An Ab Initio Study of Connections between Tensorial Elastic Properties and Chemical Bonds in Σ5(210) Grain Boundaries in Ni3Si




Abstract
:1. Introduction
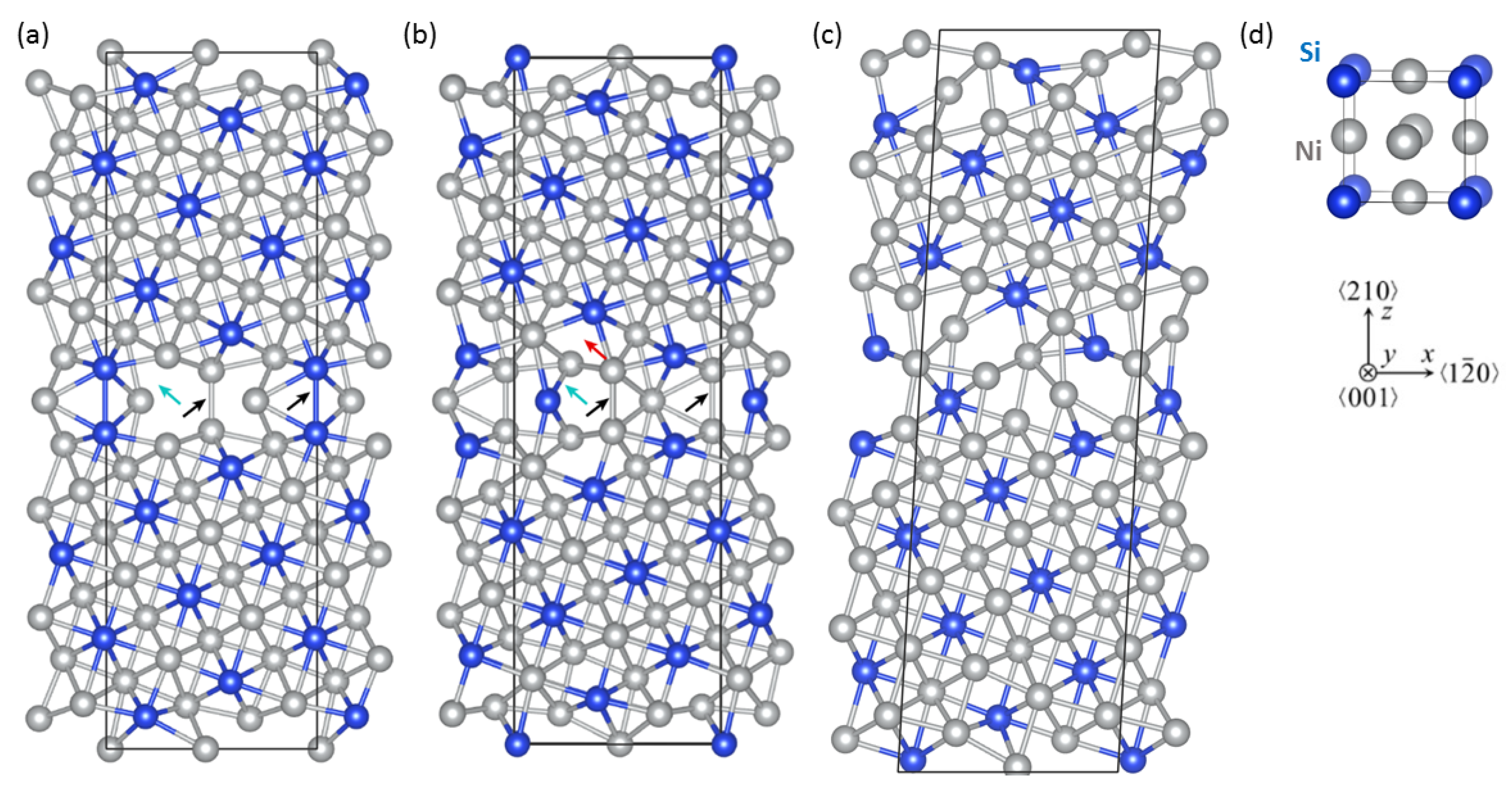
2. Methods
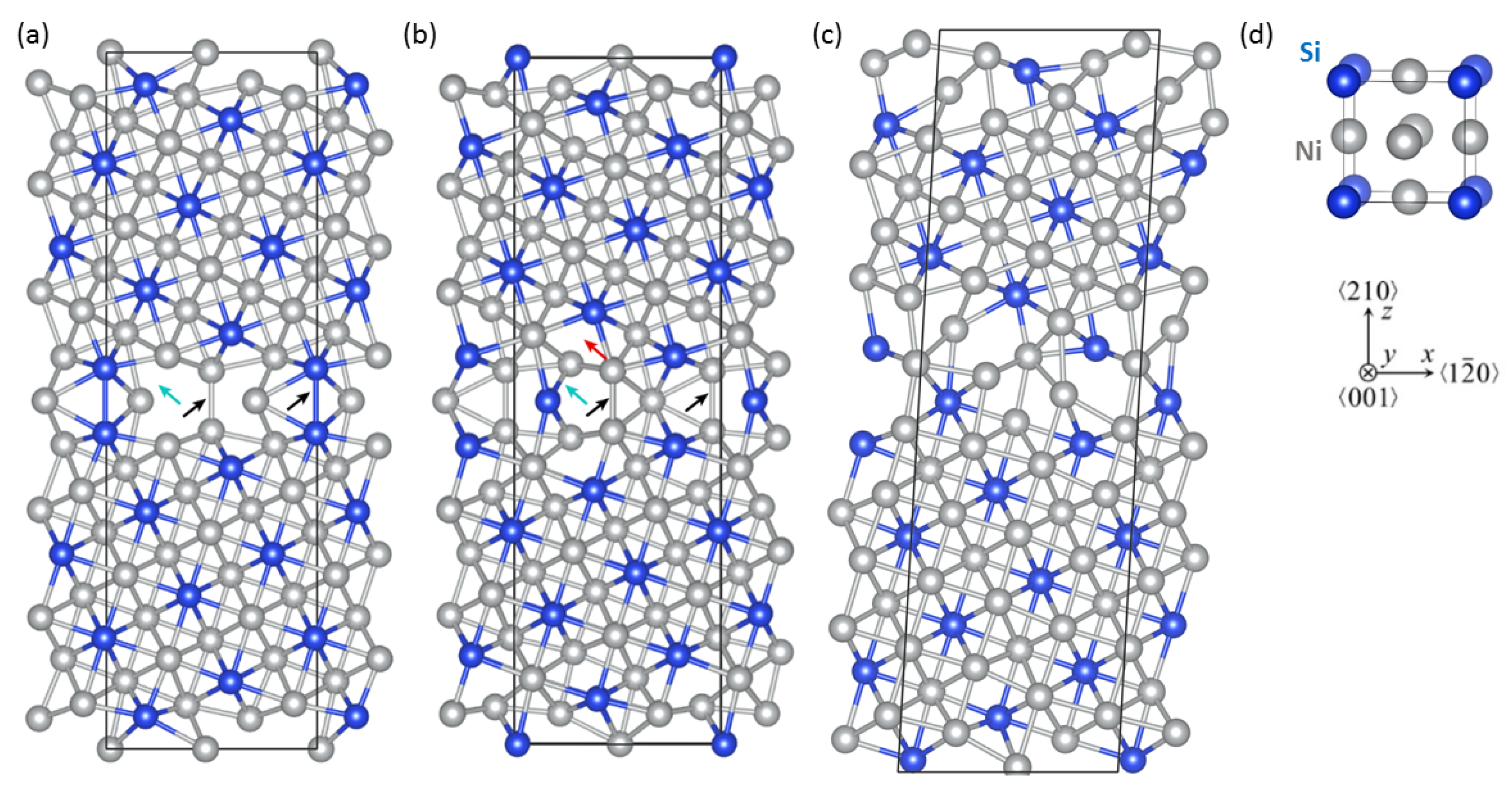
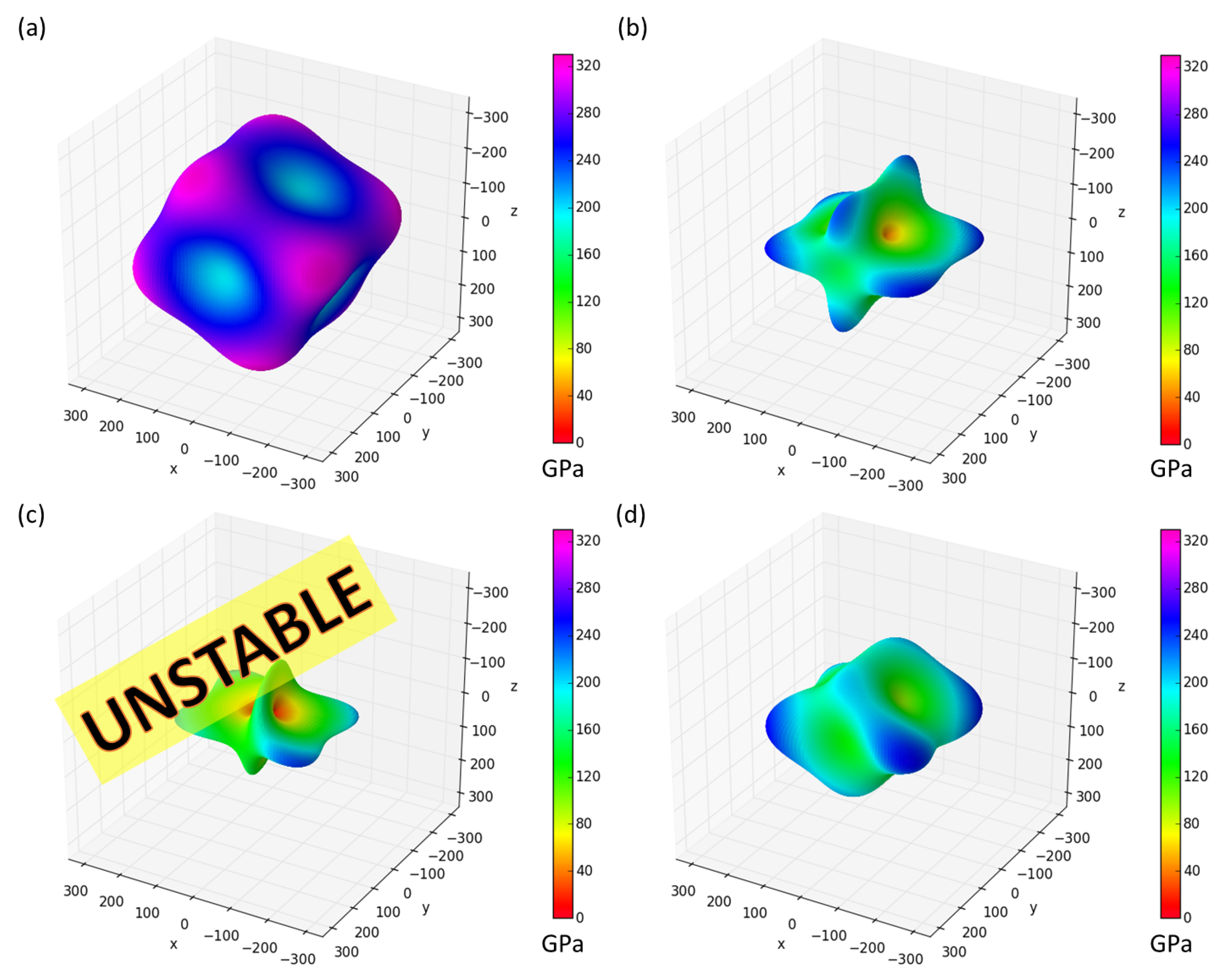
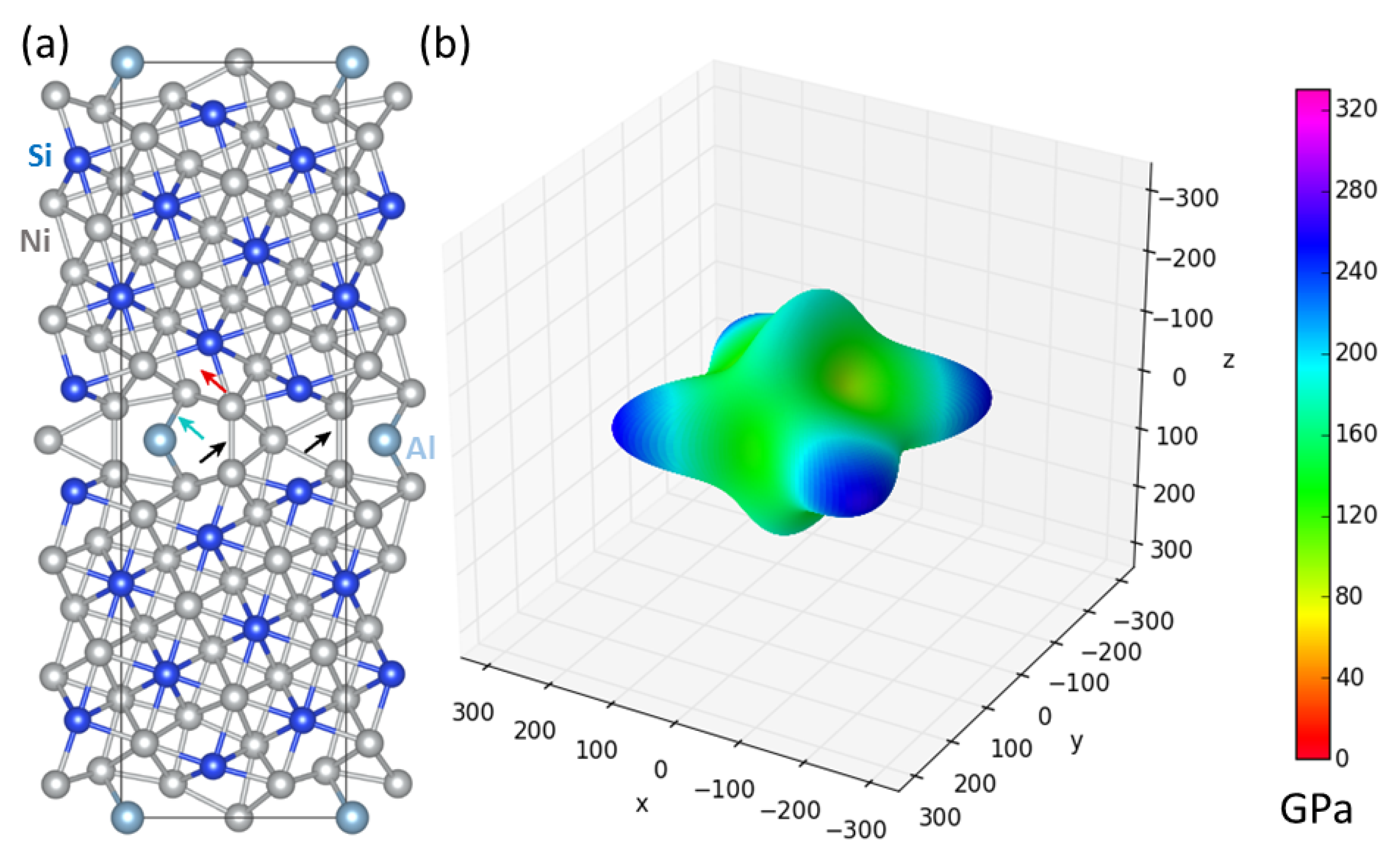
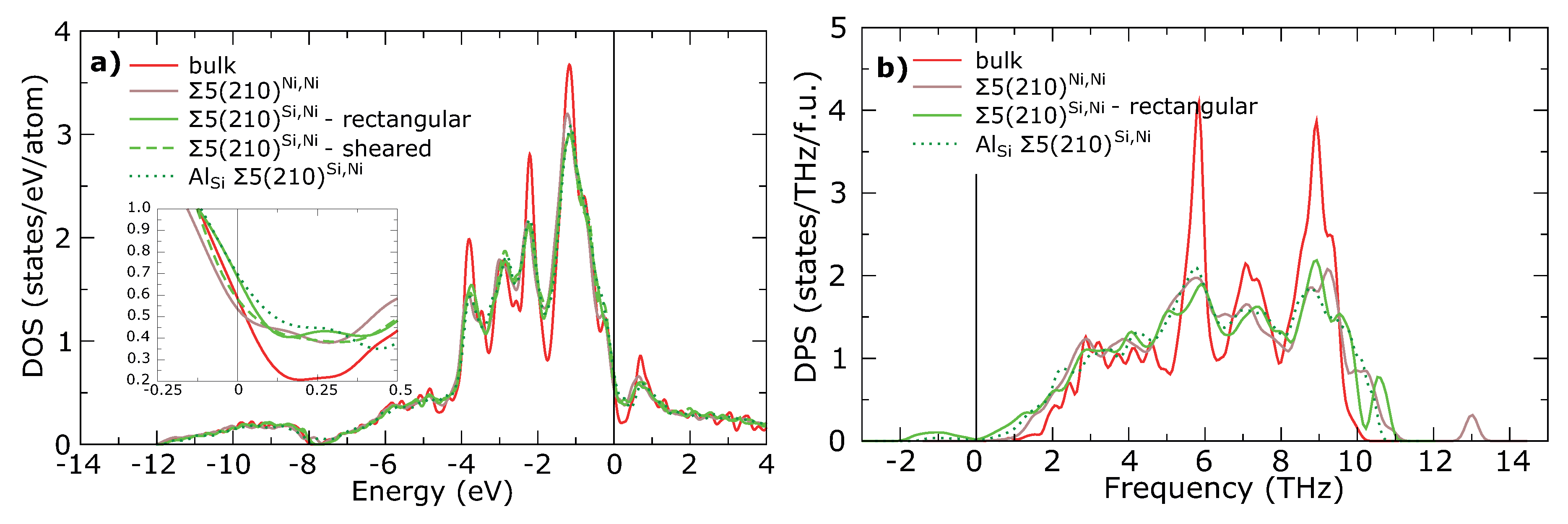
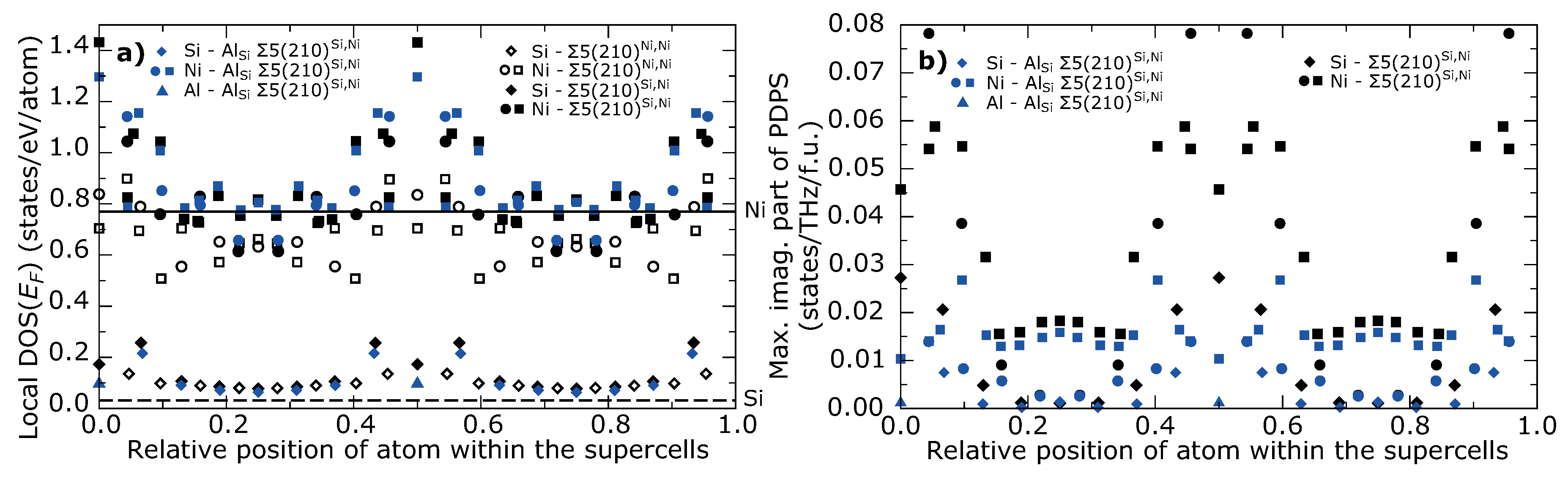
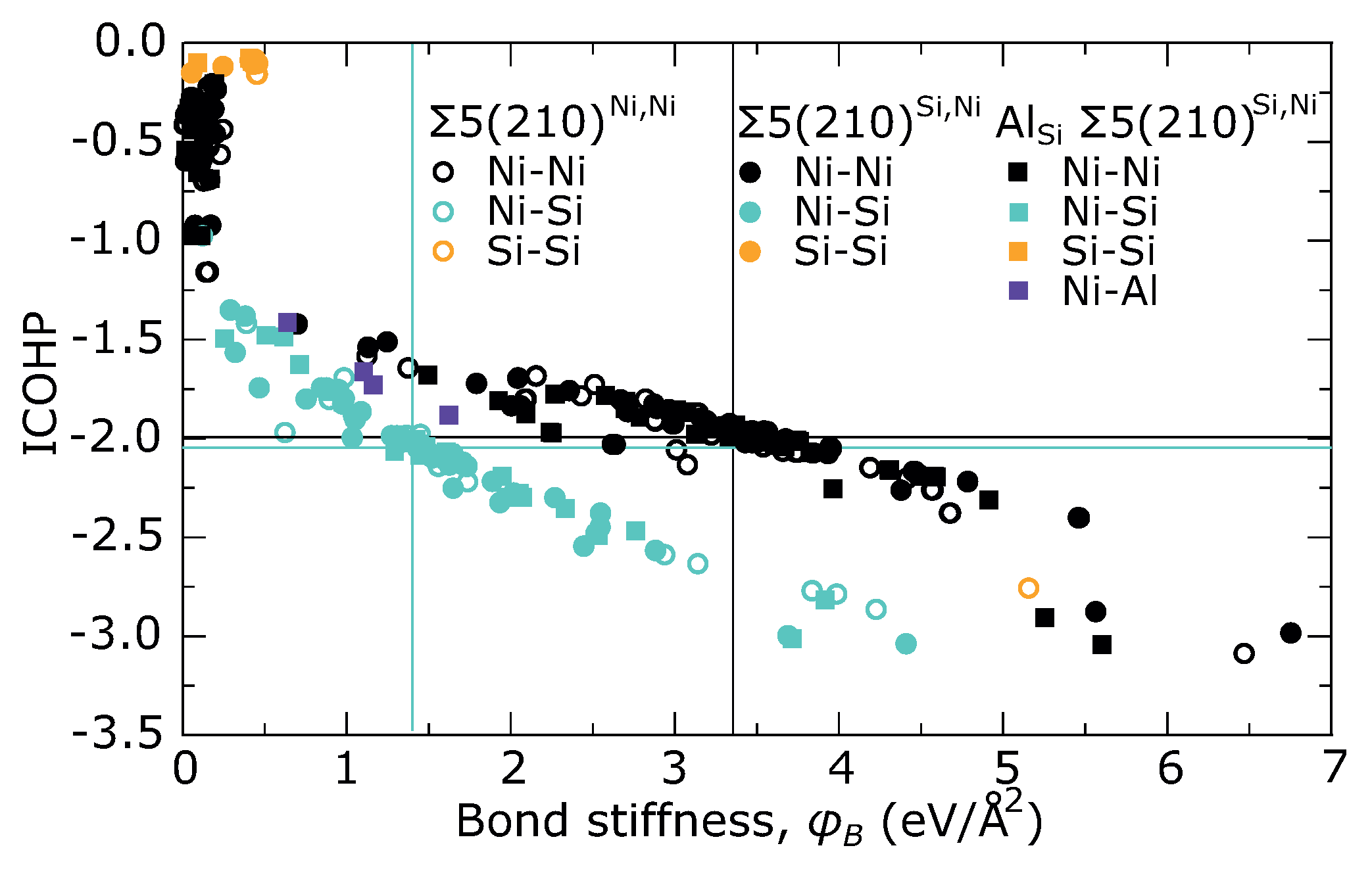
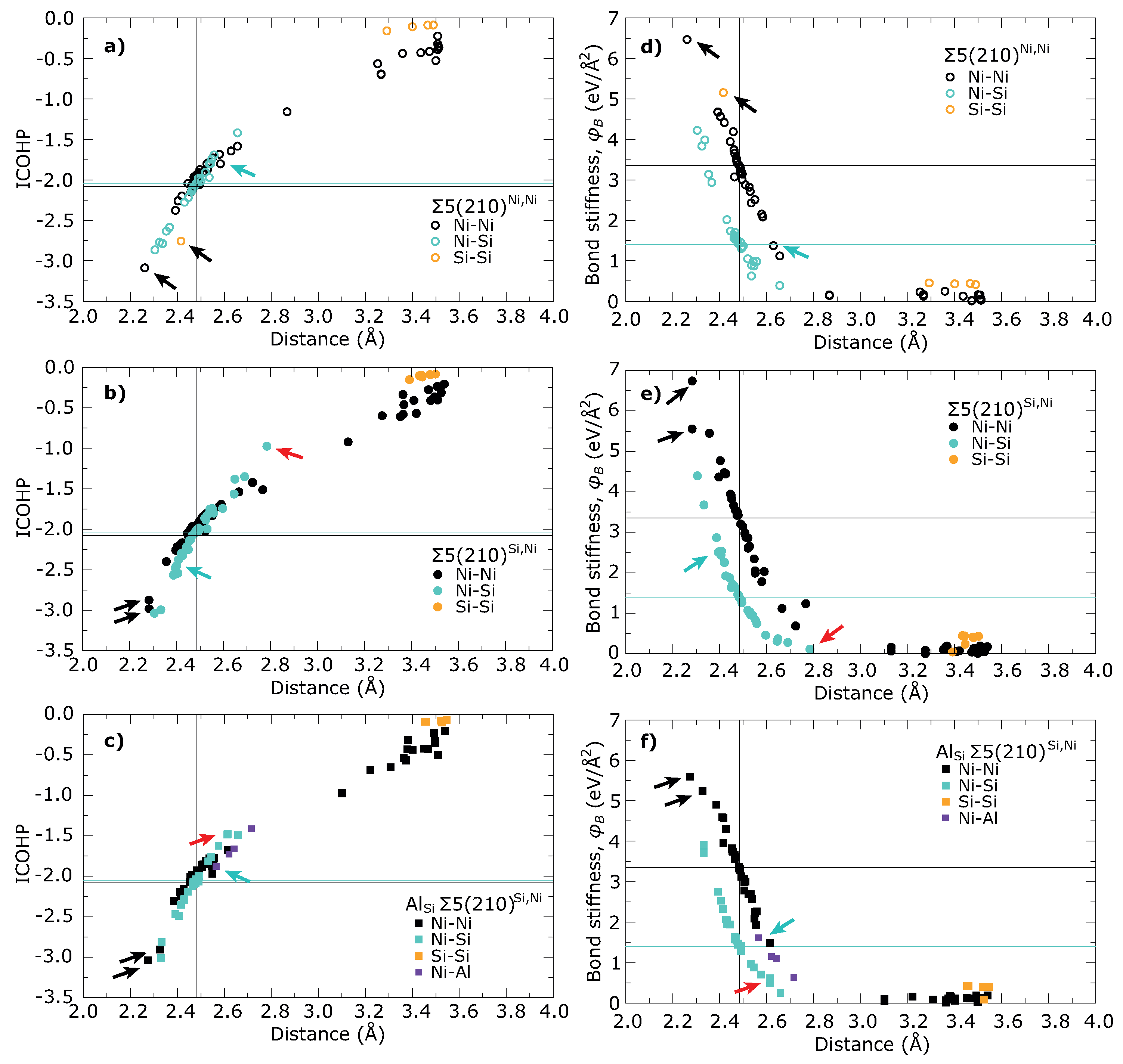
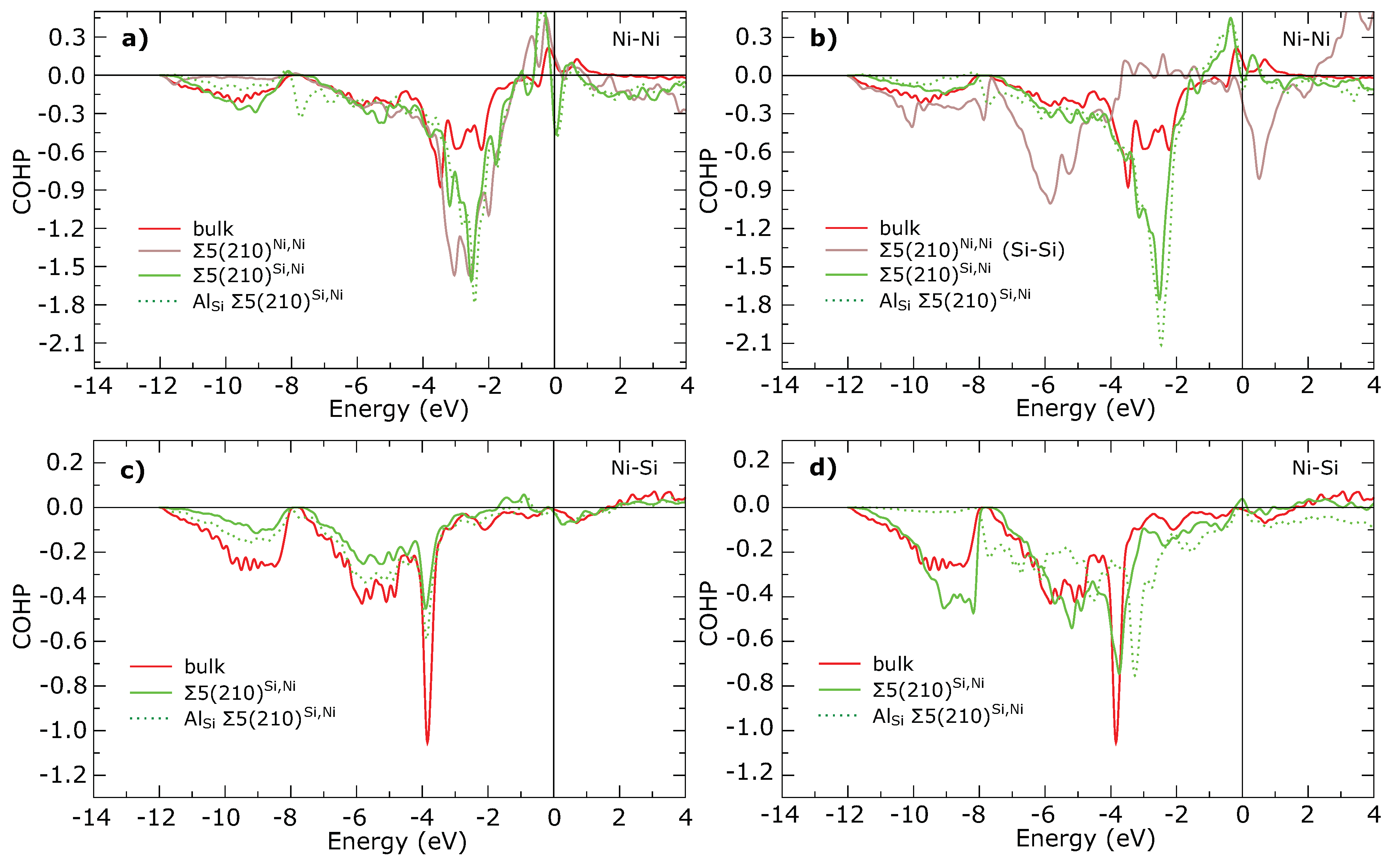
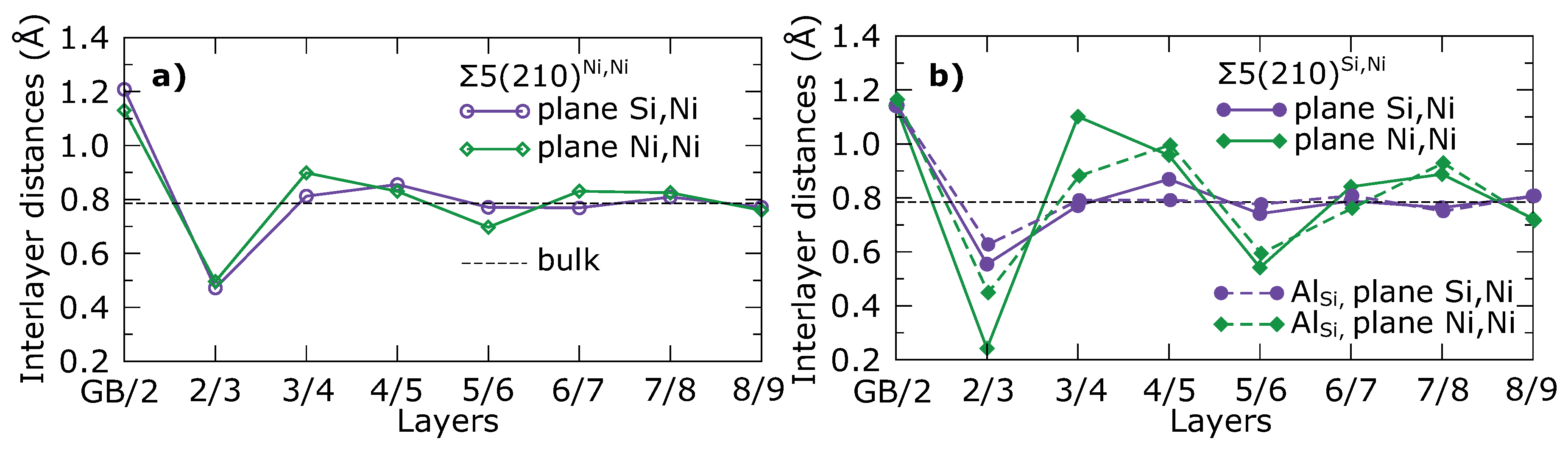
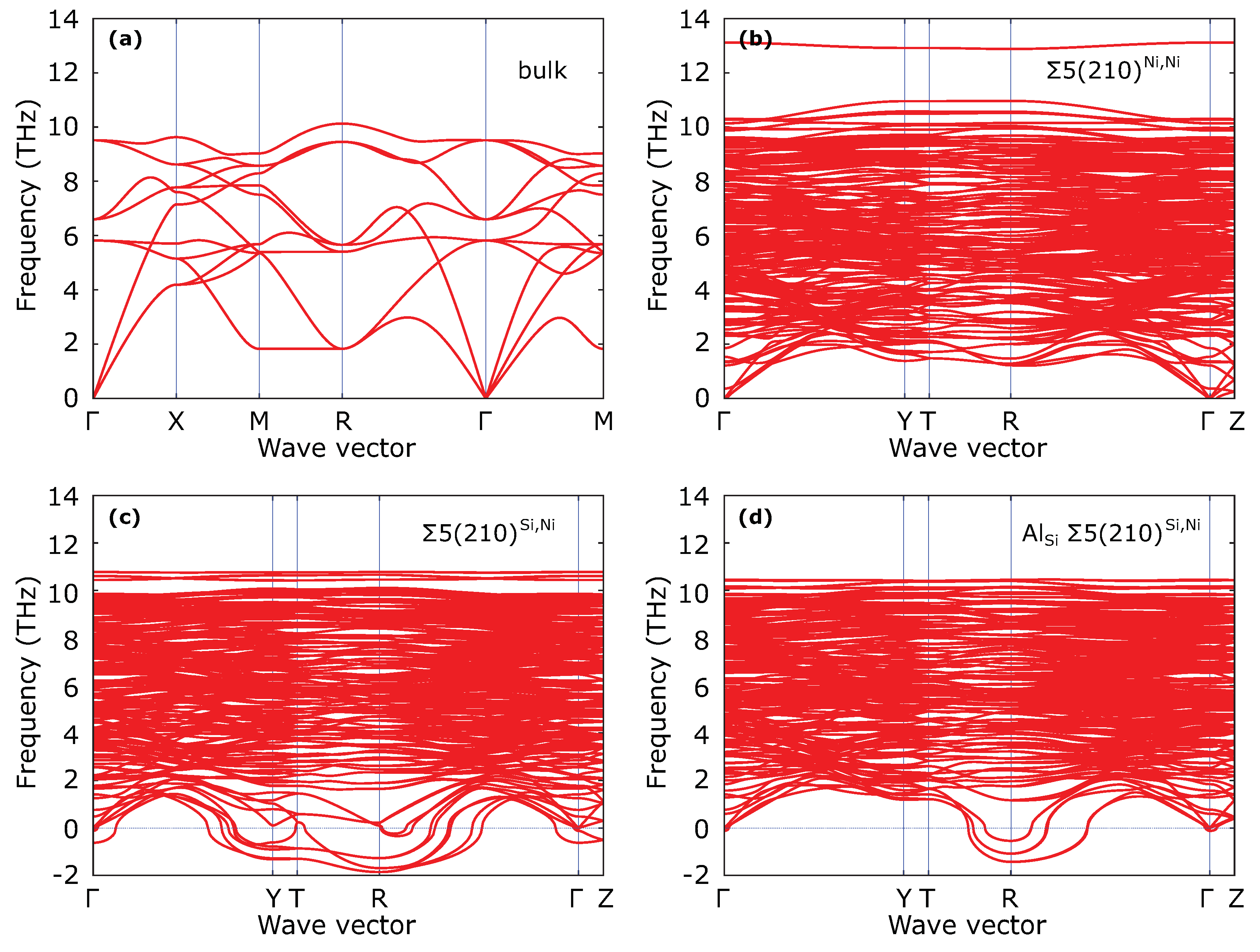
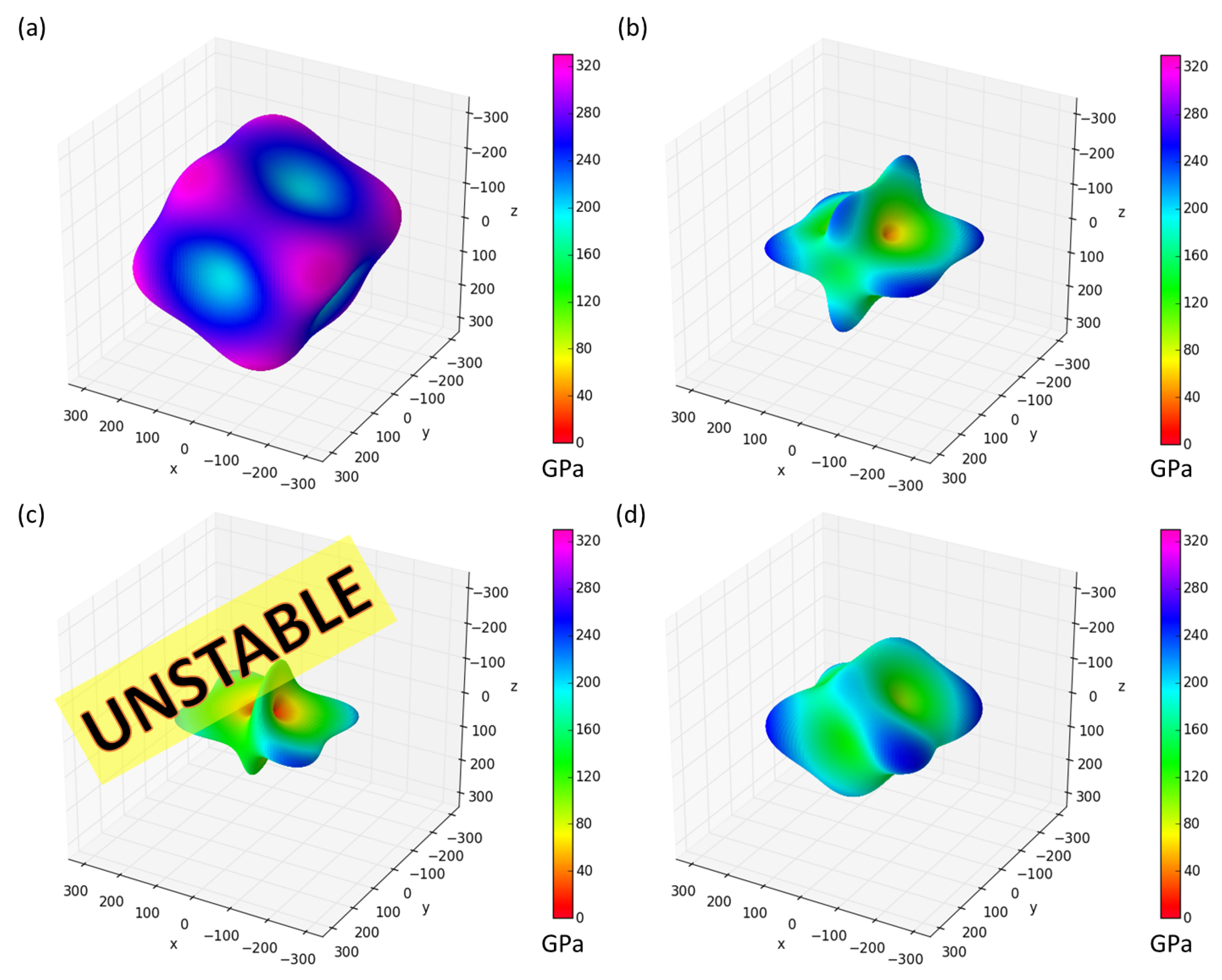
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ni3Si States: | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| bulk 〈10〉 〈001〉 〈210〉 | 0 | 26.5 | 0 | 0 | 0 | 0 | 0 | −26.5 | 1 | 0 | 0 | 0 |
| ΣSi,Ni rectangular | 0 | −3 | 0 | 1 | 13 | −3 | 1 | 16 | 7 | −12 | −1 | −4 |
| ΣSi,Ni sheared | 0 | −5 | 0 | −1 | 6 | 1 | 2 | −4 | 0 | 1 | 9 | −2 |

References
- Duscher, G.; Chisholm, M.F.; Alber, U.; Ruhle, M. Bismuth-induced embrittlement of copper grain boundaries. Nat. Mater. 2004, 3, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.H.; Deng, S.H.; Wang, T.M.; Kohyama, M.; Yamamoto, R. Theoretical tensile strength of an Al grain boundary. Phys. Rev. B 2004, 69, 134106. [Google Scholar] [CrossRef]
- Kohyama, M. Ab initio study of the tensile strength and fracture of coincidence tilt boundaries in cubic SiC: Polar interfaces of the {122} Σ9 boundary. Phys. Rev. B 2002, 65, 184107. [Google Scholar] [CrossRef]
- Ogata, S.; Umeno, Y.; Kohyama, M. First-principles approaches to intrinsic strength and deformation of materials: Perfect crystals, nano-structures, surfaces and interfaces. Model. Simul. Mater. Sci. Eng. 2009, 17, 013001. [Google Scholar] [CrossRef]
- Pokluda, J.; Černý, M.; Šob, M.; Umeno, Y. Ab initio calculations of mechanical properties: Methods and applications. Prog. Mater. Sci. 2015, 73, 127–158. [Google Scholar] [CrossRef]
- Tang, M.; Carter, W.C.; Cannon, R.M. Diffuse interface model for structural transitions of grain boundaries. Phys. Rev. B 2006, 73, 024102. [Google Scholar] [CrossRef]
- Rohrer, G.S. Grain boundary energy anisotropy: A review. J. Mater. Sci. 2011, 46, 5881–5895. [Google Scholar] [CrossRef]
- Cantwell, P.R.; Tang, M.; Dillon, S.J.; Luo, J.; Rohrer, G.S.; Harmer, M.P. Grain boundary complexions. Acta Mater. 2014, 62, 1–48. [Google Scholar] [CrossRef]
- Rohrer, G.S. Measuring and Interpreting the Structure of Grain-Boundary Networks. J. Am. Ceram. Soc. 2011, 94, 633–646. [Google Scholar] [CrossRef]
