with space group I
3d is the toughest. The remaining two substances are not very different, which proves that the symmetry of the structure has little effect on this property.
3.2. Result of the Electronic Properties
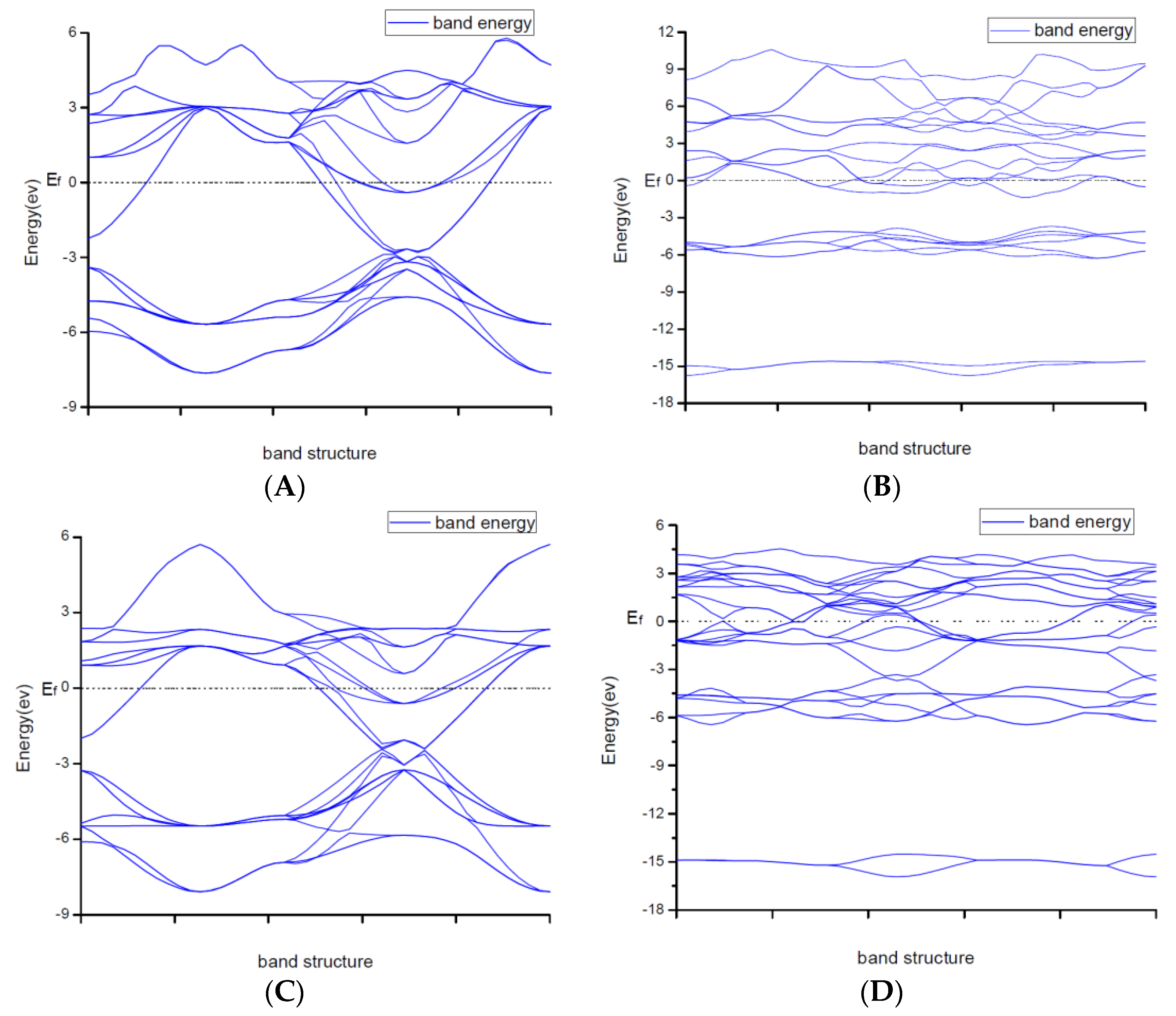
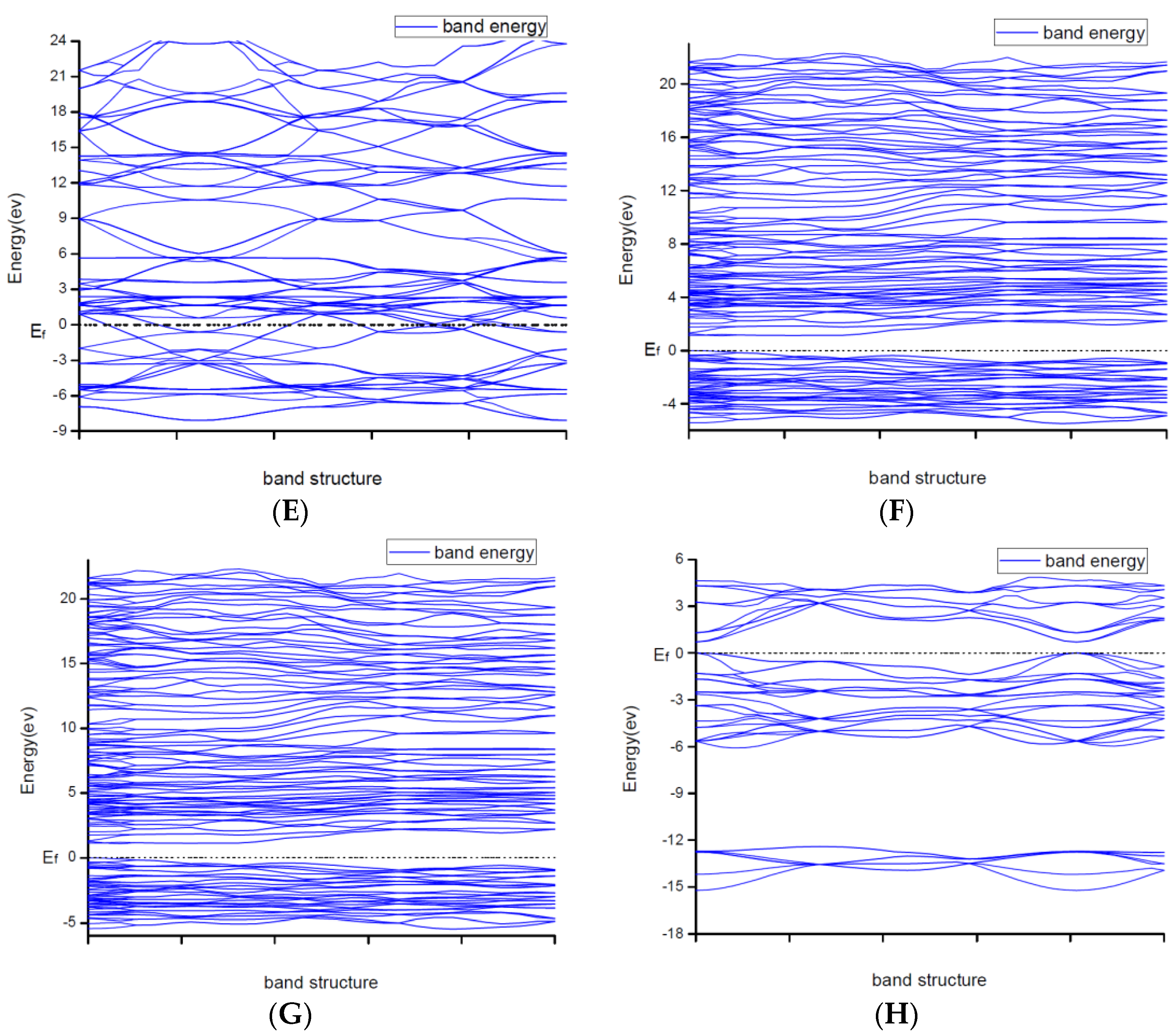
Due to explaining the macroscopic properties, we investigated the electronic band structures, density of states and difference of the charge density of all structures under zero pressure. Figure 4 shows the calculated band structures along the high symmetry directions in the Brillouin zone. It is worth mentioning that DFT cannot give reliable results for the energy gap because DFT does not consider the correlation effect of electrons in 3d orbit and 4f orbitin some strongly correlated systems. As a result, band gaps became smaller. So, the judgement on whether the system is metallic or insulating and on the value of the gap was semi-quantitative.
From
Figure 2, the conduction band minimum and valence band maximum of TiN, ZrN with space group Fm
m and Ti
2N with two kinds of structures are located at the G-point with a very small band gap. The conduction band minimum and valence band maximum of ZrN with space group P6
3mc is located at the M-point. The conduction band minimum and valence band maximum of Zr
3N
4 with space group I
3d is located at the G-point. From the results, their band gaps are less than 1 eV. So, for these substances, electrons can easily gain energy at room temperature and jump to the transfer band to conduct electricity. The conduction band minimum and valence band maximum of Zr
3N
4 with space groups Pna2
1 and Pnam are located at the Z-point with band gap between1eVand 3eV. These two substances are between conductors and insulators. Therefore, they are electrically conductive as long as the appropriate energy is given or the gaps between their energy are changed. It indicates that the electrical conductivity of ZrN with space group Fm
m is better than that of ZrN with space group P6
3mc. For TiN and ZrN with the same space group, TiNhas smaller electron effective mass and larger atomic non-localization than ZrN due to the bigger width of band structure. For three different structures of Zr
3N
4, symmetry and asymmetry have little influence on the electronic properties. The electrical conductivities of Zr
3N
4 with space groups Pna2
1 and Pnam are better than that of Zr
3N
4 with space group I
3d. Ti
2N with space group I4
1/amdz has a smaller electron effective mass and a degree of non-localization with stronger atomic orbital extension than Ti
2N with space group P4
2/mnm.
