Keggin-Type Heteropoly Salts as Bifunctional Catalysts in Aerobic Baeyer-Villiger Oxidation

 and
and
Abstract
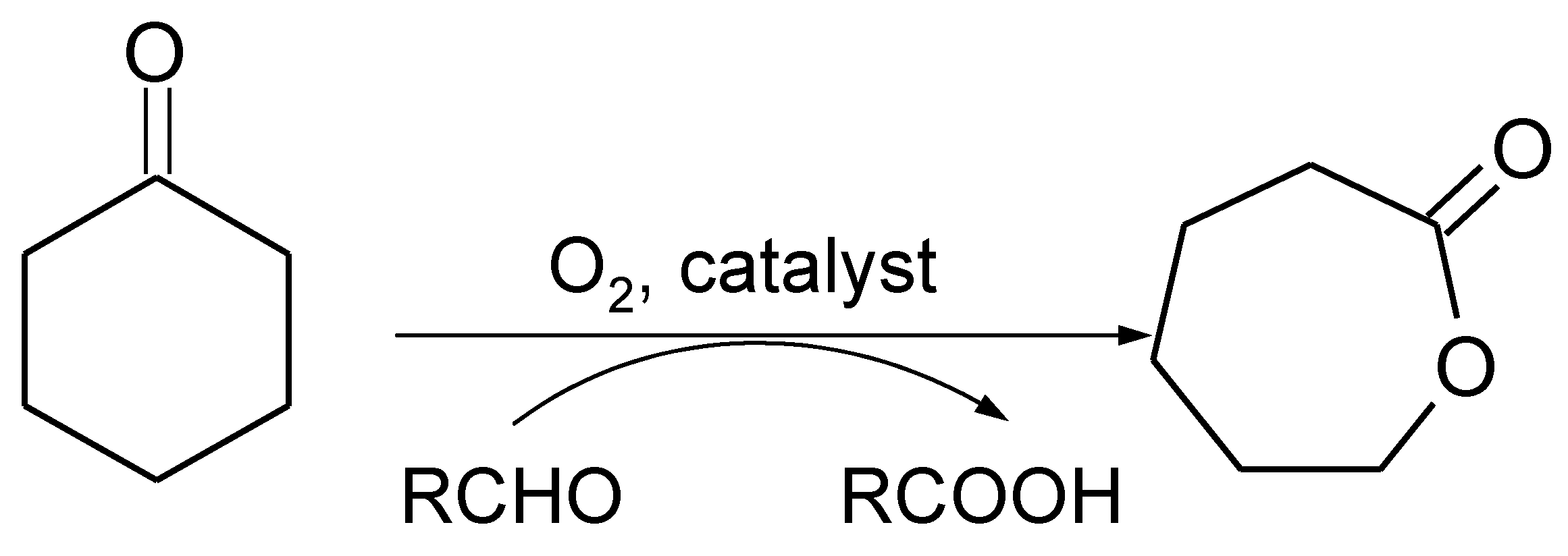
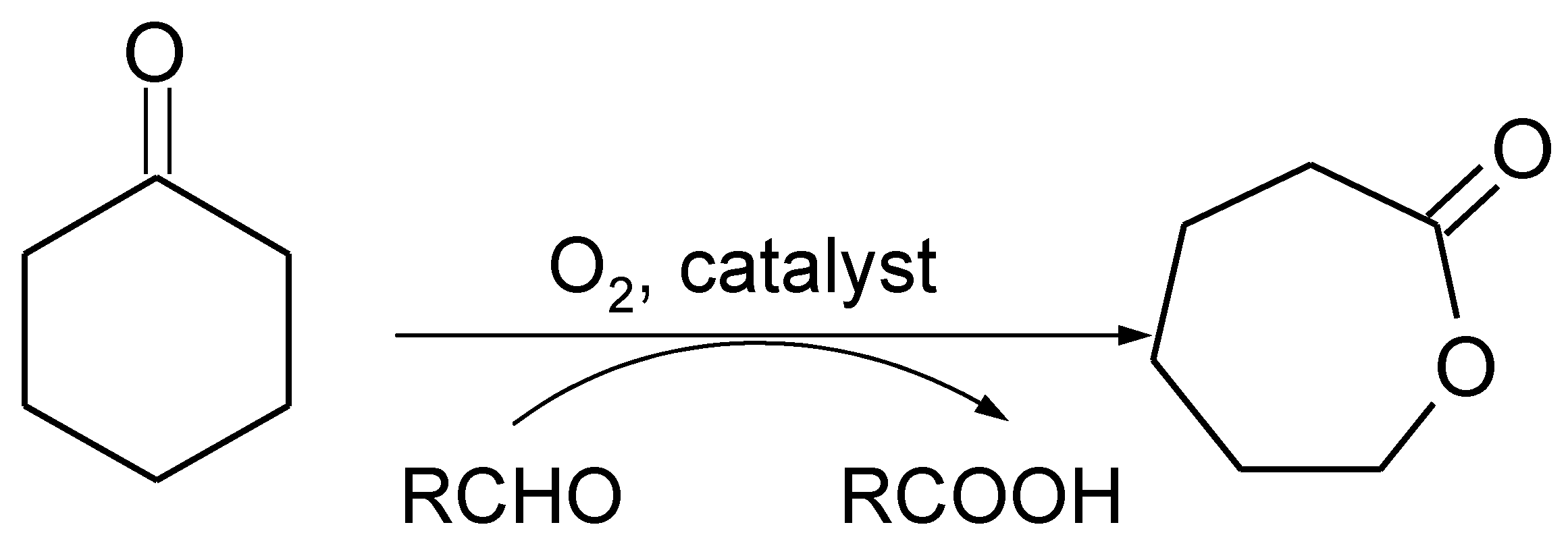
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
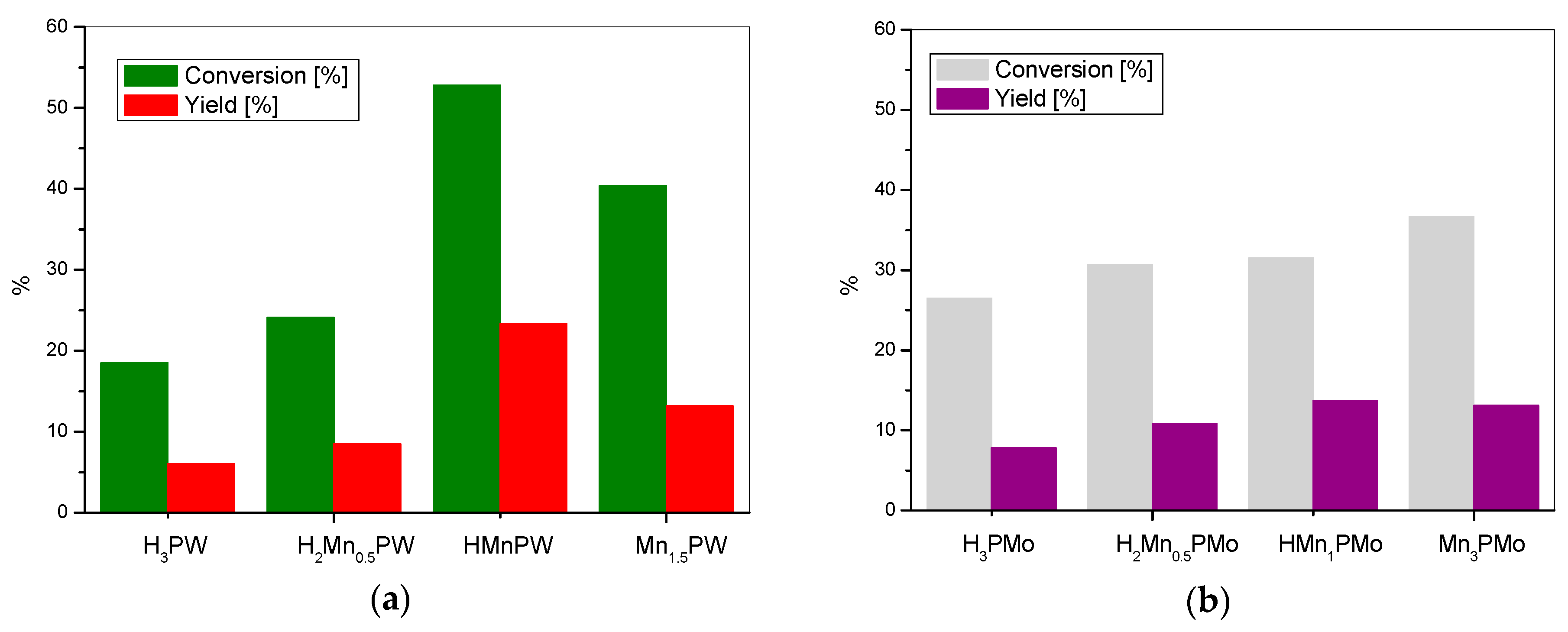
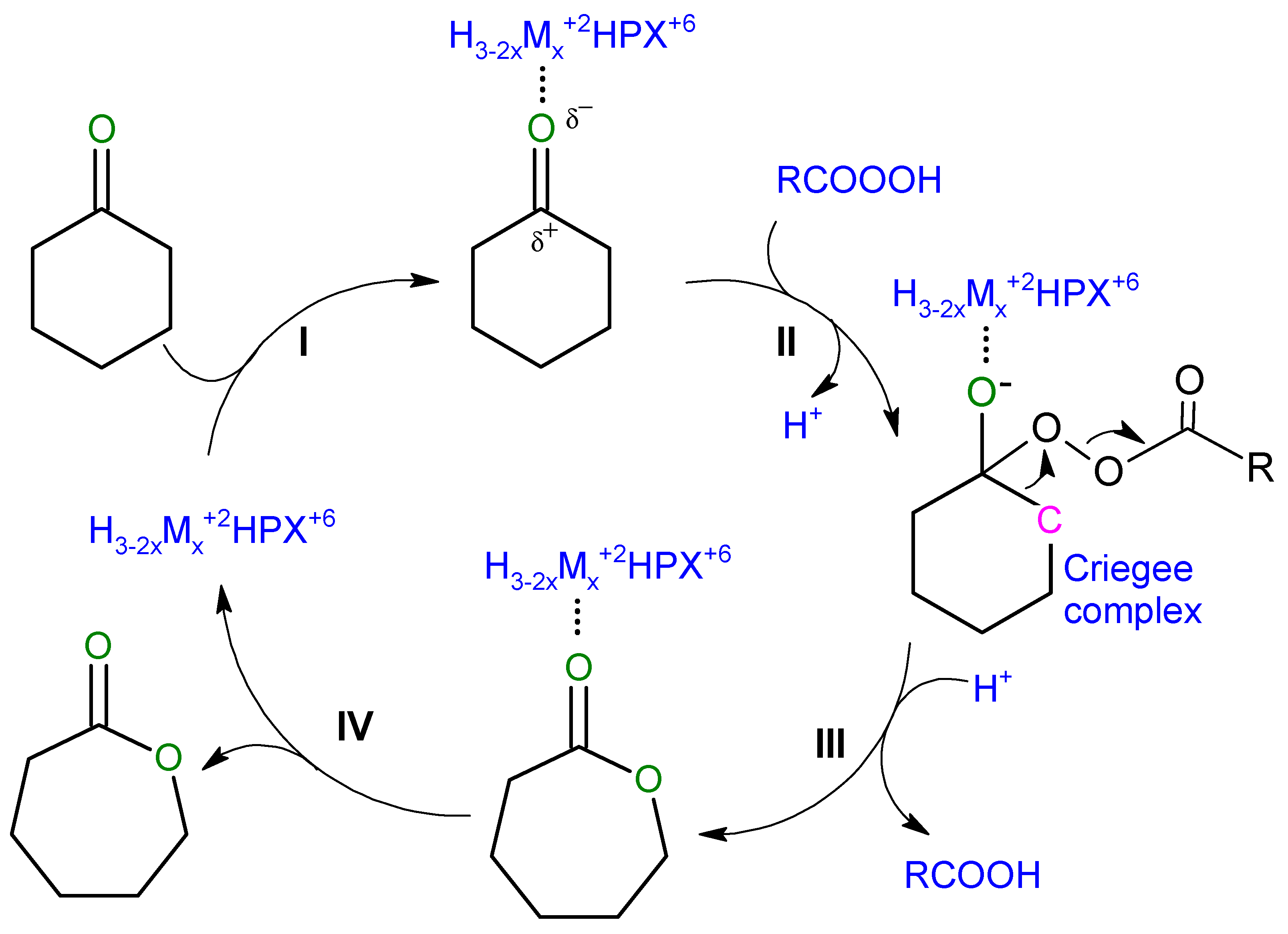
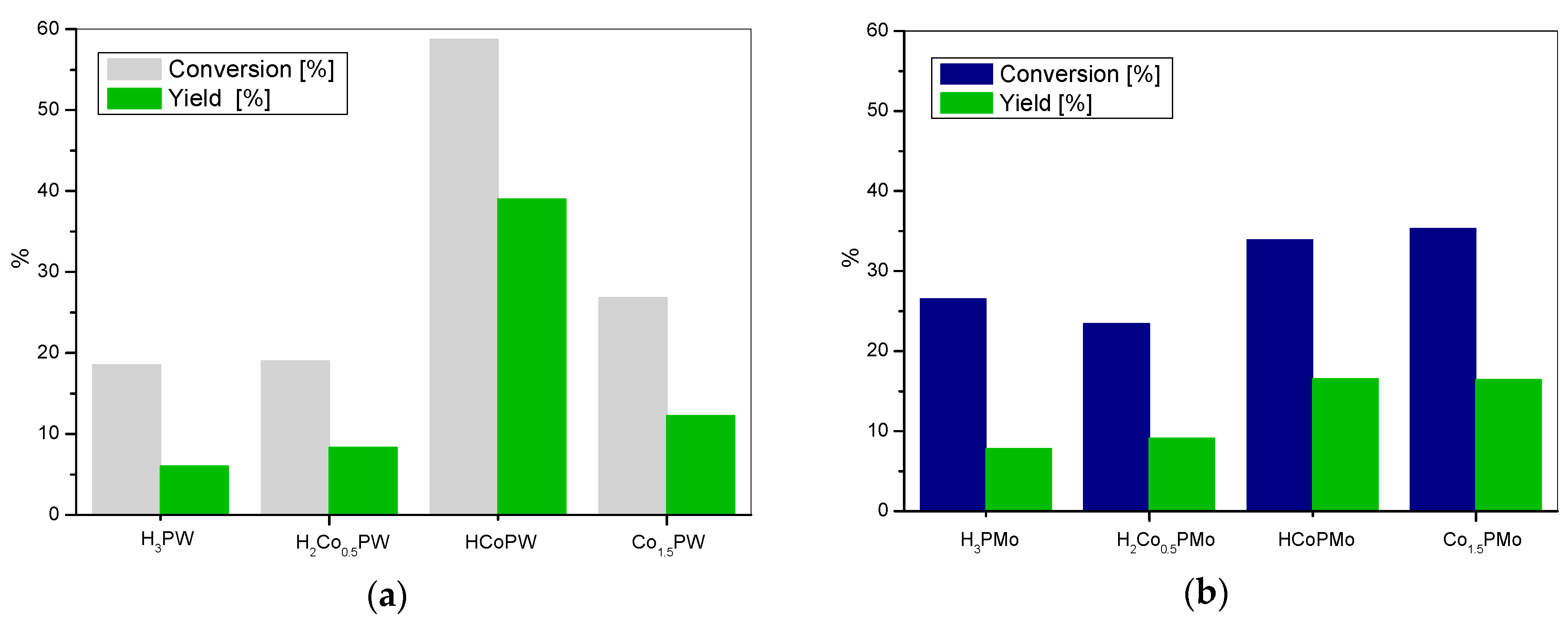
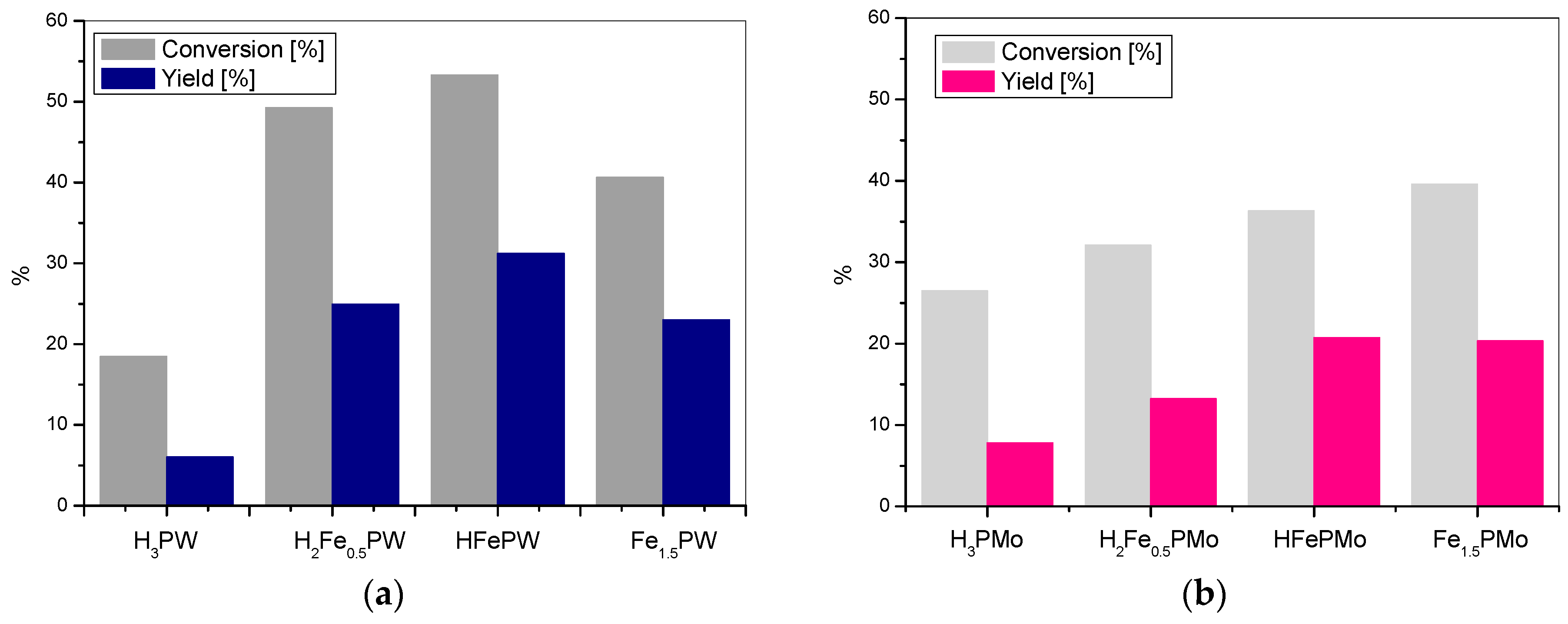
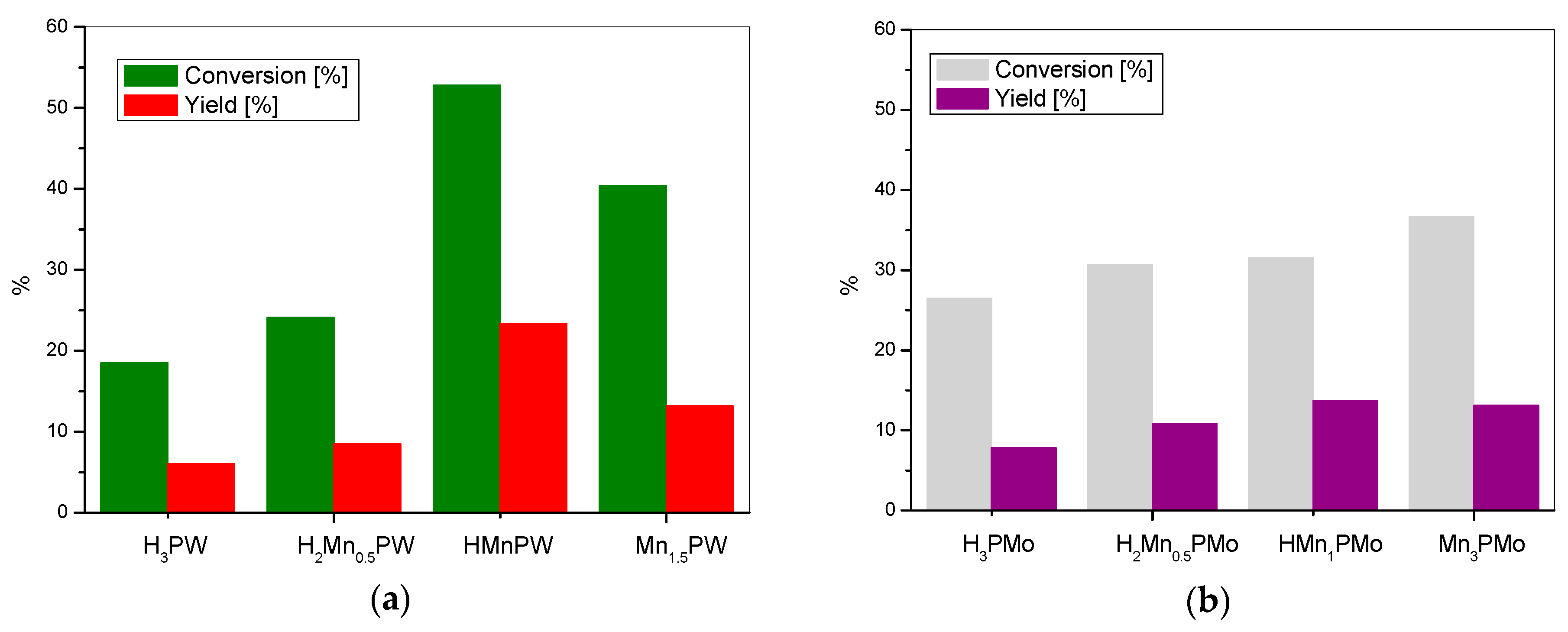
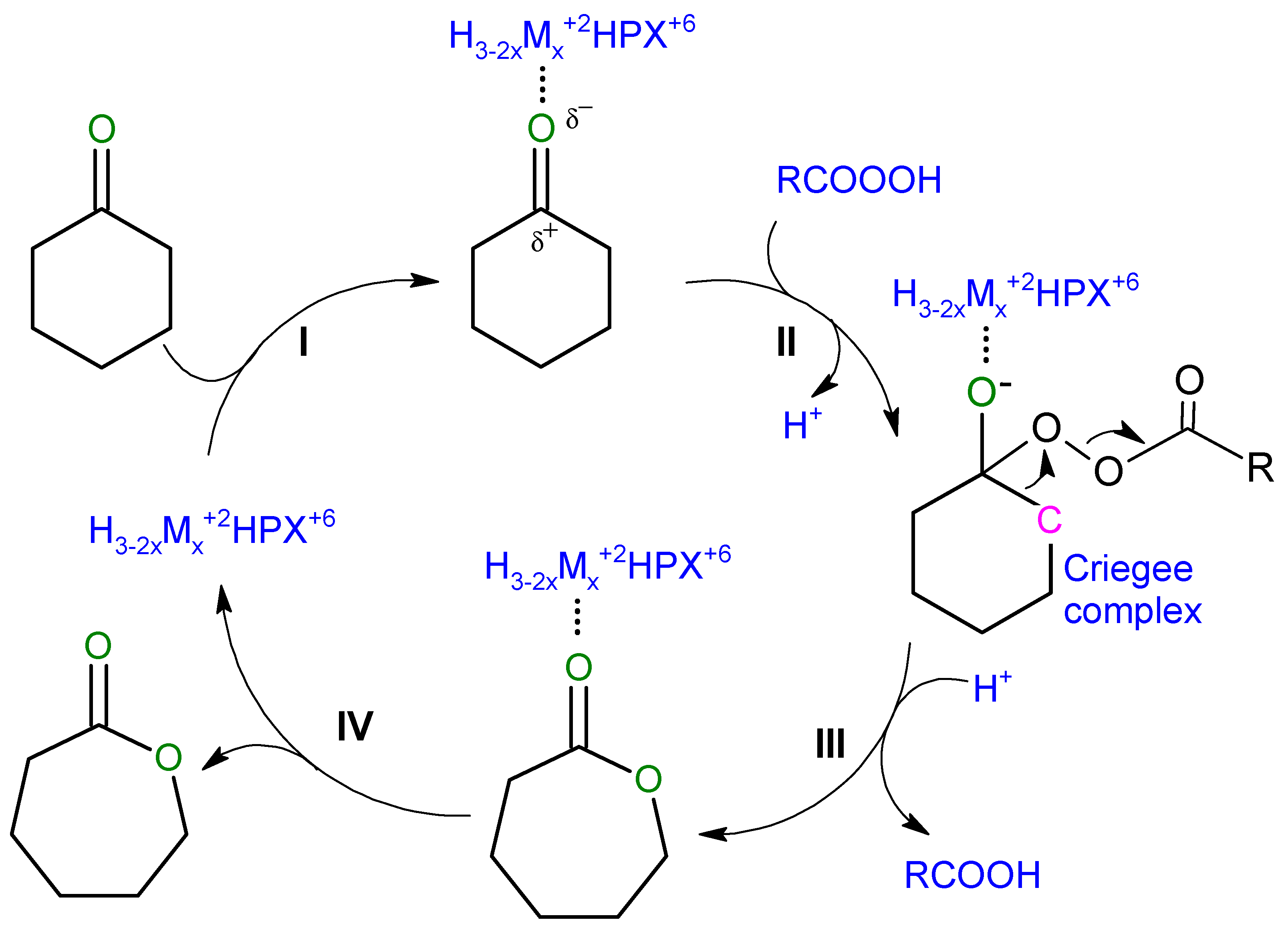
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kozhevnikov, I.V. Heteropoly Acids and Related-Compounds as Catalysts for Fine Chemical Synthesis. Catal. Rev. 1995, 37, 311–352. [Google Scholar] [CrossRef]
- Combswalker, L.A.; Hill, C.L. Stabilization of the Defect (Lacunary) Complex PMo11O39(7−)—Isolation, Purification, Stability Characteristics, and Metalation Chemistry. Inorg. Chem. 1991, 30, 4016–4026. [Google Scholar] [CrossRef]
- Krow, G.R. The Baeyer–Villiger Oxidation of Ketones and Aldehydes. Org. React. 1993, 43, 251–798. [Google Scholar]
- Strukul, G. Transition metal catalysis in the Baeyer-Villiger oxidation of ketones. Angew. Chem. Int. Ed. 1998, 37, 1199–1209. [Google Scholar] [CrossRef]
- Ma, Q.G.; Zhao, J.R.; Xing, W.Z.; Peng, X.H. Baeyer-Viiliger Oxidation of Cyclic Ketones Using Aqueous Hydrogen Peroxide Catalyzed by Heteropolyacids in Solvent-free System. J. Adv. Oxid. Technol. 2014, 17, 212–216. [Google Scholar] [CrossRef]
