Sintering Behavior and Mechanical Properties of Mullite Fibers/Hydroxyapatite Ceramic

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
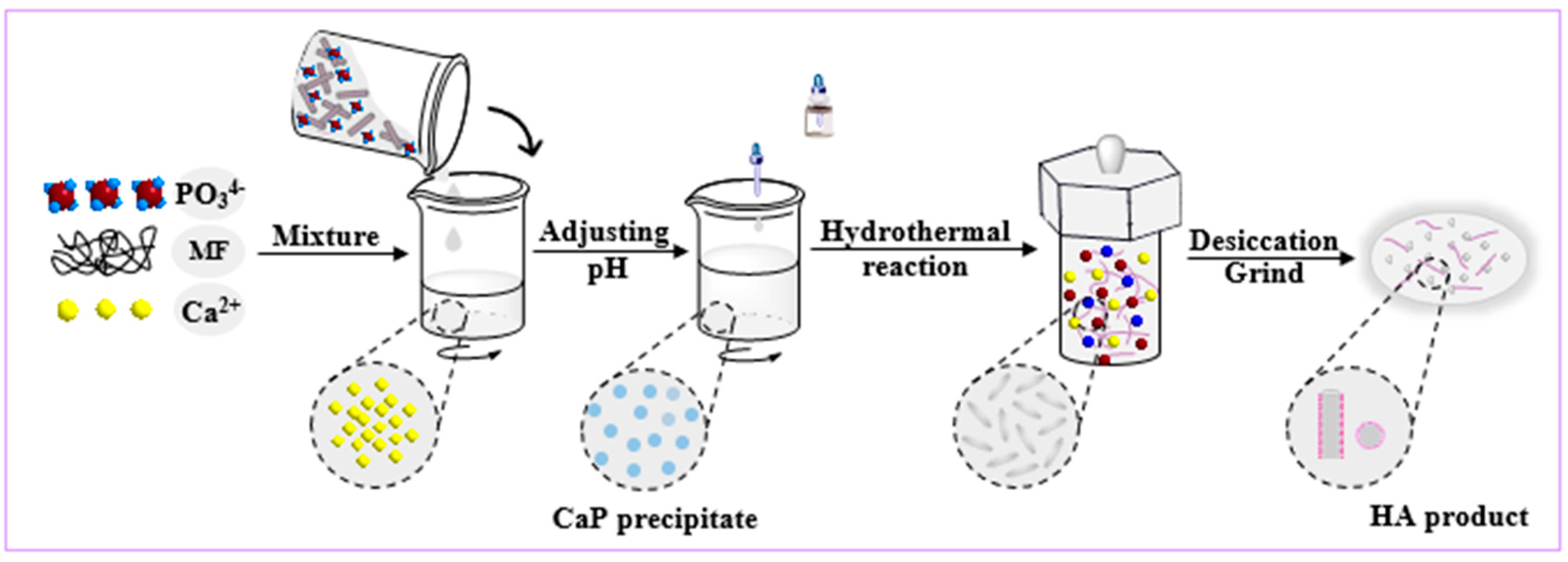
2.2. Methods
2.3. Characterization
3. Results and Discussion
3.1. Morphological Analysis
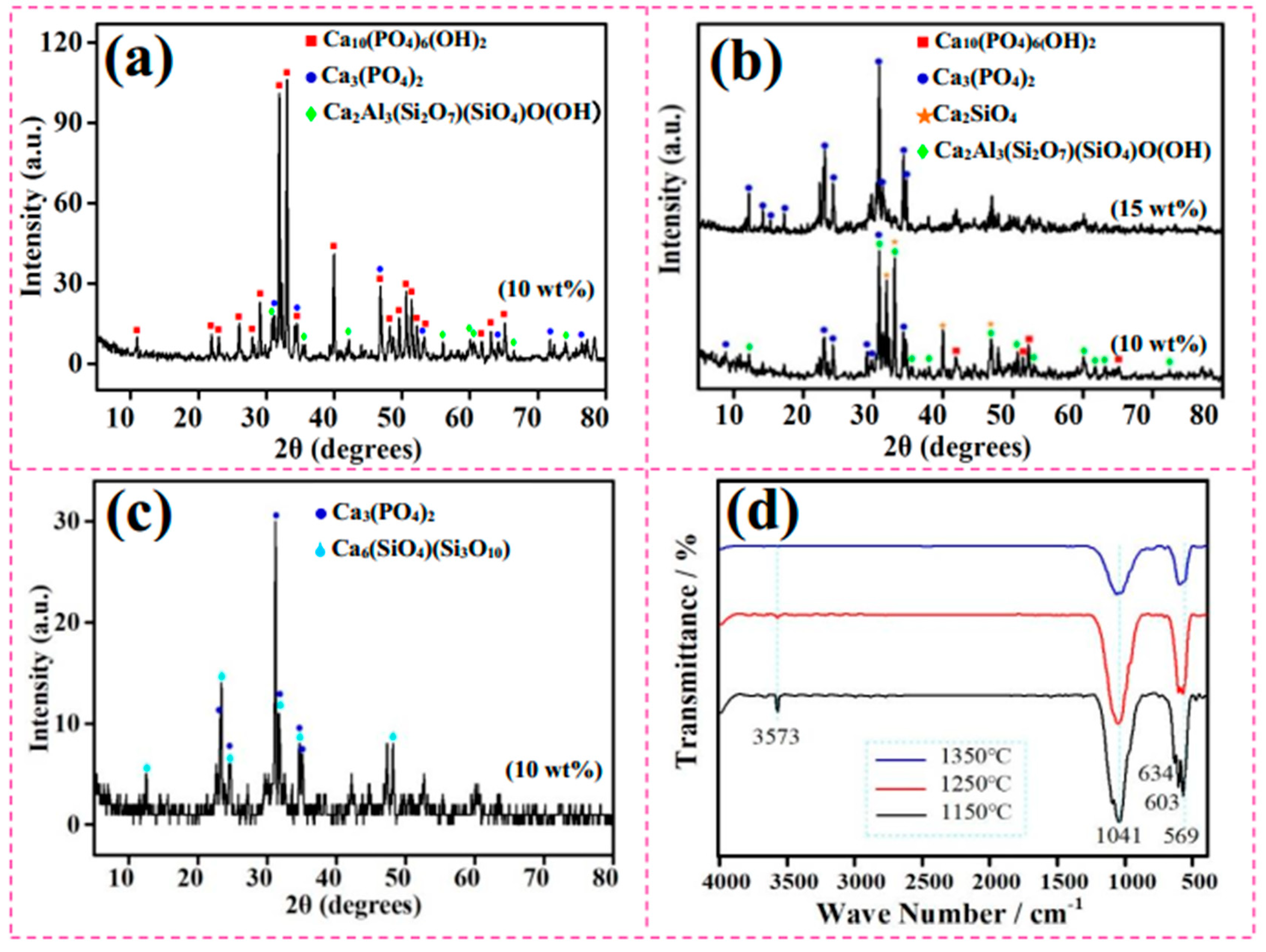
3.2. Sintered Phases
3.3. Thermal Analysis
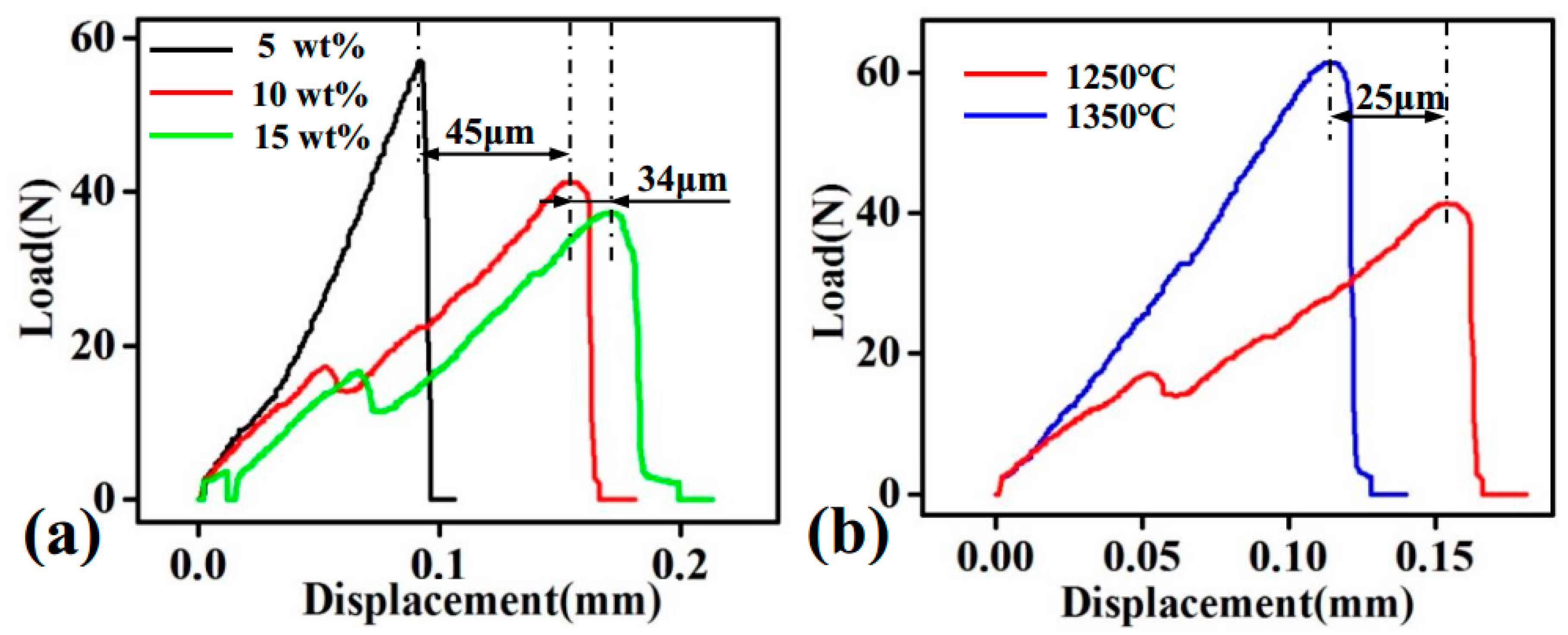
3.4. Density, Vickers Hardness, and Bending Strength
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Samavedi, S.; Whittington, A.R.; Goldstein, A.S. Calcium phosphate ceramics in bone tissue engineering: A review of properties and their influence on cell behavior. Acta Biomater. 2013, 9, 8037–8045. [Google Scholar] [CrossRef] [PubMed]
- Koutsopoulos, S. Kinetic Study on the Crystal Growth of Hydroxyapatite. Langmuir 2001, 17, 8092–8097. [Google Scholar] [CrossRef]
- Zhang, X.; Chang, W.; Lee, P.; Wang, Y.; Yang, M.; Li, J.; Kumbar, S.; Yu, X. Polymer-Ceramic Spiral Structured Scaffolds for Bone Tissue Engineering: Effect of Hydroxyapatite Composition on Human Fetal Osteoblasts. PLoS ONE 2014, 9, e85871. [Google Scholar] [CrossRef] [PubMed]
- Fidancevska, E.; Ruseska, G.; Bossert, J.; Lin, Y.M.; Boccaccini, A.R. Fabrication and characterization of porous bioceramic composites based on hydroxyapatite and titania. Mater. Chem. Phys. 2007, 103, 95–100. [Google Scholar] [CrossRef]
- Reilly, D.T.; Burstein, A.H. The Mechanical Properties of Cortical Bone. JBJS 1976, 56A, 1001–1022. [Google Scholar] [CrossRef]
- Ravikumar, K.; Mallik, P.K.; Basu, B. Twinning induced enhancement of fracture toughness in ultrafine grained Hydroxyapatite-Calcium Titanate composites. J. Eur. Ceram. Soc. 2016, 36, 805–815. [Google Scholar]
- Suchanek, W.; Yoshimura, M. Processing and properties of hydroxyapatite-based biomaterials for use as hard tissue replacement implants. J. Mater. Res. 1998, 13, 94–117. [Google Scholar] [CrossRef]