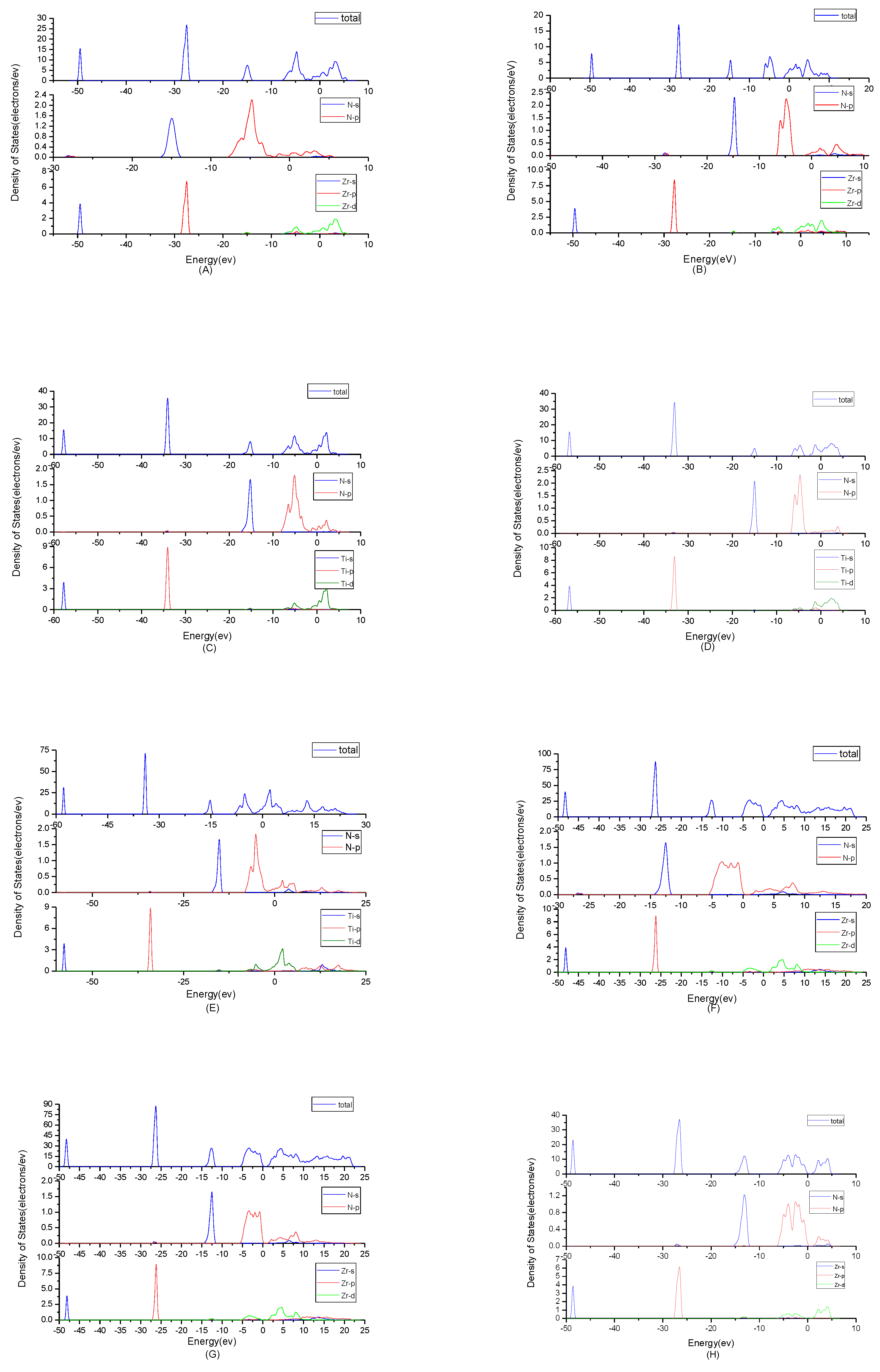
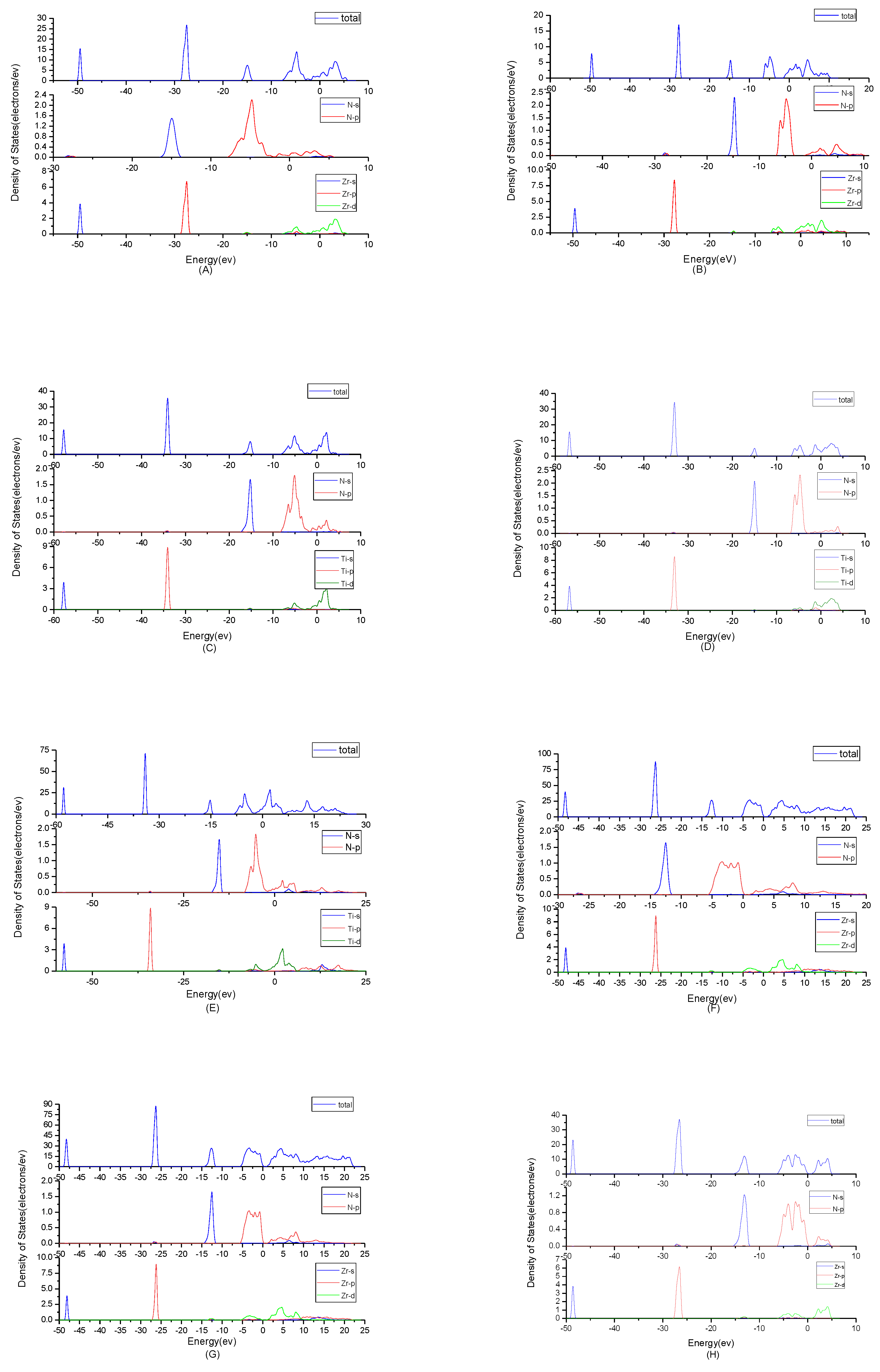
To find origins of band structures, the total density of states and partial density of states were calculated as shown in
Figure 3. PBE cannot accurately show the weak interaction between molecules, because it will make the values of d track and f track be slightly lower than the results of dispersion correction and there will besome differences in the hybridization at the top of the conduction band. But the corrected value is very small and will not affect the conclusion of this work. At the same time, the focus of this study is near the Fermi level, where the situation will not change substantially due to the effect of dispersion. By observing the density of states of ZrN, the results showed that the contributions of the atoms to the energy band are similar. N2p and Zr4d have obvious peaks at −5 eV, and they have a state density resonance, so N2p and Zr4d are bonded. Zr4d, Zr4P and Zr5s have a state density contribution to N2p, so they also form covalent bonds. Meanwhile, ZrN with space group P6
3mc has a greater span of density of state and stronger domain. Accordingly, it has stronger bonds. For TiN, N2p have obvious peaks at Ti3d; they are bonded. In addition, Ti4p and Ti4s have a part in the contribution of state density to N2p. Ti’s s orbit and p orbit are the main sources of the valence band. N’s s orbits, p orbits and Ti’s have a small contribution. The d orbit of Ti and the p orbit of N are the main sources of the guide band. As for Ti