- Balbinot, L.; Schuchardt, U.; Vera, C.; Sepulveda, J. Oxidation of cyclohexanol to epsilon-caprolactone with aqueous hydrogen peroxide on H3PW12O40 and Cs2.5H0.5PW12O40. Catal. Commun. 2008, 9, 1878–1881. [Google Scholar] [CrossRef]
- Yang, Z.W.; Xu, X.Q.; Li, T.J.; Zhang, N.N.; Zhao, X.; Chen, W.L.; Liang, X.X.; He, X.L.; Ma, H.C. Preparation and Catalytic Property of Multi-walled Carbon Nanotubes Supported Keggin-Typed Tungstosilicic Acid for the Baeyer-Villiger Oxidation of Ketones. Catal. Lett. 2015, 145, 1955–1960. [Google Scholar] [CrossRef]
- Ten Brink, G.J.; Arends, I.W.C.E.; Sheldon, R.A. The Baeyer-Villiger reaction: New developments toward greener procedures. Chem. Rev. 2004, 104, 4105–4123. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takahashi, K.; Kato, K.; Takai, T.; Inoki, S.; Mukaiyama, T. The Baeyer-Villiger Oxidation of Ketones Catalyzed by Nickel(II) Complexes with Combined Use of Molecular-Oxygen and Aldehyde. Chem. Lett. 1991, 641–644. [Google Scholar] [CrossRef]
- Raja, R.; Thomas, J.M.; Sankar, G. Baeyer-Villiger oxidations with a difference: Molecular sieve redox catalysts for the low-temperature conversion of ketones to lactones. Chem. Commun. 1999, 525–526. [Google Scholar] [CrossRef]