- Viswanath, B.; Ravishankar, N. Interfacial reactions in hydroxyapatite/alumina nanocomposites. Scr. Mater. 2006, 55, 863–866. [Google Scholar] [CrossRef]
- Radha, G.; Balakumar, S.; Venkatesan, B.; Vellaichamy, E. Evaluation of hemocompatibility and in vitro immersion on microwave-assisted hydroxyapatite-alumina nanocomposites. Mater. Sci. Eng. C 2015, 50, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Gautam, C.R.; Tamuk, M.; Manpoong, C.W.; Gautam, S.S.; Kumar, S.; Singh, A.K.; Mishra, V.K. Microwave synthesis of hydroxyapatite bioceramic and tribological studies of its composites with SrCO3 and ZrO2. J. Mater. Sci. 2016, 51, 4973–4983. [Google Scholar] [CrossRef]
- Ponta, O.; Ciceo-Lucacel, R.; Vulpoi, A.; Radu, T.; Simon, V.; Simon, S. Synthesis and characterisation of nanostructured silica-powellite-HAP composites. J. Mater. Sci. 2015, 50, 577–586. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, J.; Zhao, S.; Zhu, Y. Three-dimensional printing of cerium-incorporated mesoporous calcium-silicate scaffolds for bone repair. J. Mater. Sci. 2016, 51, 836–844. [Google Scholar] [CrossRef]
- Sadik, C.; Amrani, I.E.E.; Albizane, A. Recent advances in silica-alumina refractory: A review. J. Asian Ceram. Soc. 2014, 2, 83–96. [Google Scholar] [CrossRef]
- Duval, D.J.; Risbud, S.H.; Shackelford, J.F. Mullite Structure, Properties and Processing. In Ceramic and Glass Materials; Shackelford, J.F., Doremus, R.H., Eds.; Springer: Boston, MA, USA, 2008; pp. 27–39. [Google Scholar]
- Anggono, J. Mullite Ceramics: Its Properties, Structure, and Synthesis. J. Tek. Mesin 2005, 7, 1–10. [Google Scholar]
- Dubey, A.K.; Sitesh, G.; Nath, S.; Basu, B. Spark plasma sintering to restrict sintering reactions and enhance properties of hydroxyapatite-mullite biocomposites. Ceram. Int. 2011, 37, 2755–2761. [Google Scholar] [CrossRef]