- Raabe, D.; Herbig, M.; Sandlöbes, S.; Li, Y.; Tytko, D.; Kuzmina, M.; Ponge, D.; Choi, P.P. Grain boundary segregation engineering in metallic alloys: A pathway to the design of interfaces. Curr. Opin. Sol. State Mater. Sci. 2014, 18, 253–261. [Google Scholar] [CrossRef]
- Dillon, S.J.; Harmer, M.P.; Luo, J. Grain Boundary Complexions in Ceramics and Metals: An Overview. JOM 2009, 61, 38–44. [Google Scholar] [CrossRef]
- Shi, X.; Luo, J. Developing grain boundary diagrams as a materials science tool: A case study of nickel-doped molybdenum. Phys. Rev. B 2011, 84, 014105. [Google Scholar] [CrossRef]
- Kundu, A.; Asl, K.M.; Luo, J.; Harmer, M.P. Identification of a bilayer grain boundary complexion in Bi-doped Cu. Scr. Mater. 2013, 68, 146–149. [Google Scholar] [CrossRef]
- Bojarski, S.A.; Ma, S.; Lenthe, W.; Harmer, M.P.; Rohrer, G.S. Changes in the Grain Boundary Character and Energy Distributions Resulting from a Complexion Transition in Ca-Doped Yttria. Metall. Mater. Trans. A 2012, 43A, 3532–3538. [Google Scholar] [CrossRef]
- Rickman, J.M.; Chan, H.M.; Harmer, M.P.; Luo, J. Grain-boundary layering transitions in a model bicrystal. Surf. Sci. 2013, 618, 88–93. [Google Scholar] [CrossRef]
- Bojarski, S.A.; Harmer, M.P.; Rohrer, G.S. Influence of grain boundary energy on the nucleation of complexion transitions. Scr. Mater. 2014, 88, 1–4. [Google Scholar] [CrossRef]
- Frazier, W.E.; Rohrer, G.S.; Rollett, A.D. Abnormal grain growth in the Potts model incorporating grain boundary complexion transitions that increase the mobility of individual boundaries. Acta Mater. 2015, 96, 390–398. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Luo, J. Developing grain boundary diagrams for multicomponent alloys. Acta Mater. 2015, 91, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Moghadam, M.M.; Rickman, J.M.; Harmer, M.P.; Chan, H.M. The role of boundary variability in polycrystalline grain-boundary diffusion. J. Appl. Phys. 2015, 117, 045311. [Google Scholar] [CrossRef]
- Lu, G.H.; Zhang, Y.; Deng, S.; Wang, T.; Kohyama, M.; Yamamoto, R.; Liu, F.; Horikawa, K.; Kanno, M. Origin of intergranular embrittlement of Al alloys induced by Na and Ca segregation: Grain boundary weakening. Phys. Rev. B 2006, 73, 224115. [Google Scholar] [CrossRef]
- Yan, M.; Šob, M.; Luzzi, D.E.; Vitek, V.; Ackland, G.; Methfessel, M.; Rodriguez, C. Interatomic forces and atomic-structure of grain-boundaries in copper-bismuth alloys. Phys. Rev. B 1993, 47, 5571–5582. [Google Scholar] [CrossRef]
- Braithwaite, J.S.; Rez, P. Grain boundary impurities in iron. Acta Mater. 2005, 53, 2715–2726. [Google Scholar] [CrossRef]
- Christensen, M.; Wahnstrom, G. Co-phase penetration of WC(1010)/WC(110) grain boundaries from first principles. Phys. Rev. B 2003, 67, 115415. [Google Scholar] [CrossRef]