- Murahashi, S.I.; Oda, Y.; Naota, T. Fe2O3-Catalyzed Baeyer-Villiger Oxidation of Ketones with Molecular Oxygen in the Presence of Aldehydes. Tetrahedron Lett. 1992, 33, 7557–7560. [Google Scholar] [CrossRef]
- Lakk-Bogath, D.; Speier, G.; Kaizer, J. Oxoiron(IV)-mediated Baeyer-Villiger oxidation of cyclohexanones generated by dioxygen with co-oxidation of aldehydes. New J. Chem. 2015, 39, 8245–8248. [Google Scholar] [CrossRef]
- Ma, Y.L.; Liang, Z.Y.; Feng, S.X.; Zhang, Y.D. Baeyer-Villiger oxidation of cyclohexanone by molecular oxygen with Fe-Sn-O mixed oxides as catalysts. Appl. Organomet. Chem. 2015, 29, 450–455. [Google Scholar] [CrossRef]
- Subramanian, H.; Nettleton, E.G.; Budhi, S.; Koodali, R.T. Baeyer–Villiger oxidation of cyclic ketones using Fe containing MCM-48 cubic mesoporous materials. J. Mol. Catal. A 2010, 330, 66–72. [Google Scholar] [CrossRef]
- Rahman, S.; Enjamuri, N.; Gomes, R.; Bhaumik, A.; Sen, D.; Pandey, J.K.; Mazumdar, S.; Chowdhury, B. Aerobic Baeyer–Villiger oxidation of cyclic ketones over periodic mesoporous silica Cu/Fe/Ni/Co-HMS-X. Appl. Catal. A 2015, 505, 515–523. [Google Scholar] [CrossRef]
- Hamamoto, M.; Nakayama, K.; Nishiyama, Y.; Ishii, Y. Oxidation of Organic Substrates by Molecular-Oxygen Aldehyde Heteropolyoxometalate System. J. Org. Chem. 1993, 58, 6421–6425. [Google Scholar] [CrossRef]