- Ebadzadeh, T.; Behnamghader, A.; Nemati, R. Preparation of porous hydroxyapatite ceramics containing mullite by reaction sintering of clay, alumina and hydroxyapatite. Ceram. Int. 2011, 37, 2887–2889. [Google Scholar] [CrossRef]
- Yetmez, M.; Erkmen, Z.E.; Kalkandelen, C.; Ficai, A.; Oktar, F.N. Sintering effects of mullite-doping on mechanical properties of bovine hydroxyapatite. Mater. Sci. Eng. C 2017, 77, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Biswas, K.; Wang, K.; Bordia, R.K.; Basu, B. Sintering, Phase Stability, and Properties of Calcium Phosphate-Mullite Composites. J. Am. Ceram. Soc. 2010, 93, 1639–1649. [Google Scholar] [CrossRef]
- Nath, S.; Biswas, K.; Basu, B. Phase stability and microstructure development in hydroxyapatite-mullite system. Scr. Mater. 2008, 58, 1054–1057. [Google Scholar] [CrossRef]
- Cordell, J.M.; Vogl, M.L.; Johnson, A.J.W. The influence of micropore size on the mechanical properties of bulk hydroxyapatite and hydroxyapatite scaffolds. J. Mech. Behav. Biomed. Mater. 2009, 2, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Muralithran, G.; Ramesh, S. The effects of sintering temperature on the properties of hydroxyapatite. Ceram. Int. 2000, 26, 221–230. [Google Scholar] [CrossRef]
- Shen, M.; Qiu, K.; Zhang, L.; Huang, Z.; Wang, Z.; Liu, J. Influence of Coal Blending on Ash Fusibility in Reducing Atmosphere. Energies 2015, 8, 4735–4754. [Google Scholar] [CrossRef] [Green Version]
- Huggins, F.E.; Kosmack, D.A.; Huffman, G.P. Correlation between ash-fusion temperatures and ternary equilibrium phase diagrams. Fuel 1981, 60, 577–584. [Google Scholar] [CrossRef]
- Blakeslee, K.C.; Condrate, R.A., Sr. Vibrational Spectra of Hydrothermally Prepared Hydroxyapatites. J. Am. Ceram. Soc. 1971, 54, 559–563. [Google Scholar] [CrossRef]