- Du, Y.A.; Ismer, L.; Rogal, J.; Hickel, T.; Neugebauer, J.; Drautz, R. First-principles study on the interaction of H interstitials with grain boundaries in α- and γ-Fe. Phys. Rev. B 2011, 84, 144121. [Google Scholar] [CrossRef]
- Asta, M.; Hoyt, J.J. Thermodynamic properties of coherent interfaces in f.c.c.-based Ag-Al alloys: A first-principles study. Acta Mater. 2000, 48, 1089–1096. [Google Scholar] [CrossRef]
- Thomson, D.I.; Heine, V.; Payne, M.C.; Marzari, N.; Finnis, M.W. Insight into gallium behavior in aluminum grain boundaries from calculation on Σ11 (113) boundary. Acta Mater. 2000, 48, 3623–3632. [Google Scholar] [CrossRef]
- Wachowicz, E.; Ossowski, T.; Kiejna, A. Cohesive and magnetic properties of grain boundaries in bcc Fe with Cr additions. Phys. Rev. B 2010, 81, 094104. [Google Scholar] [CrossRef]
- Wachowicz, E.; Kiejna, A. Effect of impurities on grain boundary cohesion in bcc iron. Comput. Mater. Sci. 2008, 43, 736–743. [Google Scholar] [CrossRef]
- Sutton, A.P.; Balluffi, R.W. Interfaces in Crystalline Materials; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Lejček, P. Grain Boundary Segregation in Metals; Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Liu, C.T.; George, E.P.; Oliver, W.C. Grain-boundary fracture and boron effect in Ni3Si alloys. Intermetallics 1996, 4, 77–83. [Google Scholar] [CrossRef]
- Vitek, V. Micromechanisms of intergranular brittle fracture in intermetallic compounds. J. Phys. III France 1991, 1, 1085–1097. [Google Scholar] [CrossRef]
- Kruisman, J.J.; Vitek, V.; Hosson, J.D. Atomic structure of stoichiometric and non-stoichiometric grain boundaries in A3B compounds with L12 structure. Acta Metall. 1988, 36, 2729–2741. [Google Scholar] [CrossRef]
- Briant, C.L. Intermetallic Compounds: Principles; JohnWiley and Sons, Ltd.: New York, NY, USA, 1994; Volume 1, p. 895. [Google Scholar]
- Stoloff, N.; Liu, C.; Deevi, S. Emerging applications of intermetallics. Intermetallics 2000, 8, 1313–1320. [Google Scholar] [CrossRef]
- Takasugi, T.; Izumi, O. Electronic and structural studies of grain boundary strength and fracture in L12 ordered alloys—I. On binary A3B alloys. Acta Metall. 1985, 33, 1247–1258. [Google Scholar] [CrossRef]
- Taub, A.; Briant, C. Composition dependence of ductility in boron-doped, nickel-base L12 alloys. Acta Metall. 1987, 35, 1597–1603. [Google Scholar] [CrossRef]
- Messmer, R.; Briant, C. The role of chemical bonding in grain boundary embrittlement. Acta Metall. 1982, 30, 457–467. [Google Scholar] [CrossRef]
- Liu, C.T.; White, C.L.; Horton, J.A. Effect of boron on grain-boundaries in Ni3Al. Acta Metall. 1985, 33, 213–229. [Google Scholar] [CrossRef]
- Schulson, E.; Briggs, L.; Baker, I. The strength and ductility of Ni3Si. Acta Metall. Mater. 1990, 38, 207–213. [Google Scholar] [CrossRef]