2N, Ti
4p, Ti4s and Ti3d are bonded respectively to N2s and N2p in the two structures. The s, p orbit of Ti (s
Ti and p
Ti) and N (s
N and p
N) are the main sources of the valence band. d
Ti orbit has a small contribution. For Ti
2N with space group P4
2/mnm, the d orbit of Ti (d
Ti) and the p orbit of N (p
N) are the main sources of the guide band. For another structure of Ti
2N, the s
Ti, d
Ti and p
Ti orbits are the main sources of the guide band. The s
N orbits and p
N orbits have some contributions. Finally, according to the density of states of Zr
3N
4, the differences between Zr
3N
4 with space group Pna2
1 and Zr
3N
4 with space group Pnam are minimal; this situation shows that the symmetry of the space group has little effect on it. For Zr
3N
4 with space group I
3d, s
N orbits, p
Zr orbits and d
Zr orbits form covalent bonds. The 2p
N forms covalent bonds with 5s
Zr, 4p
Zrand 4d
Zr. For the other two structures, 2p
N forms covalent bonds with 5s
Zr, 4d
Zr and 4p
Zr and 2s
N forms covalent bonds with 4d
Zr and 4p
Zr. At the same time, Zr
3N
4 with space group Pna2
1 and Zr
3N
4 with space group Pnam have a greater span of density of state and stronger domain. Therefore, they have stronger bonds.