- Weis, F.A.; Lazor, P.; Skogby, H.; Stalder, R.; Eriksson, L. Polarized IR and Raman spectra of zoisite: Insights into OH-dipole orientation and the luminescence. Eur. J. Miner. 2016, 28, 537–543. [Google Scholar] [CrossRef]
- Legeros, R.Z.; Bonel, G.; Legros, R. Types of “H2O” in human enamel and in precipitated apatites. Calcif. Tissue Int. 1978, 26, 111–118. [Google Scholar] [CrossRef]
- Karakassides, M.A.; Gournis, D.; Petridis, D. An infrared reflectance study of Si-O vibrations in thermally treated alkali-saturated montmorillonites. Clay Miner. 1999, 34, 429–438. [Google Scholar] [CrossRef]
- Frost, R.L.; Palmer, S.J.; Reddy, B.J. Near-infrared and mid-IR spectroscopy of selected humite minerals. Vib. Spectrosc. 2007, 44, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Marchi, J.; Morais, D.S.; Schneider, J.; Bressiani, J.C.; Bressiani, A.H.A. Characterization of rare earth aluminosilicate glasses. J. Non-Cryst. Solids 2005, 351, 863–868. [Google Scholar] [CrossRef]
- Veres, R.; Vanea, E.; Gruian, C.; Baia, L.; Simon, V. The effects of peg assisted synthesis and zinc addition on gamma irradiated bioactive glasses. Compos. Part B 2014, 66, 83–88. [Google Scholar] [CrossRef]
- Li, K.; Guo, Q.; Zhang, L.; Zhang, Y.; Liu, S.; Guo, K.; Li, S. Synthesis and characterization of Si-substituted hydroxyapatite bioactive coating for SiC-coated carbon/carbon composites. Ceram. Int. 2017, 43, 1410–1414. [Google Scholar]
- Tang, X.L.; Xiao, X.F.; Liu, R.F. Structural characterization of silicon-substituted hydroxyapatite synthesized by a hydrothermal method. Mater. Lett. 2005, 59, 3841–3846. [Google Scholar] [CrossRef]
- Bai, J. Fabrication and properties of porous mullite ceramics from calcined carbonaceous kaolin and α-Al2O3. Ceram. Int. 2010, 36, 673–678. [Google Scholar] [CrossRef]
- Chen, Y.F.; Wang, M.C.; Hon, M.H. Kinetics of secondary mullite formation in kaolin-Al2O3 ceramics. Scr. Mater. 2004, 51, 231–235. [Google Scholar] [CrossRef]