- Aoki, K.; Izumi, O. Improvement in room temperature ductility of the L12 type intermetallic compound Ni3Al by boron addition. J. Jpn. Inst. Met. 1979, 43, 1190–1196. [Google Scholar] [CrossRef]
- Takasugi, T.; Nagashima, M.; Izumi, O. Strengthening and ductilization of Ni3Si by the addition of Ti elements. Acta Metall. Mater. 1990, 38, 747–755. [Google Scholar] [CrossRef]
- Heatherly, L.; George, E.; Liu, C.; Kumar, K. An Auger investigation of the grain-boundary chemistry in Ni3(Si,Ti) alloys. Mater. Sci. Eng. A 1998, 245, 80–87. [Google Scholar] [CrossRef]
- Friák, M.; Všianská, M.; Holec, D.; Zelený, M.; Šob, M. Tensorial elastic properties and stability of interface states associated with Σ5 (210) grain boundaries in Ni3 (Al, Si). Sci. Technol. Adv. Mater. 2017, 18, 273. [Google Scholar] [CrossRef] [PubMed]
- Friák, M.; Všianská, M.; Holec, D.; Šob, M. Quantum-mechanical study of tensorial elastic and high-temperature thermodynamic properties of grain boundary states in superalloy-phase Ni3Al. IOP Conf. Ser. Mater. Sci. Eng. 2017, 219, 012019. [Google Scholar] [CrossRef]
- Slater, J.C. Introduction to Chemical Physics; McGraw-Hill: New York, NY, USA, 1939. [Google Scholar]
- Lu, G.; Kioussis, N.; Wu, R.; Ciftan, M. First-principles studies of the Σ5 tilt grain boundary in Ni3Al. Phys. Rev. B 1999, 59, 891. [Google Scholar] [CrossRef]
- Muller, D.A.; Singh, D.J.; Silcox, J. Connections between the electron-energy-loss spectra, the local electronic structure, and the physical properties of a material: A study of nickel aluminum alloys. Phys. Rev. B 1998, 57, 8181–8202. [Google Scholar] [CrossRef]
- Mrovec, M.; Groeger, R.; Bailey, A.G.; Nguyen-Manh, D.; Elsässer, C.; Vitek, V. Bond-order potential for simulations of extended defects in tungsten. Phys. Rev. B 2007, 75, 104119. [Google Scholar] [CrossRef]
- Lojkowski, W.; Fecht, H.J. The structure of intercrystalline interfaces. Prog. Mater. Sci. 2000, 45, 339–568. [Google Scholar] [CrossRef]
- Kohyama, M. Computational studies of grain boundaries in covalent materials. Model. Simul. Mater. Sci. Eng. 2002, 10, R31–R59. [Google Scholar] [CrossRef]
- Ochs, T.; Beck, O.; Elsässer, C.; Meyer, B. Symmetrical tilt grain boundaries in body-centred cubic transition metals: An ab initio local-density-functional study. Philos. Mag. A 2000, 80, 2405–2423. [Google Scholar] [CrossRef]
- Všianská, M.; Šob, M. The effect of segregated sp-impurities on grain-boundary and surface structure, magnetism and embrittlement in nickel. Prog. Mat. Sci. 2011, 56, 817. [Google Scholar] [CrossRef]
- Všianská, M.; Šob, M. Magnetically dead layers at sp-impurity-decorated grain boundaries and surfaces in nickel. Phys. Rev. B 2011, 84, 014418. [Google Scholar] [CrossRef]
