Mesoporous Silicon Particles Favor the Induction of Long-Lived Humoral Responses in Mice to a Peptide-Based Vaccine

Abstract
:
1. Introduction
2. Results
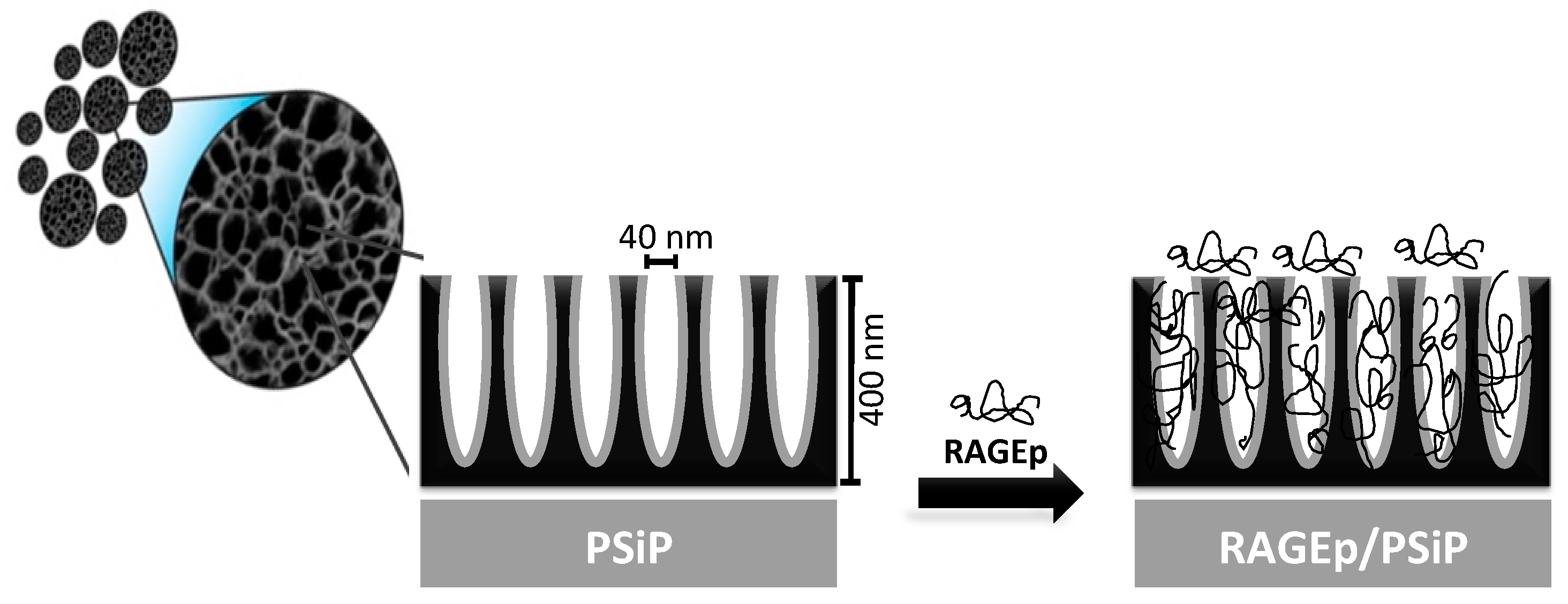
2.1. Characterization of PSiP Particles
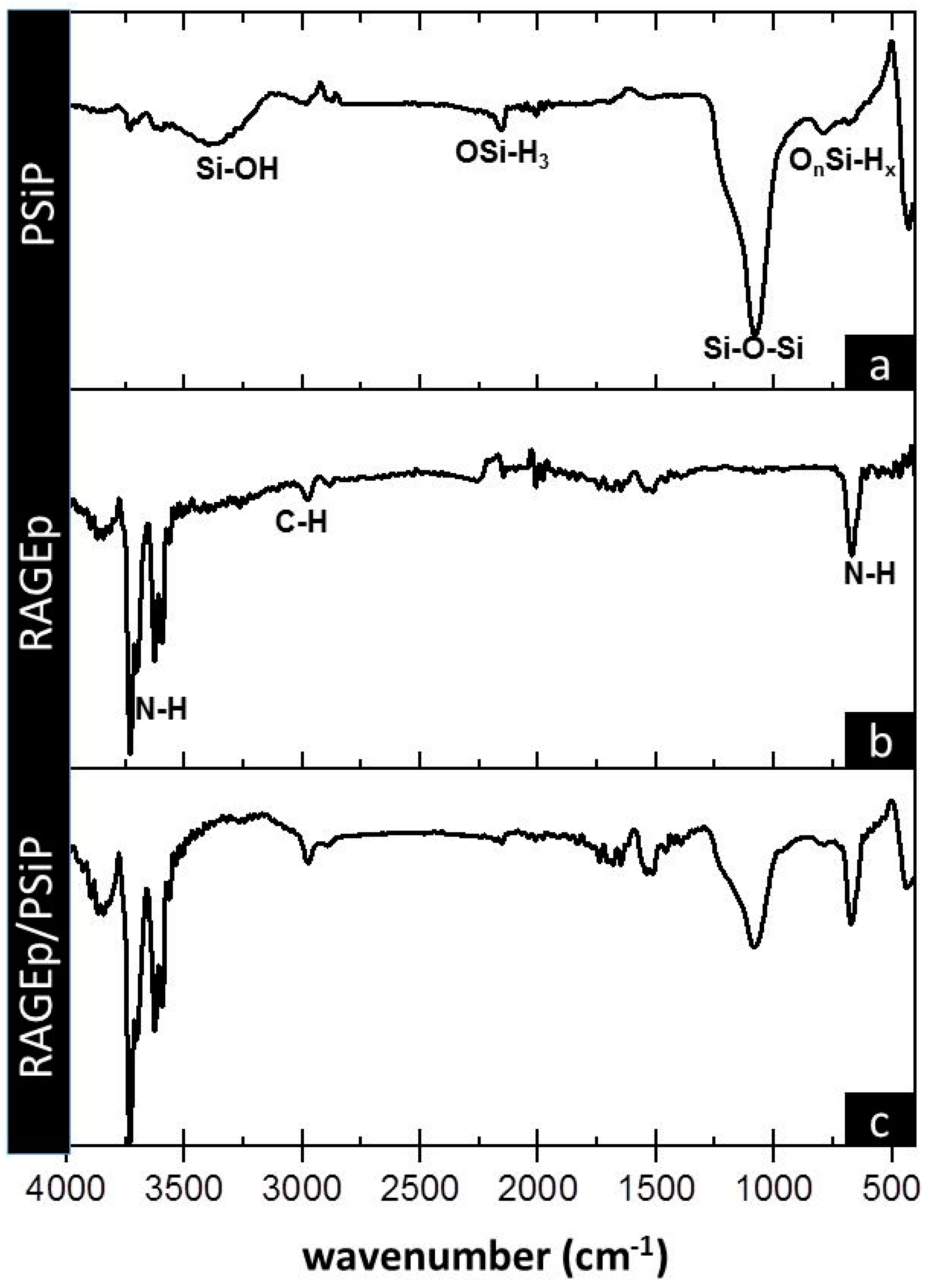
2.2. Evaluation of RAGEp Adsorption onto the PSIP Surface
2.3. The RAGEp/PSIP Vaccine Induces Long-Lasting Humoral Response in BALB/c Mice
3. Discussion
4. Materials and Methods
4.1. Electrochemical Etching Synthesis of PSiP
4.2. PSiP Characterization
4.3. Adsorption of the Synthetic RAGE Peptide onto the PSiP Surface
4.4. Immunogenicity Assay
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key Roles of Adjuvants in Modern Vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.P.; Burkhard, P. Vaccine Technologies: From Whole Organisms to Rationally Designed Protein Assemblies. Biochem. Pharmacol. 2016, 120, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Italiani, P. From Antigen Delivery System to Adjuvanticy: The Board Application of Nanoparticles in Vaccinology. Vaccines 2015, 3, 930–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques Neto, L.M.; Kipnis, A.; Junqueira-Kipnis, A.P. Role of Metallic Nanoparticles in Vaccinology: Implications for Infectious Disease Vaccine Development. Front. Immunol. 2017, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Tovar, G.; Wong-Arce, A.; Campos-Portillo, M.; Palestino, G.; Rosales-Mendoza, S. The Potential of Porous Silicon Particles for Multi-Epitopic Vaccine Development. Mesoporous Biomater. 2016, 3, 83–92. [Google Scholar] [CrossRef]
- Salonen, J.; Kaukonen, A.M.; Hirvonen, J.; Lehto, V.-P. Mesoporous Silicon in Drug Delivery Applications. J. Pharm. Sci. 2008, 97, 632–653. [Google Scholar] [CrossRef] [PubMed]
- Anglin, E.J.; Cheng, L.; Freeman, W.R.; Sailor, M.J. Porous Silicon in Drug Delivery Devices and Materials. Adv. Drug Deliv. Rev. 2008, 60, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Ruff, L.E.; Qin, Z.; Corr, M.; Hedrick, S.M.; Sailor, M.J. Multivalent Porous Silicon Nanoparticles Enhance the Immune Activation Potency of Agonistic CD40 Antibody. Adv. Mater. 2012, 24, 3981–3987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-H.; Gu, L.; von Maltzahn, G.; Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Biodegradable Luminescent Porous Silicon Nanoparticles for In Vivo Applications. Nat. Mater. 2009, 8, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.