- Agathopoulos, S.; Nikolopoulos, P.; Salomoni, A.; Tucci, A.; Stamenkovic, I. Preparation and properties of binary oxide bioceramics. J. Mater. Sci. Mater. Med. 1996, 7, 629–636. [Google Scholar] [CrossRef]
- Oktar, F.N.; Agathopoulos, S.; Ozyegin, L.S.; Gunduz, O.; Demirkol, N.; Bozkurt, Y.; Salman, S. Mechanical properties of bovine hydroxyapatite (BHA) composites doped with SiO2, MgO, Al2O3, and ZrO2. J. Mater. Sci. Mater. Med. 2007, 18, 2137–2143. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Dey, A.; Mukhopadhyay, A.K.; Basu, B. Nanoindentation response of novel hydroxyapatite-mullite composites. Mater. Sci. Eng. A 2009, 513, 197–201. [Google Scholar] [CrossRef]
- Witek, S.R.; Miller, G.A.; Harmer, M.P. Effects of CaO on the Strength and Toughness of AIN. J. Am. Ceram. Soc. 2010, 72, 469–473. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, X.; Xin, H.; Zhang, L.; Yang, J.; Jiang, G. Controllable preparation of SiC coating protecting carbon fiber from oxidation damage during sintering process and SiC coated carbon fiber reinforced hydroxyapatite composites. Appl. Surf. Sci. 2018, 450, 265–273. [Google Scholar] [CrossRef]
- Murakami, S.; Kato, K.; Enari, Y.; Kamitakahara, M.; Watanabe, N.; Ioku, K. Hydrothermal synthesis of porous hydroxyapatite ceramics composed of rod-shaped particles and evaluation of their fracture behavior. Ceram. Int. 2012, 38, 1649–1654. [Google Scholar] [CrossRef]
- Yan, S.; He, P.; Jia, D.; Yang, Z.; Duan, X.; Wang, S.; Zhou, Y. Effect of fiber content on the microstructure and mechanical properties of carbon fiber felt reinforced geopolymer composites. Ceram. Int. 2016, 42, 7837–7843. [Google Scholar] [CrossRef]