- Lejček, P.; Šob, M.; Paidar, V. Interfacial segregation and grain boundary embrittlement: An overview and critical assessment of experimental data and calculated results. Prog. Mater. Sci. 2017, 87, 83–139. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blöchl, P.E. Crystal Orbital Hamilton Populations (COHP). Energy-Resolved Visualization of Chemical Bonding in Solids based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis as Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. Analytic Projection from Plane-Wave and PAW Wavefunctions and Application to Chemical-Bonding Analysis in Solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Zhu, J.; Ye, H.Q. Calculations of single-crystal elastic constants made simple. Comput. Phys. Commun. 2010, 181, 671–675. [Google Scholar] [CrossRef]
- Zhou, L.; Holec, D.; Mayrhofer, P.H. Ab initio study of the alloying effect of transition metals on structure, stability and ductility of CrN. J. Appl. Phys. 2013, 113, 043511. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Parlinski, K.; Li, Z.Q.; Kawazoe, Y. First-Principles Determination of the Soft Mode in Cubic ZrO2. Phys. Rev. Lett. 1997, 78, 4063–4066. [Google Scholar] [CrossRef]
- Deringer, V.L.; Stoffel, R.P.; Wuttig, M.; Dronskowski, R. Vibrational properties and bonding nature of Sb2Se3 and their implications for chalcogenide materials. Chem. Sci. 2015, 6, 5255–5262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Bakker, H. Atomic disorder and phase transformation in L12-structure Ni3Si by ball milling. Acta Metall. Mater. 1994, 42, 3009–3017. [Google Scholar] [CrossRef]
- Kumar, A.; Wang, J.; Tomé, C.N. First-principles study of energy and atomic solubility of twinning-associated boundaries in hexagonal metals. Acta Mater. 2015, 85, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Prikhodko, S.V.; Ma, Y.; Ardell, A.J.; Isaak, D.G.; Carnes, J.D.; Moser, S. Elastic constants of face-centered cubic and L12 Ni–Si alloys: Composition and temperature dependence. Metall. Mater. Trans. A 2003, 34, 1863–1868. [Google Scholar] [CrossRef]
- Fu, C.L.; Ye, Y.Y.; Yoo, M.H. Theoretical investigation of the elastic constants and shear fault energies of Ni3Si. Philos. Mag. Lett. 1993, 67, 179–185. [Google Scholar] [CrossRef]
- Iotova, D.; Kioussis, N.; Lim, S.P. Electronic structure and elastic properties of the Ni3X (X = Mn, Al, Ga, Si, Ge) intermetallics. Phys. Rev. B 1996, 54, 14413–14422. [Google Scholar] [CrossRef]