- Pamin, K.; Prończuk, M.; Basąg, S.; Kubiak, W.; Sojka, Z.; Połtowicz, J. A new hybrid porphyrin-heteropolyacid material: Synthesis, characterization and investigation as catalyst in Baeyer-Villiger oxidation. Synergistic effect. Inorg. Chem. Commun. 2015, 59, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Leisch, H.; Morley, K.; Lau, P.C.K. Baeyer-Villiger Monooxygenases: More Than Just Green Chemistry. Chem. Rev. 2011, 111, 4165–4222. [Google Scholar] [CrossRef] [PubMed]
- De Gonzalo, G.; Mihovilovic, M.D.; Fraaije, M.W. Recent Developments in the Application of Baeyer–Villiger Monooxygenases as Biocatalysts. ChemBioChem 2010, 11, 2208–2231. [Google Scholar] [CrossRef] [PubMed]
- González-Martínez, D.; Rodríguez-Mata, M.; Méndez-Sánchez, D.; Gotor, V.; Gotor-Fernández, V. Lactonization reactions through hydrolase-catalyzed peracid formation. Use of lipases for chemoenzymatic Baeyer–Villiger oxidations of cyclobutanones. J. Mol. Catal. B Enzym. 2015, 114, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Karcz, R.; Niemiec, P.; Pamin, K.; Połtowicz, J.; Kryściak-Czerwenka, J.; Napruszewska, B.D.; Michalik-Zym, A.; Witko, M.; Tokarz-Sobieraj, R.; Serwicka, E.M. Effect of cobalt location in Keggin-type heteropoly catalysts on aerobic oxidation of cyclooctane: Experimental and theoretical study. Appl. Catal. A 2017, 542, 317–326. [Google Scholar] [CrossRef]