-A.; Herranz, B.; Santos, H.A. Nanostructured Porous Si-Based Nanoparticles for Targeted Drug Delivery. Biomatter 2012, 2, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Serda, R.E.; Gu, J.; Bhavane, R.C.; Liu, X.; Chiappini, C.; Decuzzi, P.; Ferrari, M. The Association of Silicon Microparticles with Endothelial Cells in Drug Delivery to the Vasculature. Biomaterials 2009, 30, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.-A.; Shrestha, N.; Mäkilä, E.; Araújo, F.; Correia, A.; Ramos, T.; Sarmento, B.; Salonen, J.; Hirvonen, J.; Santos, H.A. A Prospective Cancer Chemo-Immunotherapy Approach Mediated by Synergistic CD326 Targeted Porous Silicon Nanovectors. Nano Res. 2015, 8, 1505–1521. [Google Scholar] [CrossRef]
- Gu, L.; Hall, D.J.; Qin, Z.; Anglin, E.; Joo, J.; Mooney, D.J.; Howell, S.B.; Sailor, M.J. In Vivo Time-Gated Fluorescence Imaging with Biodegradable Luminescent Porous Silicon Nanoparticles. Nat. Commun. 2013, 4, 2326. [Google Scholar] [CrossRef] [PubMed]
- Cooray, M.C.D.; Liu, Y.; Langford, S.J.; Bond, A.M.; Zhang, J. One Pot Synthesis of poly(5-Hydroxyl-1,4-Naphthoquinone) Stabilized Gold Nanoparticles Using the Monomer as the Reducing Agent for Nonenzymatic Electrochemical Detection of Glucose. Anal. Chim. Acta 2015, 856, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.-L.; Cai, S.-J.; Li, S.; He, X.-W.; Li, W.-Y.; Li, Y.-H.; Zhang, Y.-K. One-Pot Microwave Synthesis of Water-Dispersible, High Fluorescence Silicon Nanoparticles and Their Imaging Applications In Vitro and In Vivo. Anal. Chem. 2016, 88, 11631–11638. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.J.; Liu, X.; Curley, S.A.; Ferrari, M.; Serda, R.E. Porous Silicon Advances in Drug Delivery and Immunotherapy. Curr. Opin. Pharmacol. 2013, 13, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Periáñez, A.; Abos Gracia, B.; López Relaño, J.; Diez-Rivero, C.M.; Reche, P.A.; Martínez-Naves, E.; Matveyeva, E.; Gómez Del Moral, M. Mesoporous Silicon Microparticles Enhance MHC Class I Cross-Antigen Presentation by Human Dendritic Cells. Clin. Dev. Immunol. 2013, 2013, 362163. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Mai, J.; Xu, R.; Perez, J.E.T.; Guevara, M.L.; Shen, Q.; Mu, C.; Tung, H.-Y.; Corry, D.B.; Evans, S.E.; et al. Porous Silicon Microparticle Potentiates Anti-Tumor Immunity by Enhancing Cross-Presentation and Inducing Type I Interferon Response. Cell Rep. 2015, 11, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, Classification, and Expression of RAGE Gene Splice Variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.J. Is RAGE Still a Therapeutic Target for Alzheimer’s Disease? Future Med. Chem. 2012, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Kumeria, T. Electrochemical Etching Methods for Producing Porous Silicon; Springer: Cham, Switzerland, 2015; pp. 1–36. [Google Scholar]