- Liu, L.; Chen, L.; Jiang, Y.; He, C.; Xu, G.; Wen, Y. Temperature Effects on the Elastic Constants, Stacking Fault Energy and Twinnability of Ni3Si and Ni3Ge: A First-Principles Study. Crystals 2018, 8, 364. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Černý, M. Elastic stability of magnetic crystals under isotropic compression and tension. Mater. Sci. Eng. A 2007, 462, 432–435. [Google Scholar] [CrossRef]
- Titrian, H.; Aydin, U.; Friák, M.; Ma, D.; Raabe, D.; Neugebauer, J. Self-consistent Scale-bridging Approach to Compute the Elasticity of Multi-phase Polycrystalline Materials. MRS Proc. 2013, 1524, mrsf12-1524-rr06-03. [Google Scholar] [CrossRef]
- Friák, M.; Counts, W.; Ma, D.; Sander, B.; Holec, D.; Raabe, D.; Neugebauer, J. Theory-Guided Materials Design of Multi-Phase Ti-Nb Alloys with Bone-Matching Elastic Properties. Materials 2012, 5, 1853–1872. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.F.; Friák, M.; Lymperakis, L.; Titrian, H.; Aydin, U.; Janus, A.; Fabritius, H.O.; Ziegler, A.; Nikolov, S.; Hemzalová, P.; et al. Ab initio study of single-crystalline and polycrystalline elastic properties of Mg-substituted calcite crystals. J. Mech. Behav. Biomed. Mater. 2013, 20, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Gaillac, R.; Pullumbi, P.; Coudert, F.X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Lutsko, J.F. Structurally induced supermodulus effect in superlattices. Phys. Rev. Lett. 1988, 60, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Kluge, M.D.; Wolf, D.; Lutsko, J.F.; Phillpot, S.R. Formalism for the calculation of local elastic constants at grain boundaries by means of atomistic simulation. J. Appl. Phys. 1990, 67, 2370–2379. [Google Scholar] [CrossRef]
- Hughbanks, T.; Hoffmann, R. Chains of trans-edge-sharing molybdenum octahedra: Metal-metal bonding in extended systems. J. Am. Chem. Soc. 1983, 105, 3528–3537. [Google Scholar] [CrossRef]
- Bylander, D.M.; Kleinman, L.; Mednick, K. Self-consistent energy bands and bonding of NiSi2. Phys. Rev. B 1982, 25, 1090–1095. [Google Scholar] [CrossRef]
- Yoo, M.; Fu, C.; Horton, J. Crack-tip dislocations and fracture behavior in Ni3Al and Ni3Si. Mater. Sci. Eng. A 1994, 176, 431–437. [Google Scholar] [CrossRef]
- Pang, X.Y.; Janisch, R.; Hartmaier, A. Interplanar potential for tension-shear coupling at grain boundaries derived from ab initio calculations. Model. Simul. Mater. Sci. Eng. 2016, 24, 015007. [Google Scholar] [CrossRef]
- Razumovskiy, V.I.; Ruban, A.; Razumovskii, I.; Lozovoi, A.; Butrim, V.; Vekilov, Y. The effect of alloying elements on grain boundary and bulk cohesion in aluminum alloys: An ab initio study. Scr. Mater. 2011, 65, 926–929. [Google Scholar] [CrossRef]
- Tahir, A.M.; Janisch, R.; Hartmaier, A. Hydrogen embrittlement of a carbon segregated Σ 5(310)[001] symmetrical tilt grain boundary in α-Fe. Mater. Sci. Eng. A 2014, 612, 462–467. [Google Scholar] [CrossRef]