- Emeis, C.A. Determination of Integrated Molar Extinction Coefficients for Infrared Absorption Bands of Pyridine Adsorbed on Solid Acid Catalysts. J. Catal. 1993, 141, 347–354. [Google Scholar] [CrossRef]
- Tsigdinos, G.A. Preparation and Characterization of 12-Molybdophosphoric and 12-Molybdosilicic Acids and Their Metal Salts. Ind. Eng. Chem. Prod. Res. Dev. 1974, 13, 267–274. [Google Scholar] [CrossRef]
- Pamin, K.; Połtowicz, J.; Prończuk, M.; Basąg, S.; Maciejewska, J.; Kryściak-Czerwenka, J.; Tokarz-Sobieraj, R. Hydroxylation of phenol by hydrogen peroxide catalyzed by heteropoly compounds in presence of glycerol as green solvent. Catal. Today 2015, 257, 80–85. [Google Scholar] [CrossRef]
- Rocchiccioli-Deltcheff, C.; Thouvenot, R.; Franck, R. Spectres i.r. et Raman d’hétéropolyanions α-XM12O40n− de structure de type Keggin (X = BIII, SiIV, GeIV, PV, AsV et M = WVI et MoVI). Spectrochim. Acta A 1976, 32, 587–597. [Google Scholar] [CrossRef]
- Rocchiccioli-Deltcheff, C.; Thouvenot, R. Metal complexes of heteropolyanions α-XM11O39n− with X = SiIV or PV and M = MoVI or WVI: Study of structural modifications of ligand by infrared and Raman spectrometry. J. Chem. Res. Miniprint 1977, 549–571. [Google Scholar] [CrossRef]