- He, Z.; Ma, J.; Wang, C. Constitutive modeling of the densification and the grain growth of hydroxyapatite ceramics. Biomaterials 2005, 26, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Chrissanthopoulos, A.; Bouropoulos, N.; Yannopoulos, S.N. Vibrational spectroscopic and computational studies of sol-gel derived CaO-MgO-SiO2 binary and ternary bioactive glasses. Vib. Spectrosc. 2008, 48, 118–125. [Google Scholar] [CrossRef]
- Kikuyama, H. A Study of the Dissociation State and the SiO2 Etching Reaction for HF Solutions of Extremely Low Concentration. J. Electrochem. Soc. 1994, 141, 366–374. [Google Scholar] [CrossRef]
- Laidler, K.J. The development of the Arrhenius equation. J. Chem. Educ. 1984, 61, 494–498. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Performance Parameters | Fiber Content (wt %)–Sintering Temperature (°C) | |||
|---|---|---|---|---|
| 5–1250 | 10–1250 | 15–1250 | 10–350 | |
| Theoretical density (g/cm3) | 3.1648 | 3.1645 | 3.1643 | 3.1645 |
| Sintered density (g/cm3) | 2.1134 | 1.9040 | 1.8400 | 2.1534 |
| Relative density (%) | 66.78 | 60.17 | 58.15 | 68.05 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Liu, Q.; Yang, J.; Zhang, W.; Wang, Y. Sintering Behavior and Mechanical Properties of Mullite Fibers/Hydroxyapatite Ceramic. Materials 2018, 11, 1859. https://doi.org/10.3390/ma11101859
Zhao X, Liu Q, Yang J, Zhang W, Wang Y. Sintering Behavior and Mechanical Properties of Mullite Fibers/Hydroxyapatite Ceramic. Materials. 2018; 11(10):1859. https://doi.org/10.3390/ma11101859
Chicago/Turabian StyleZhao, Xueni, Qingyao Liu, Jianjun Yang, Weigang Zhang, and Yao Wang. 2018. "Sintering Behavior and Mechanical Properties of Mullite Fibers/Hydroxyapatite Ceramic" Materials 11, no. 10: 1859. https://doi.org/10.3390/ma11101859
APA StyleZhao, X., Liu, Q., Yang, J., Zhang, W., & Wang, Y. (2018). Sintering Behavior and Mechanical Properties of Mullite Fibers/Hydroxyapatite Ceramic. Materials, 11(10), 1859. https://doi.org/10.3390/ma11101859