- Tahir, A.M.; Janisch, R.; Hartmaier, A. Ab initio calculation of traction separation laws for a grain boundary in molybdenum with segregated C impurites. Model. Simul. Mater. Sci. Eng. 2013, 21, 075005. [Google Scholar] [CrossRef] [Green Version]
- Razumovskiy, V.I.; Vekilov, Y.K.; Razumovskii, I.M.; Ruban, A.V.; Butrim, V.N.; Mironenko, V.N. Effect of alloying elements and impurities on interface properties in aluminum alloys. Phys. Solid State 2011, 53, 2189–2193. [Google Scholar] [CrossRef]
- Hristova, E.; Janisch, R.; Drautz, R.; Hartmaier, A. Solubility of carbon in α-iron under volumetric strain and close to the Σ5(310)[001] grain boundary: Comparison of DFT and empirical potential methods. Comput. Mater. Sci. 2011, 50, 1088–1096. [Google Scholar] [CrossRef]
- Janisch, R.; Ahmed, N.; Hartmaier, A. Ab initio tensile tests of Al bulk crystals and grain boundaries: Universality of mechanical behavior. Phys. Rev. B 2010, 81, 184108. [Google Scholar] [CrossRef]
- Razumovskiy, V.I.; Lozovoi, A.Y.; Razumovskii, I.M. First-principles-aided design of a new Ni-base superalloy: Influence of transition metal alloying elements on grain boundary and bulk cohesion. Acta Mater. 2015, 82, 369–377. [Google Scholar] [CrossRef]
- Scheiber, D.; Razumovskiy, V.I.; Puschnig, P.; Pippan, R.; Romaner, L. Ab initio description of segregation and cohesion of grain boundaries in W–25at.% Re alloys. Acta Mater. 2015, 88, 180–189. [Google Scholar] [CrossRef]
- Janisch, R.; Elsässer, C. Interstitial impurities at grain boundaries in metals: Insight from atomistic calculations. Int. J. Mater. Res. 2009, 100, 1488–1493. [Google Scholar] [CrossRef]
- Janisch, R.; Elsässer, C. Growth and mechanical properties of a MoC precipitate at a Mo grain boundary: An ab initio density functional theory study. Phys. Rev. B 2008, 77, 094118. [Google Scholar] [CrossRef]
- Gemming, S.; Janisch, R.; Schreiber, M.; Spaldin, N.A. Density-functional investigation of the (113)[-110] twin grain boundary in Co-doped anatase TiO2 and its influence on magnetism in dilute magnetic semiconductors. Phys. Rev. B 2007, 76, 045204. [Google Scholar] [CrossRef]
- Janisch, R.; Elsässer, C. Segregated light elements at grain boundaries in niobium and molybdenum. Phys. Rev. B 2003, 67, 224101. [Google Scholar] [CrossRef]
- Šob, M.; Wang, L.G.; Vitek, V. Local stability of higher-energy phases in metallic materials and its relation to the structure of extended defects. Comput. Mater. Sci. 1997, 8, 100–106. [Google Scholar] [CrossRef]
- Wang, L.G.; Šob, M.; Zhang, Z.Y. Instability of higher-energy phases in simple and transition metals. J. Phys. Chem. Solids 2003, 64, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Šob, M.; Wang, L.G.; Vitek, V. Ab initio calculation of the ideal tensile strength in copper and nickel aluminide. Kovove Mater. Met. Mater. 1998, 36, 145–152. [Google Scholar]