- Eguchi, K.; Toyozawa, Y.; Yamazoe, N.; Seiyama, T. An infrared study on the reduction processes of dodecamolybdophosphates. J. Catal. 1983, 83, 32–41. [Google Scholar] [CrossRef]
- Renz, M.; Meunier, B. 100 years of Baeyer-Villiger oxidations. Eur. J. Org. Chem. 1999, 1999, 737–750. [Google Scholar] [CrossRef]
- Corma, A. Attempts to Fill the Gap Between Enzymatic, Homogeneous, and Heterogeneous Catalysis. Catal. Rev. 2004, 46, 369–417. [Google Scholar] [CrossRef]
- Weiss, J.J. Nature of Iron-Oxygen Bond in Oxyhaemoglobin. Nature 1964, 202, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.; Młodnicka, T.; Połtowicz, J. Metal-Dependent Reactivity of Some Metalloporphyrins in Oxidation with Dioxygen. J. Mol. Catal. 1989, 54, 451–461. [Google Scholar] [CrossRef]
- Weschler, C.J.; Hoffman, B.M.; Basolo, F. Synthetic Oxygen Carrier—Dioxygen Adduct of a Manganese Porphyrin. J. Am. Chem. Soc. 1975, 97, 5278–5280. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | W/Mo (wt. %) | P (wt. %) | Co/Mn/Fe (wt. %) | |||
|---|---|---|---|---|---|---|
| Calculated | Found | Calculated | Found | Calculated | Found | |
| H3PW | 98.6 | 98.6 | 1.4 | 1.4 | - | - |
| H3PMo | 97.4 | 97.4 | 2.6 | 2.6 | - | - |
| H2Co0.5PW | 97.3 | 97.3 | 1.4 | 1.5 | 1.3 | 1.2 |
| HCoPW | 96.1 | 96.2 | 1.3 | 1.4 | 2.6 | 2.4 |
| Co1.5PW | 94.9 | 95.11 | 1.3 | 1.4 | 3.8 | 3.4 |