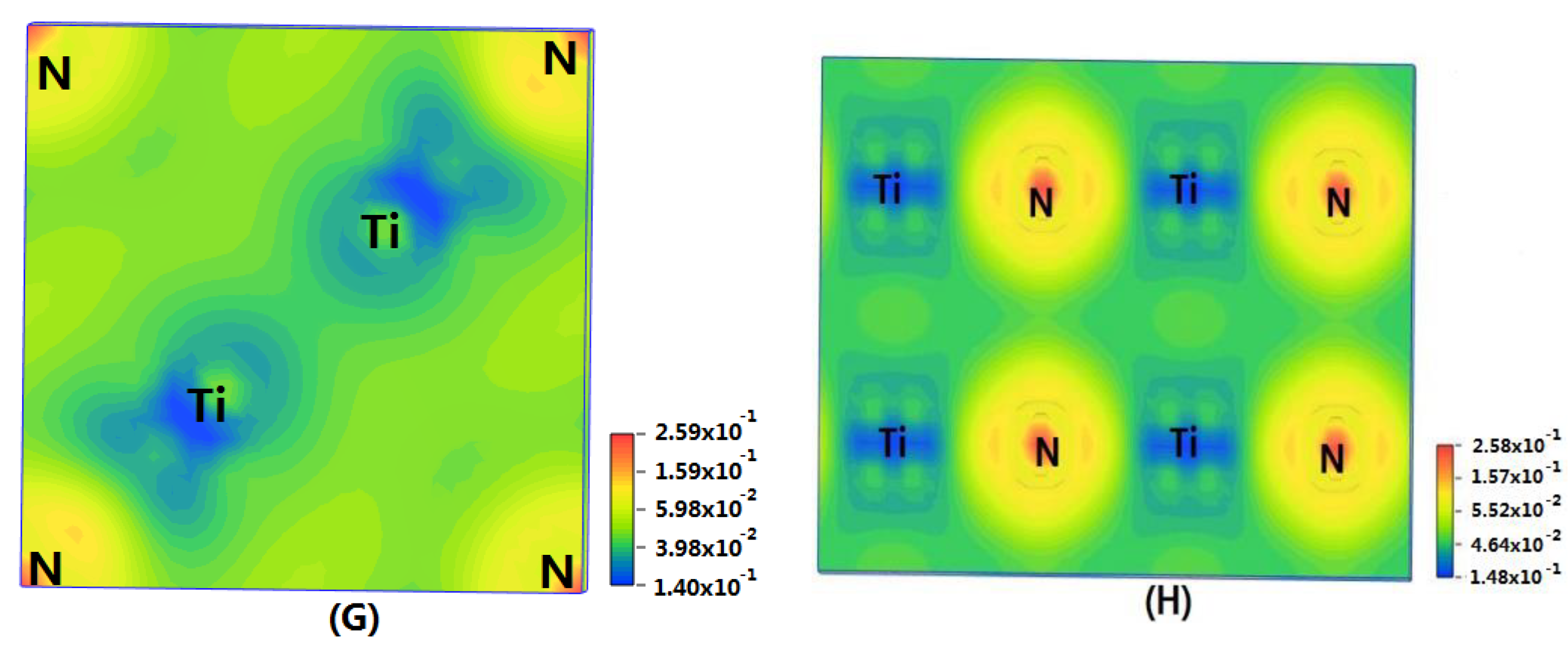
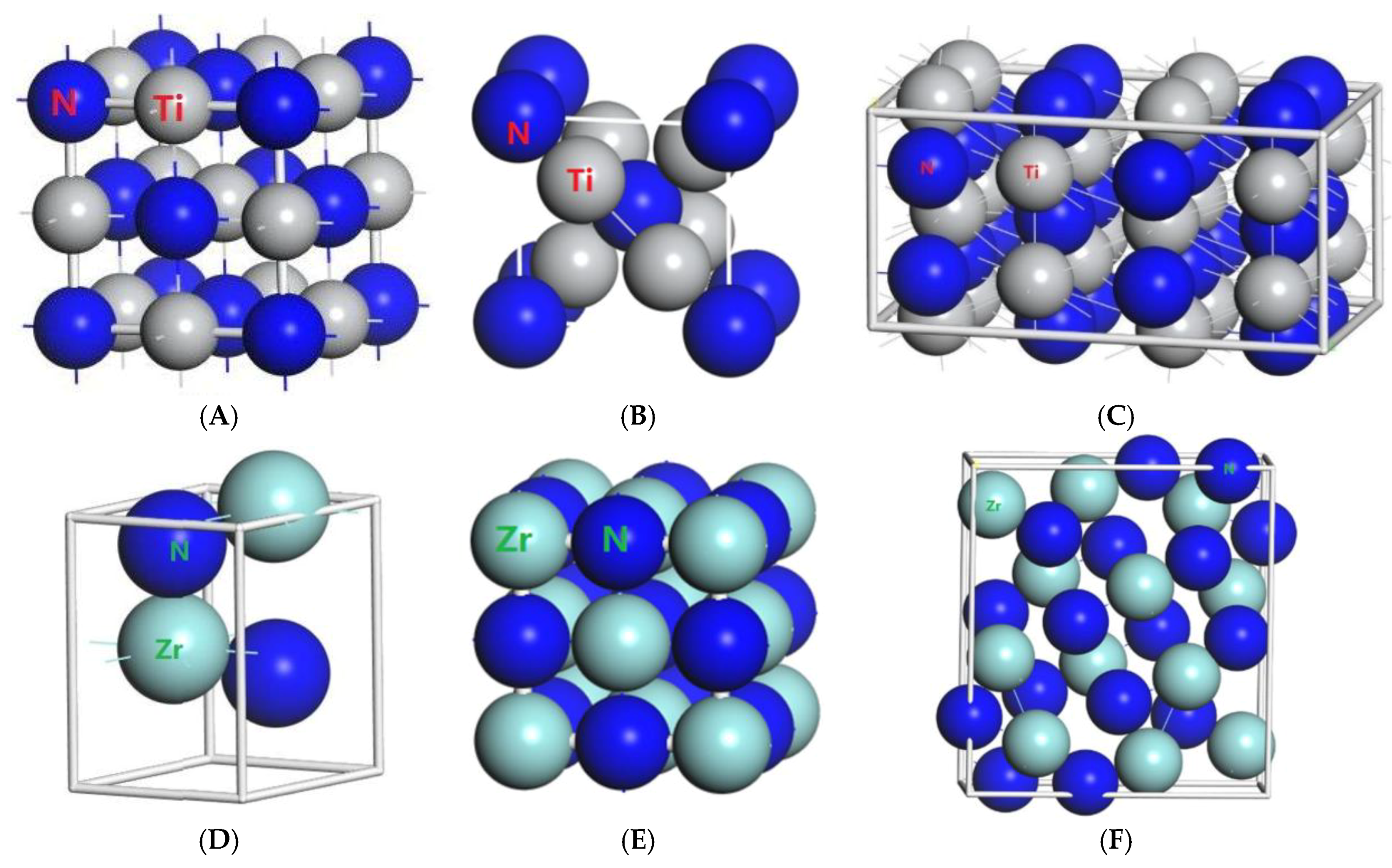
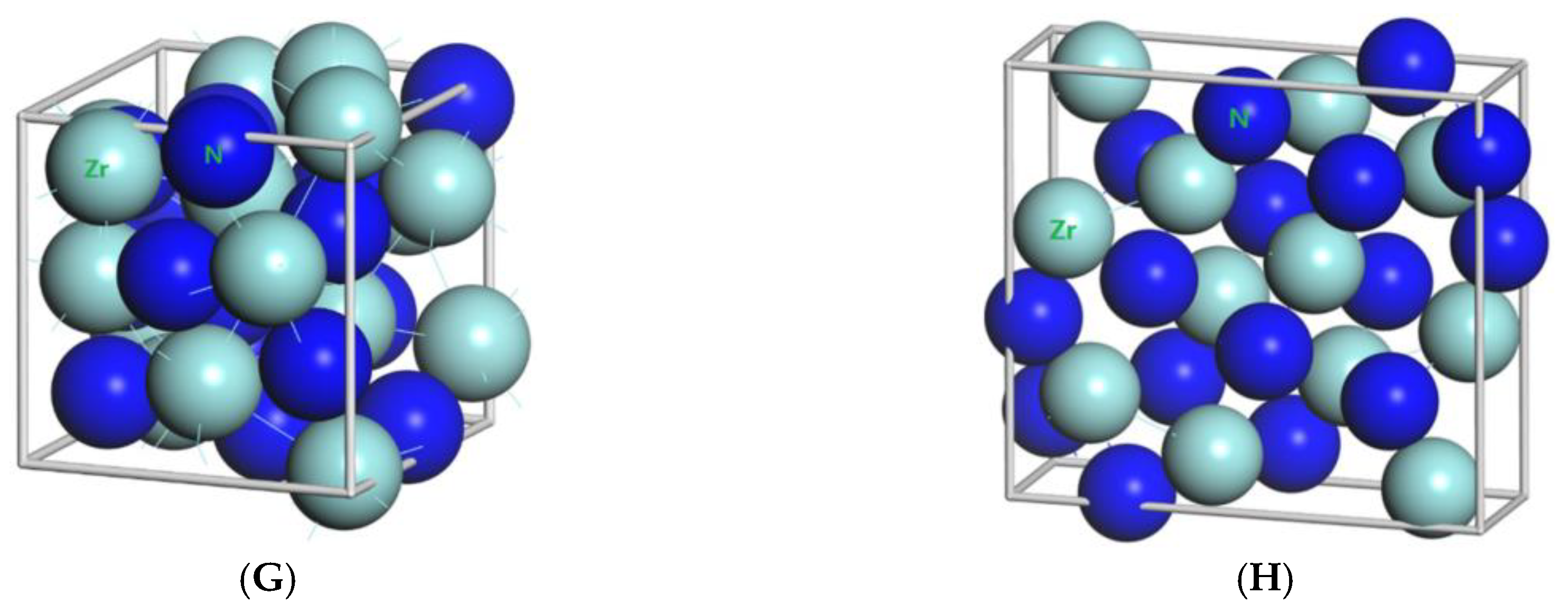
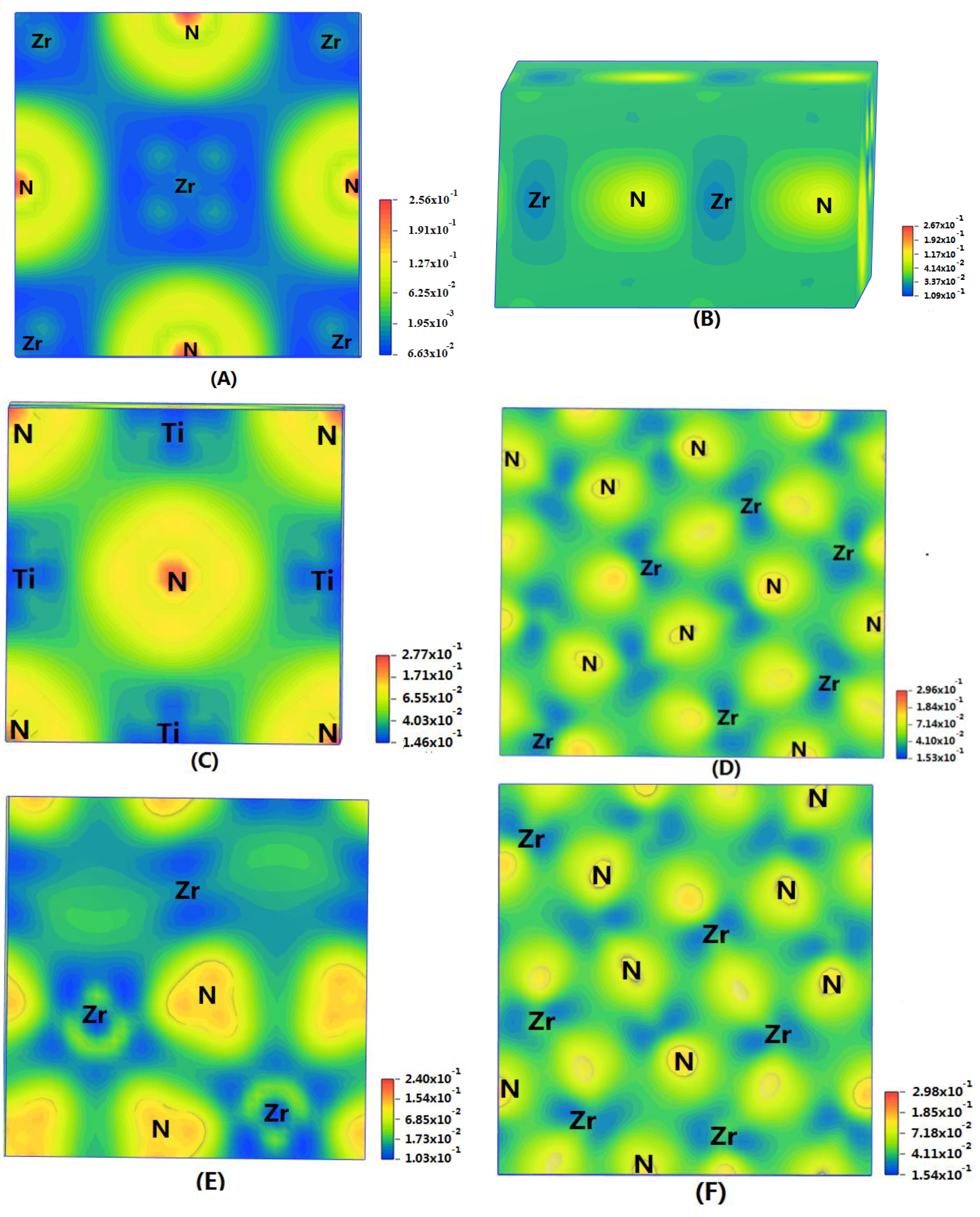
In order to show the bonding between atoms of different crystals more intuitively, difference charge density maps are shown in
Figure 4. According to plot A and plot C, for TiN and ZrNwith space group Fm
m, the charge densities between Ti and N are larger than that of Zr and N, which means that the effects between Ti–N are stronger. Meanwhile, ZrN with space group Fm
m has stronger Zr–N bonds than ZrN with space group P6
3mc. So, the interatomic interaction of ZrN with space group Fm
m is greater than that of ZrN with space group P6
3mc. For three different structures of Zr