- Maniya, N.H.; Patel, S.R.; Murthy, Z.V.P. Drug Delivery with Porous Silicon Films, Microparticles, and Nanoparticles. Rev. Adv. Mater. Sci. 2016, 44, 257–272. [Google Scholar]
- Wang, F.; Hui, H.; Barnes, T.J.; Barnett, C.; Prestidge, C.A. Oxidized Mesoporous Silicon Microparticles for Improved Oral Delivery of Poorly Soluble Drugs. Mol. Pharm. 2010, 7, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Chhablani, J.; Nieto, A.; Hou, H.; Wu, E.C.; Freeman, W.R.; Sailor, M.J.; Cheng, L. Oxidized Porous Silicon Particles Covalently Grafted with Daunorubicin as a Sustained Intraocular Drug Delivery System. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1268–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonio, J.; Júnior, A.; Baldo, J.B. The Behavior of Zeta Potential of Silica Suspensions. NJGC 2014, 4, 29–37. [Google Scholar] [CrossRef]
- Ogata, Y.H. Characterization of Porous Silicon by Infrared Spectroscopy. In Handbook of Porous Silicon; Canham, L., Ed.; Springer: Cham, Switzerland, 2014. [Google Scholar]
- Bywalez, R.; Karacuban, H.; Nienhaus, H.; Schulz, C.; Wiggers, H. Stabilization of Mid-Sized Silicon Nanoparticles by Functionalization with Acrylic Acid. Nanoscale Res. Lett. 2012, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Rivillon, S.; Brewer, R.T.; Chabal, Y.J. Water Reaction with Chlorine-Terminated Silicon (111) and (100) Surfaces. Appl. Phys. Lett. 2005, 87, 173118. [Google Scholar] [CrossRef]
- Limnell, T.; Riikonen, J.; Salonen, J.; Kaukonen, A.M.; Laitinen, L.; Hirvonen, J.; Lehto, V.-P. Surface Chemistry and Pore Size Affect Carrier Properties of Mesoporous Silicon Microparticles. Int. J. Pharm. 2007, 343, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Huotari, A.; Xu, W.; Mönkäre, J.; Kovalainen, M.; Herzig, K.-H.; Lehto, V.-P.; Järvinen, K. Effect of Surface Chemistry of Porous Silicon Microparticles on Glucagon-like Peptide-1 (GLP-1) Loading, Release and Biological Activity. Int. J. Pharm. 2013, 454, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, K.L.; Barnes, T.J.; Prestidge, C.A. Surface Chemistry of Porous Silicon and Implications for Drug Encapsulation and Delivery Applications. Adv. Colloid Interface Sci. 2012, 175, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Kaasalainen, M.; Mäkilä, E.; Riikonen, J.; Kovalainen, M.; Järvinen, K.; Herzig, K.-H.; Lehto, V.