| H2Co0.5PMo | 95.0 | 95.5 | 2.6 | 2.5 | 2.4 | 2.0 |
| HCoPMo | 92.8 | 93.2 | 2.5 | 2.3 | 4.8 | 4.5 |
| Co1.5PMo | 90.6 | 90.8 | 2.4 | 2.4 | 7.0 | 6.8 |
| H2Mn0.5PW | 97.4 | 97.5 | 1.4 | 1.4 | 1.2 | 1.1 |
| HMnPW | 96.3 | 96.5 | 1.3 | 1.4 | 2.4 | 2.1 |
| Mn1.5PW | 95.1 | 95.4 | 1.3 | 1.4 | 3.6 | 3.3 |
| H2Mn0.5PMo | 95.2 | 95.3 | 2.6 | 2.6 | 2.3 | 2.1 |
| HMnPMo | 93.0 | 93.3 | 2.5 | 2.6 | 4.5 | 4.2 |
| Mn1.5PMo | 91.0 | 91.3 | 2.5 | 2.4 | 6.5 | 6.3 |
| H2Fe0.5PW | 97.4 | 97.5 | 1.4 | 1.5 | 1.2 | 1.0 |
| HFePW | 96.2 | 96.4 | 1.4 | 1.4 | 2.4 | 2.3 |
| Fe1.5PW | 95.1 | 95.4 | 1.3 | 1.3 | 3.6 | 3.3 |
| H2Fe0.5PMo | 95.1 | 95.3 | 2.6 | 2.6 | 2.3 | 2.1 |
| HFePMo | 93.0 | 93.3 | 2.5 | 2.6 | 4.5 | 4.2 |
| Fe1.5PMo | 90.9 | 91.1 | 2.4 | 2.5 | 6.6 | 6.4 |
| Yield of (%) | H3PW | H2M0.5PW | HMPW | M1.5PW | H3PMo | H2M0.5PMo | HMPMo | M1.5PMo |
| M = Co | M = Co | |||||||
| ethylene | 95.9 | 97.8 | 49.0 | 20.3 | 30.0 | 14.1 | 12.5 | 8.2 |
| diethyl ether | 4.1 | 1.4 | 20.8 | - | - | - | - | - |
| acetaldehyde | - | 0.2 | 0.2 | - | 18.0 | 12.5 | 11.0 | 9.0 |
| Conversion (%) | 97.2 | 99.4 | 70.0 | 20.3 | 48.0 | 26.6 | 23.5 | 17.2 |
| Yield of (%) | H3PW | H2M0.5PW | HMPW | M1.5PW | H3PMo | H2M0.5PMo | HM1PMo | M3PMo |
| M = Fe | M = Fe | |||||||
| ethylene | 95.9 | 99.8 | 87.6 | 70.0 | 30.0 | 38.0 | 29.9 | 37.8 |
| diethyl ether | 4.1 | - | - | - | - | - | - | - |