3N
4, symmetry and asymmetry have little influence on the electronic properties. There are larger charge densities between Zr and N in Zr
3N
4 with space group Pna2
1 and Zr
3N
4 with space group Pnam than the other structure. So, there are stronger interactions between their atoms. The last two graphs (
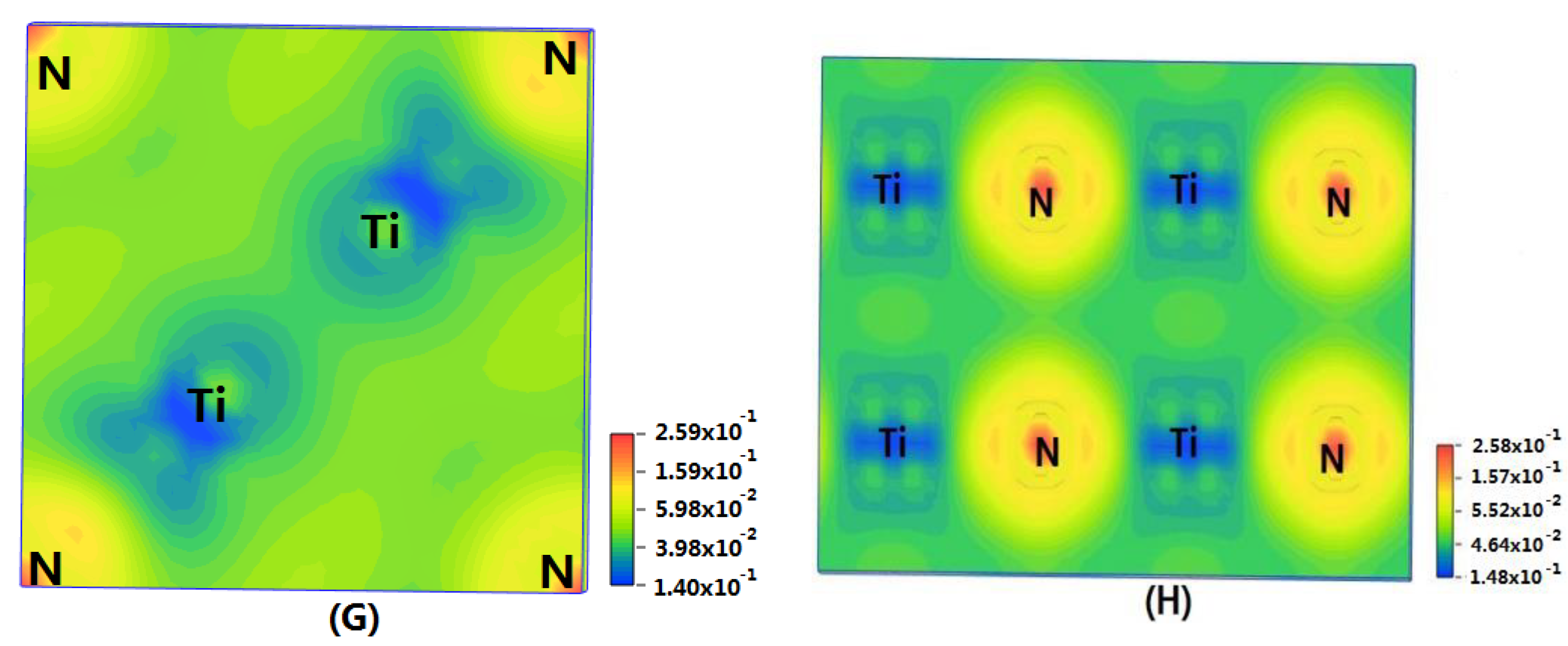
Figure 4G,H) represent the charge density of TiN with two different structures. Ti
2N with space group P4
2/mnm has a larger charge density than Ti
2N with space group I4
1/amdz. Therefore, the interatomic effect of Ti
2N with space group P4
2/mnm is stronger.
3.3. Superconducting Properties
After obtaining the elastic properties and electronic properties of the studied materials, the possible superconducting properties were discussed based on the results. According to the simplified theory of superconductivity, if a material can become a superconductor, three conditions must be satisfied. First of all, atoms that make up crystals are lighter. Second, the coefficient of elasticity of the crystal is as large as possible, and crystals are relatively tough. Third, the effective Fermi level of materials should be low. Zr and Ti are two of the 28 superconducting elements, as their nitrogen compounds satisfy the first condition. From the results of the elastic properties, TiN is tougher than ZrN when used as superconducting material. Ti2N with space group I41/amdz is better than Ti2N with space group P42/mnm. The three structures of Zr3N4 all have potential as superconducting materials; Zr3N4 with space group I3d is the best of them. From the third point of view, metal systems are more likely to superconduct. So, TiN, ZrN and Zr3N4 with space group I3d are better choices to satisfy the third condition. Combining three characteristics, Zr3N4 with space group I3d is most likely to have superconductivity in all materials. TiN is more likely to be used as a superconducting material than ZrN with same space group. Ti2N with space group I41/amdz is more suitable to be used as superconducting material than Ti2N with space group P42/mnm.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}