-P.; Salonen, J. Effect of Isotonic Solutions and Peptide Adsorption on Zeta Potential of Porous Silicon Nanoparticle Drug Delivery Formulations. Int. J. Pharm. 2012, 431, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Rivillon, S.; Chabal, Y.J.; Webb, L.J.; Michalak, D.J.; Lewis, N.S.; Halls, M.D.; Raghavachari, K. Chlorination of Hydrogen-Terminated Silicon (111) Surfaces. J. Vac. Sci. Technol. A 2005, 23, 1100–1106. [Google Scholar] [CrossRef]
- Zaman, M.; Atiyatul, A.; Gulam, Q.; Rizwan, R.; Khan, H.; Khan, R.H. Nanoparticles in Relation to Peptide and Protein Aggregation. Int. J. Nanomed. 2014, 9, 899–912. [Google Scholar]
- Maniya, N.H.; Patel, S.R.; Murthy, Z.V.P. Study on Surface Chemistry and Particle Size of Porous Silicon Prepared by Electrochemical Etching. Mater. Res. Bull. 2014, 57, 6–12. [Google Scholar] [CrossRef]
- Ikonen, T.; Nissinen, T.; Pohjalainen, E.; Sorsa, O.; Kallio, T.; Lehto, V.-P. Electrochemically Anodized Porous Silicon: Towards Simple and Affordable Anode Material for Li-Ion Batteries. Sci. Rep. 2017, 7, 7880. [Google Scholar] [CrossRef] [PubMed]
- Nadarassan, D. Biomolecule Adsorption and Release from Porous Silicon. In Handbook of Porous Silicon; Canham, L., Ed.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Arwin, H.; Gavutis, M.; Gustafsson, J.; Schultzberg, M.; Zangooie, S.; Tengvall, P. Protein Adsorption in Thin Porous Silicon Layers. Phys. Status Solidi 2000, 182, 515–520. [Google Scholar] [CrossRef]
- Cui, Z.; Zhang, J.; Xue, Y.; Duan, H. Size-Dependent Thermodynamics and Kinetics of Adsorption on Nanoparticles: A Theoretical and Experimental Study. Langmuir 2018, 34, 3197–3206. [Google Scholar] [CrossRef] [PubMed]
- Araújo, F.; Shrestha, N.; Shahbazi, M.A.; Fonte, P.; Mäkilä, E.M.; Salonen, J.J.; Hirvonen, J.T.; Granja, P.L.; Santos, H.A.; Sarmento, B. The Impact of Nanoparticles on the Mucosal Translocation and Transport of GLP-1 across the Intestinal Epithelium. Biomaterials 2014, 35, 9199–9207. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Bimbo, L.M.; Mäkilä, E.; Villanova, F.; Kaasalainen, M.; Herranz-Blanco, B.; Caramella, C.M.; Lehto, V.-P.; Salonen, J.; Herzig, K.-H.; et al. Co-Delivery of a Hydrophobic Small Molecule and a Hydrophilic Peptide by Porous Silicon Nanoparticles. J. Control. Release 2013, 170, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Moir, R.D.; Wagner, S.L. Clearance of Alzheimer’s Aβ Peptide: The Many Roads to Perdition. Neuron 2004, 43, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Herring, A.; Yasin, H.; Ambrée, O.; Sachser, N.; Paulus, W.; Keyvani, K. Environmental Enrichment Counteracts Alzheimer’s Neurovascular Dysfunction in TgCRND8 Mice. Brain Pathol. 