| acetaldehyde | - | - | - | - | 18.0 | 9.1 | 10.7 | 22.7 |
| Conversion (%) | 97.2 | 99.8 | 87.6 | 70.0 | 48.0 | 47.1 | 40.6 | 60.5 |
| Yield of (%) | H3PW | H2M0.5PW | HMPW | M1.5PW | H3PMo | H2M0.5PMo | HM1PMo | M1.5PMo |
| M = Mn | M = Mn | |||||||
| ethylene | 95.9 | 99.9 | 99.5 | 20.4 | 30.0 | 42.0 | 7.2 | 10.6 |
| diethyl ether | 4.1 | - | - | - | - | - | - | - |
| acetaldehyde | - | - | - | - | 18.0 | 9.1 | 13.1 | 23.3 |
| Conversion (%) | 97.2 | 99.5 | 99.5 | 20.9 | 48.0 | 51.1 | 20.3 | 33.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pamin, K.; Połtowicz, J.; Prończuk, M.; Kryściak-Czerwenka, J.; Karcz, R.; M. Serwicka, E. Keggin-Type Heteropoly Salts as Bifunctional Catalysts in Aerobic Baeyer-Villiger Oxidation. Materials 2018, 11, 1208. https://doi.org/10.3390/ma11071208
Pamin K, Połtowicz J, Prończuk M, Kryściak-Czerwenka J, Karcz R, M. Serwicka E. Keggin-Type Heteropoly Salts as Bifunctional Catalysts in Aerobic Baeyer-Villiger Oxidation. Materials. 2018; 11(7):1208. https://doi.org/10.3390/ma11071208
Chicago/Turabian StylePamin, Katarzyna, Jan Połtowicz, Mateusz Prończuk, Joanna Kryściak-Czerwenka, Robert Karcz, and Ewa M. Serwicka. 2018. "Keggin-Type Heteropoly Salts as Bifunctional Catalysts in Aerobic Baeyer-Villiger Oxidation" Materials 11, no. 7: 1208. https://doi.org/10.3390/ma11071208