- Šesták, P.; Friák, M.; Holec, D.; Všianská, M.; Šob, M. Strength and Brittleness of Interfaces in Fe-Al Superalloy Nanocomposites under Multiaxial Loading: An ab initio and Atomistic Study. Nanomaterials 2018, 8, 873. [Google Scholar] [CrossRef] [PubMed]
- Černý, M.; Šesták, P.; Řehák, P.; Všianská, M.; Šob, M. Ab initio tensile tests of grain boundaries in the fcc crystals of Ni and Co with segregated sp-impurities. Mater. Sci. Eng. A 2016, 669, 218–225. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]

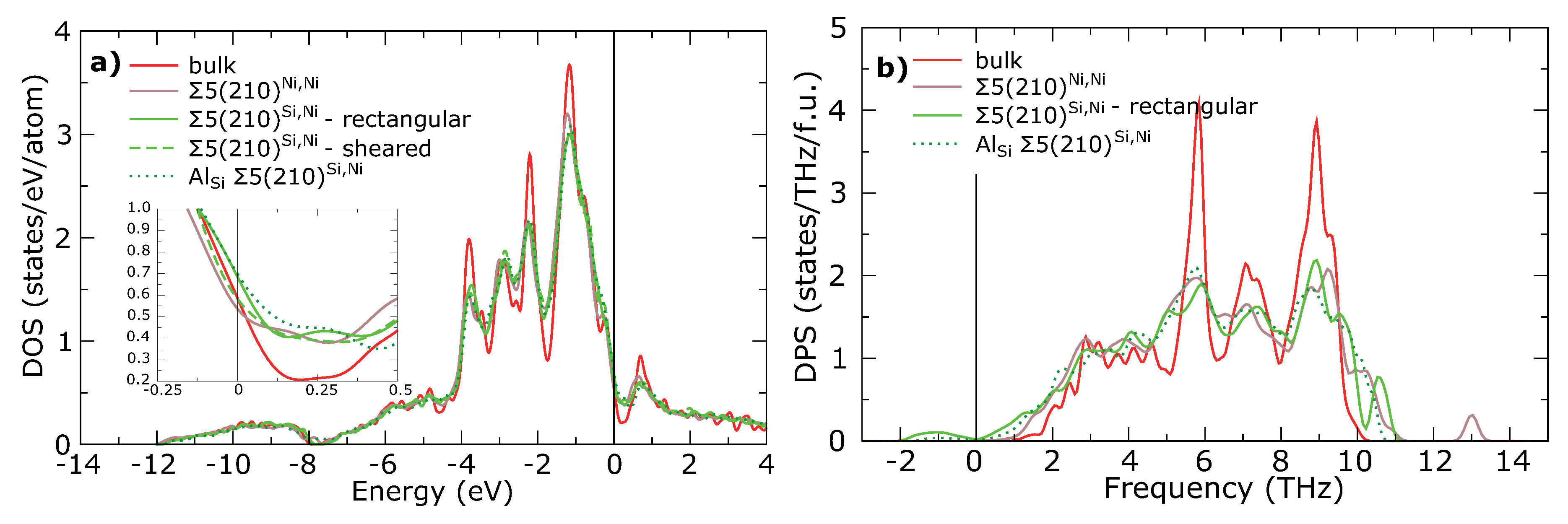

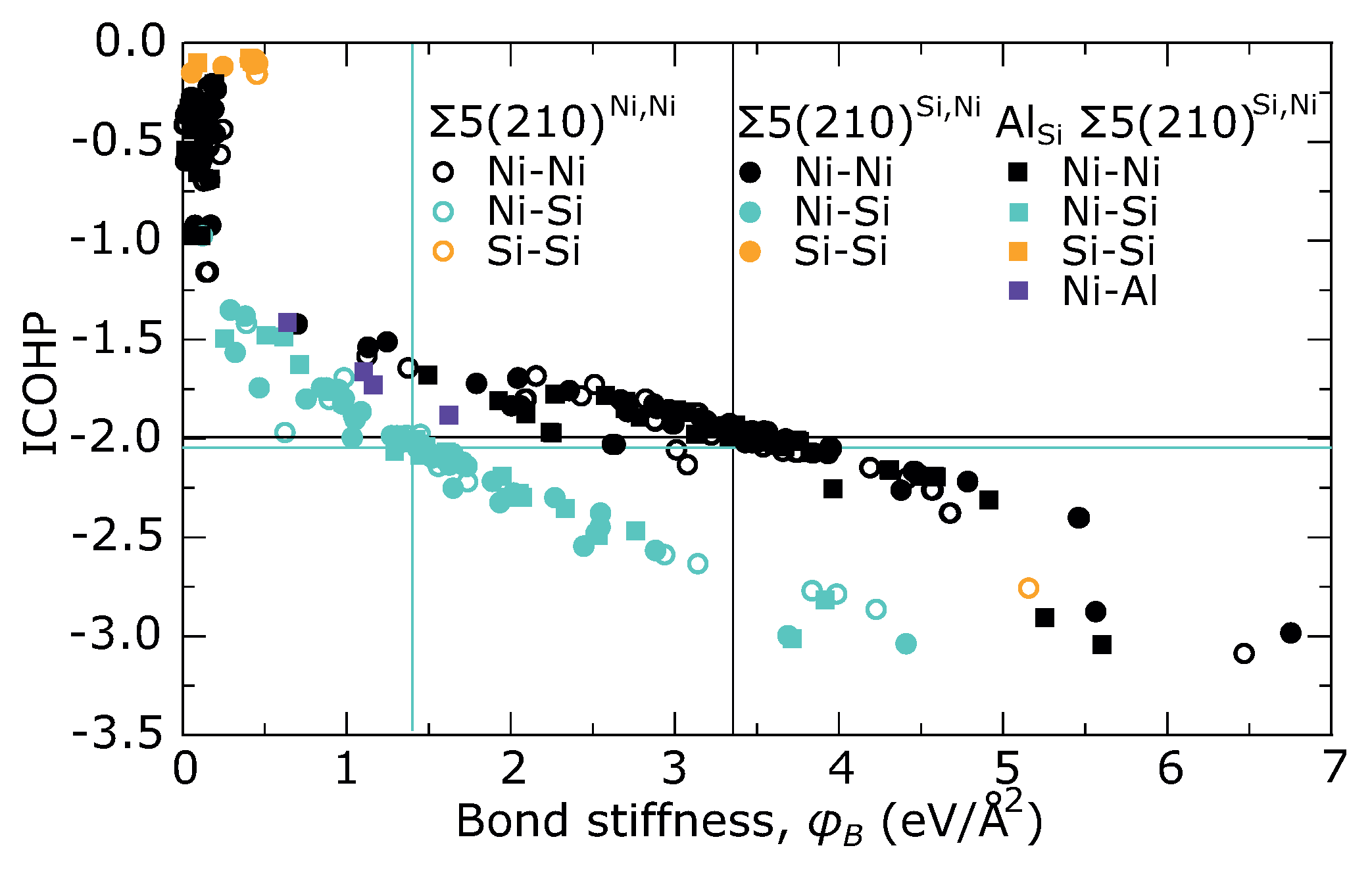
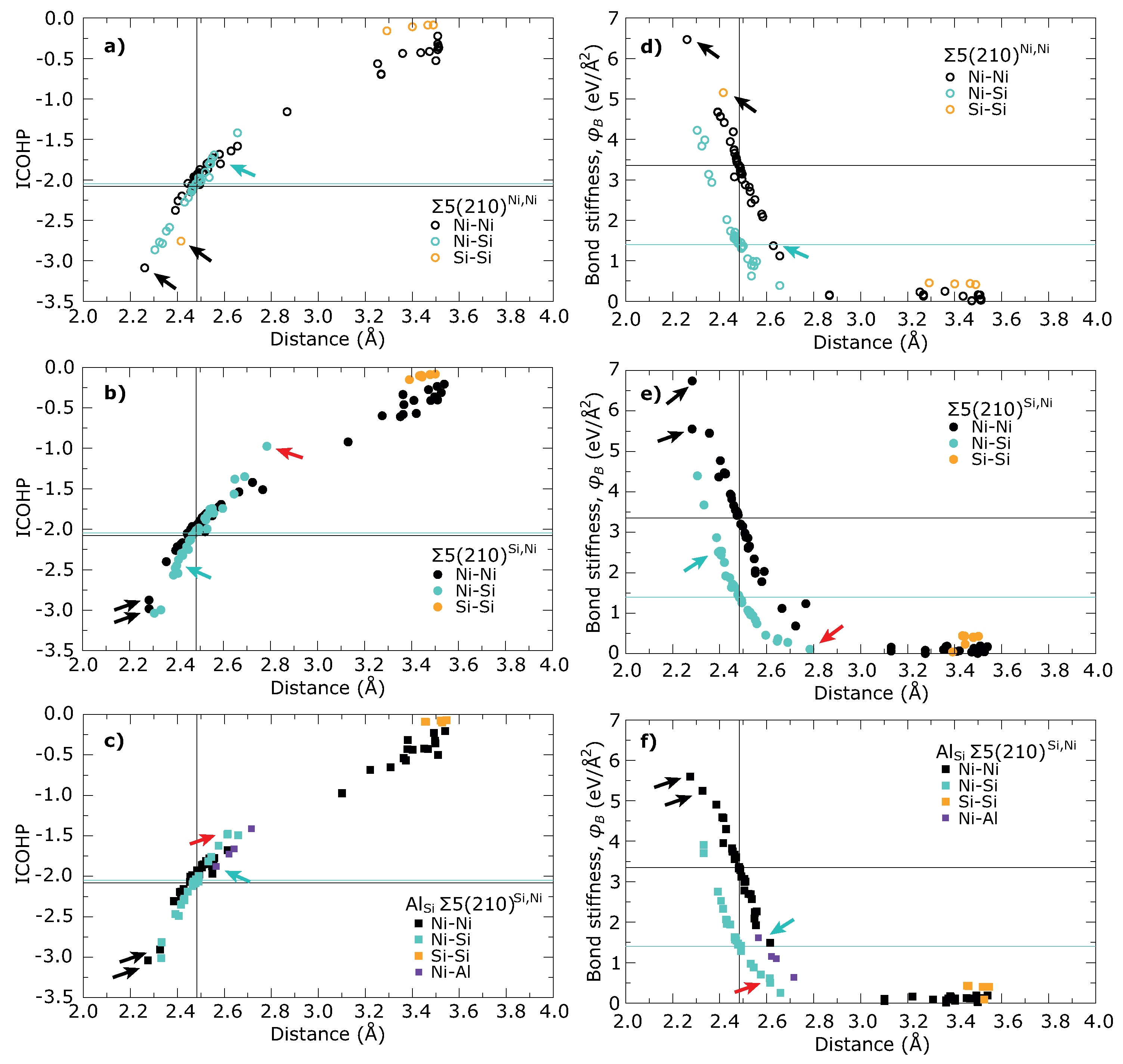

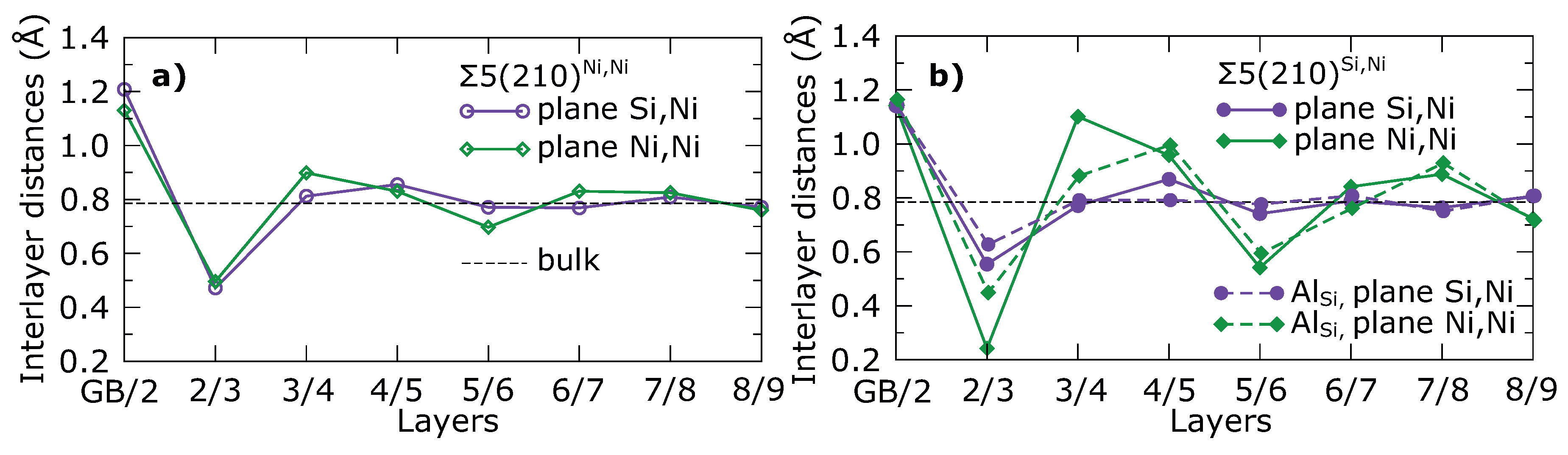
| (Å) | (Å) | (%) | (Å) | (%) | |
| x | 7.853 | 7.735 | −1.49 | 7.808 | −0.56 |
| y | 3.512 | 3.523 | 0.31 | 3.515 | 0.09 |
| Ni3Si States: | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| bulk 〈10〉 〈001〉 〈210〉 | 346 | 164 | 128 | 310 | 164 | 348 | 130 | 94 | 130 |
| 5(210) | 301 | 155 | 135 | 274 | 165 | 276 | 96 | 12 | 115 |
| 5(210) rectangular | 277 | 163 | 151 | 265 | 168 | 259 | 51 | −98 | 108 |
| 5(210) sheared | 387 | 166 | 136 | 264 | 162 | 285 | 79 | 37 | 114 |
| Al 5(210) | 282 | 172 | 131 | 256 | 152 | 283 | 61 | 30 | 115 |
| d (Å) | (eV/Å) | ICOHP | |
|---|---|---|---|
| Ni–Ni | 2.4828 | 3.35 | −2.08 |
| Ni–Si | 2.4828 | 1.40 | −2.05 |
| Si–Si | 3.5113 | 0.46 | −0.07 |
| Ni–Ni (Ni–Si plane) | 3.5113 | 0.19 | −0.60 |
| Ni–Ni (Ni plane) | 3.5113 | 0.03 | −0.41 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friák, M.; Zelený, M.; Všianská, M.; Holec, D.; Šob, M. An Ab Initio Study of Connections between Tensorial Elastic Properties and Chemical Bonds in Σ5(210) Grain Boundaries in Ni3Si. Materials 2018, 11, 2263. https://doi.org/10.3390/ma11112263
Friák M, Zelený M, Všianská M, Holec D, Šob M. An Ab Initio Study of Connections between Tensorial Elastic Properties and Chemical Bonds in Σ5(210) Grain Boundaries in Ni3Si. Materials. 2018; 11(11):2263. https://doi.org/10.3390/ma11112263
Chicago/Turabian StyleFriák, Martin, Martin Zelený, Monika Všianská, David Holec, and Mojmír Šob. 2018. "An Ab Initio Study of Connections between Tensorial Elastic Properties and Chemical Bonds in Σ5(210) Grain Boundaries in Ni3Si" Materials 11, no. 11: 2263. https://doi.org/10.3390/ma11112263
APA StyleFriák, M., Zelený, M., Všianská, M., Holec, D., & Šob, M. (2018). An Ab Initio Study of Connections between Tensorial Elastic Properties and Chemical Bonds in Σ5(210) Grain Boundaries in Ni3Si. Materials, 11(11), 2263. https://doi.org/10.3390/ma11112263