2008, 18, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Mruthinti, S.; Hill, W.D.; Buccafusco, J.J.; Terry, A.V. An Aqueous Orally Active Vaccine Targeted against a RAGE/AB Complex as a Novel Therapeutic for Alzheimer’s Disease. Neuromol. Med. 2012, 14, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Bimbo, L.M.; Sarparanta, M.; Mäkilä, E.; Laaksonen, T.; Laaksonen, P.; Salonen, J.; Linder, M.B.; Hirvonen, J.; Airaksinen, A.J.; Santos, H.A. Cellular Interactions of Surface Modified Nanoporous Silicon Particles. Nanoscale 2012, 4, 3184–3192. [Google Scholar] [CrossRef] [PubMed]
- Sarparanta, M.; Bimbo, L.M.; Rytkoänen, J.; Mäkilä, E.; Laaksonen, T.J.; Laaksonen, P.; Nyman, M.; Salonen, J.; Linder, M.B.; Hirvonen, J.; et al. Intravenous Delivery of Hydrophobin-Functionalized Porous Silicon Nanoparticles: Stability, Plasma Protein Adsorption and Biodistribution. Mol. Pharm. 2012, 9, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Tölli, M.A.; Ferreira, M.P.A.; Kinnunen, S.M.; Rysä, J.; Mäkilä, E.M.; Szabó, Z.; Serpi, R.E.; Ohukainen, P.J.; Välimäki, M.J.; Correia, A.M.R. In Vivo Biocompatibility of Porous Silicon Biomaterials for Drug Delivery to the Heart. Biomaterials 2014, 35, 8394–8405. [Google Scholar] [CrossRef] [PubMed]
- Bimbo, L.M.; Sarparanta, M.; Santos, H.A.; Airaksinen, A.J.; Mäkilä, E.; Laaksonen, T.; Peltonen, L.; Lehto, V.-P.; Hirvonen, J.; Salonen, J. Biocompatibility of Thermally Hydrocarbonized Porous Silicon Nanoparticles and Their Biodistribution in Rats. ACS Nano 2010, 4, 3023–3032. [Google Scholar] [CrossRef] [PubMed]
- Santos, H.A.; Riikonen, J.; Salonen, J.; Mäkilä, E.; Heikkilä, T.; Laaksonen, T.; Peltonen, L.; Lehto, V.P.; Hirvonen, J. In Vitro Cytotoxicity of Porous Silicon Microparticles: Effect of the Particle Concentration, Surface Chemistry and Size. Acta Biomater. 2010, 6, 2721–2731. [Google Scholar] [CrossRef] [PubMed]
- Kilpeläinen, M.; Mönkäre, J.; Vlasova, M.A.; Riikonen, J.; Lehto, V.P.; Salonen, J.; Järvinen, K.; Herzig, K.H. Nanostructured Porous Silicon Microparticles Enable Sustained Peptide (Melanotan II) Delivery. Eur. J. Pharm. Biopharm. 2011, 77, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Pastor, E.L.; Reguera-Nuñez, E.; Matveeva, E.; Garcia-Fuentes, M. Pore Size Is a Critical Parameter for Obtaining Sustained Protein Release from Electrochemically Synthesized Mesoporous Silicon Microparticles. PeerJ 2015, 3, e1277. [Google Scholar] [CrossRef] [PubMed]
- Kilpeläinen, M.; Riikonen, J.; Vlasova, M.A.; Huotari, A.; Lehto, V.P.; Salonen, J.; Herzig, K.H.; Järvinen, K. In Vivo Delivery of a Peptide, Ghrelin Antagonist, with Mesoporous Silicon Microparticles. J. Control. Release 2009, 137, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Sarparanta, M.; Mäkilä, E.; Heikkilä, T.; Salonen, J.; Kukk, E.; Lehto, V.P.; Santos, H.A.; Hirvonen, J.; Airaksinen, A.J. 18F-Labeled Modified Porous Silicon Particles for Investigation of Drug Delivery Carrier Distribution In Vivo with Positron Emission Tomography. Mol. Pharm. 2011, 8, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- A Phase 2 Study Evaluating the Efficacy and Safety of PF 04494700 in Mild to Moderate Alzheimer’s Disease—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00566397 (accessed on 17 May 2017).
- 6-Month Safety and Efficacy Study of TTP488 in Patients with Type 2 Diabetes and Persistent Albuminuria—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00287183 (accessed on 17 May 2017).
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and Expression of a Cell Surface Receptor for Advanced Glycosylation End Products of Proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumber (cm−1) | IR Signal Presenting a Change in Intensity with Respect to PSiP IR Signals | Possible Intermolecular Force |
|---|---|---|
| 3370 | O–H stretching signal of Si–O–H bond [33] | Electrostatic |
| 2150 | OSiH3 stretching band [26,27] | Ion–dipole |
| 1066 | Si–O–Si stretching signal. Broad band with a shoulder at 1215 cm−1 [26,27]. | Ion–dipole |
| 767 | OnSi–Hx deformational vibration [23,24]. | Electrostatic/ion–dipole |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro-Tovar, G.; Rocha-García, D.; Wong-Arce, A.; Palestino, G.; Rosales-Mendoza, S. Mesoporous Silicon Particles Favor the Induction of Long-Lived Humoral Responses in Mice to a Peptide-Based Vaccine. Materials 2018, 11, 1083. https://doi.org/10.3390/ma11071083
Navarro-Tovar G, Rocha-García D, Wong-Arce A, Palestino G, Rosales-Mendoza S. Mesoporous Silicon Particles Favor the Induction of Long-Lived Humoral Responses in Mice to a Peptide-Based Vaccine. Materials. 2018; 11(7):1083. https://doi.org/10.3390/ma11071083
Chicago/Turabian StyleNavarro-Tovar, Gabriela, Denisse Rocha-García, Alejandra Wong-Arce, Gabriela Palestino, and Sergio Rosales-Mendoza. 2018. "Mesoporous Silicon Particles Favor the Induction of Long-Lived Humoral Responses in Mice to a Peptide-Based Vaccine" Materials 11, no. 7: 1083. https://doi.org/10.3390/ma11071083