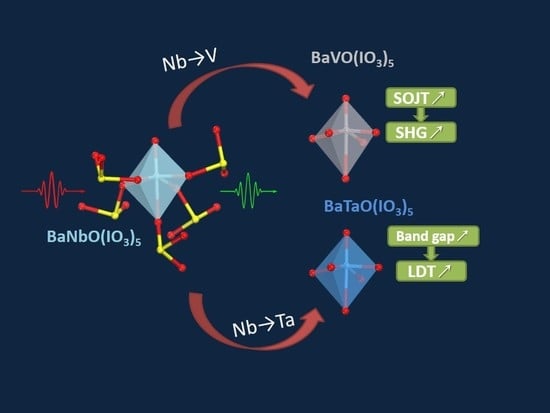
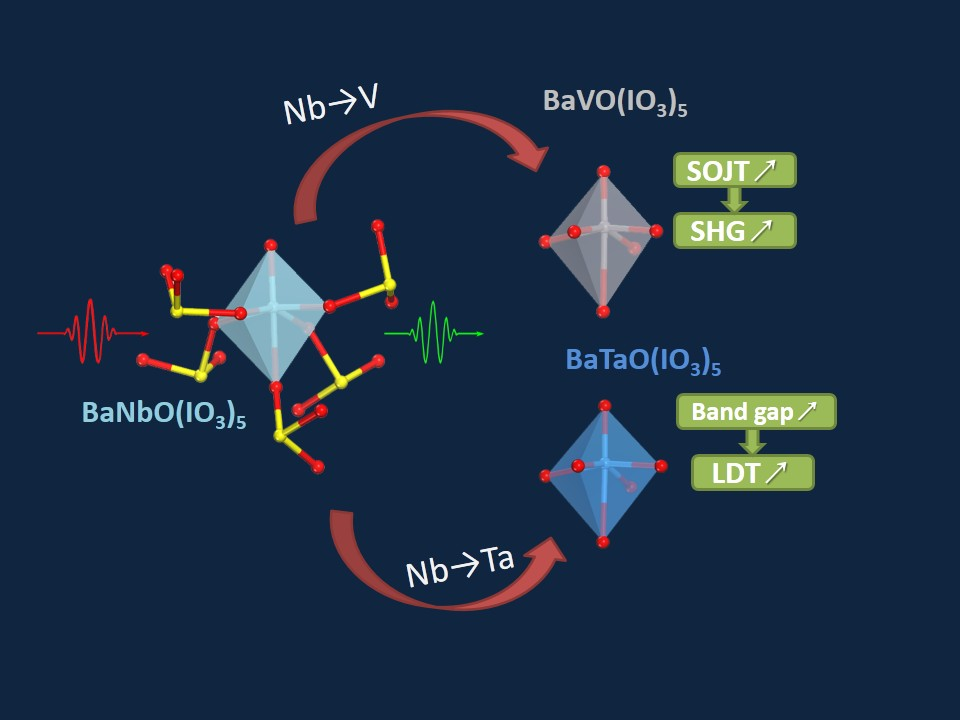
Excellent Infrared Nonlinear Optical Crystals BaMO(IO3)5 (M = V, Ta) Predicted by First Principle Calculations

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
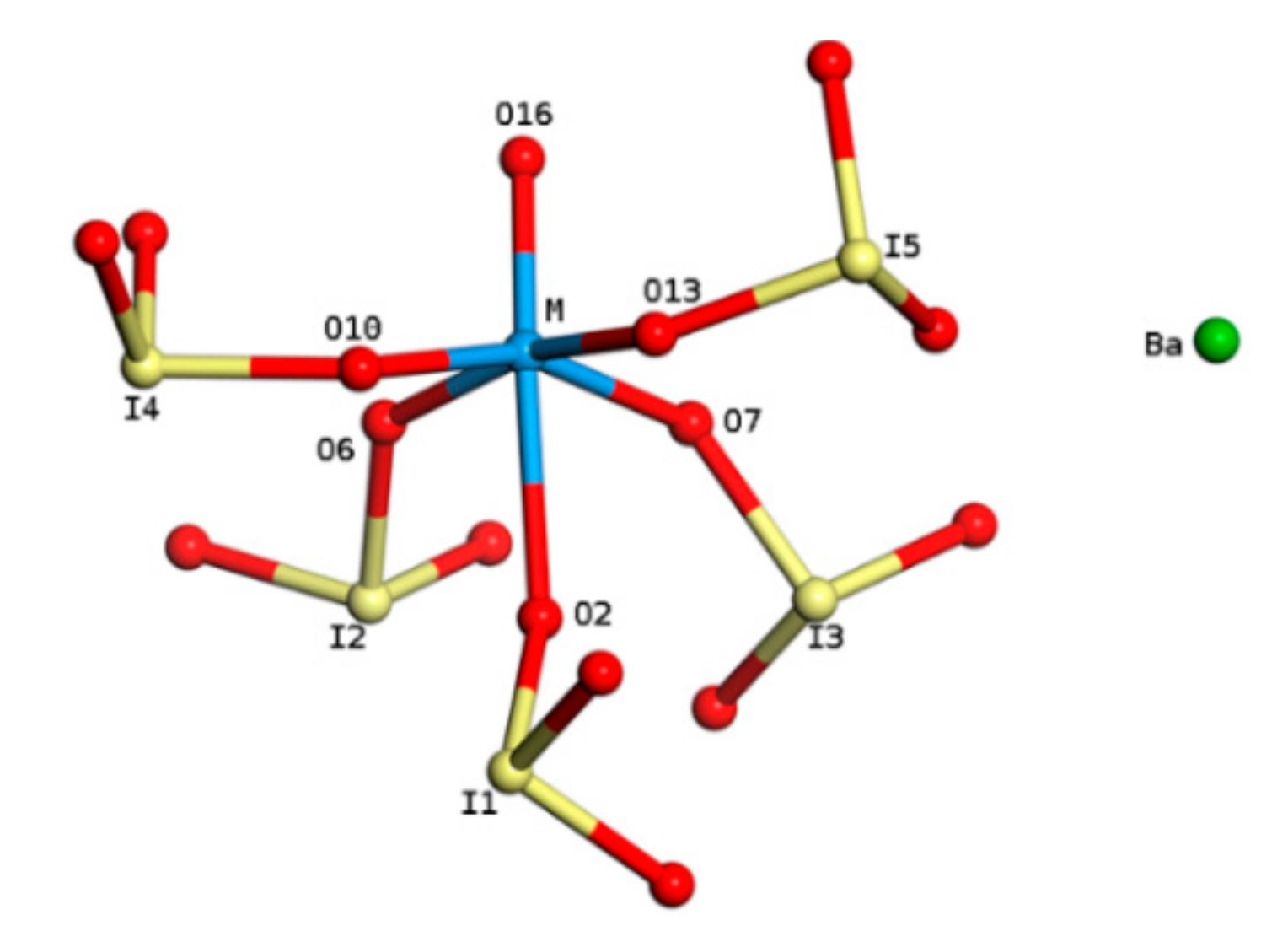
2.1. Model of BaMO(IO3)5
2.2. Properties Investigated
2.3. Computation Details
3. Results and Discussion
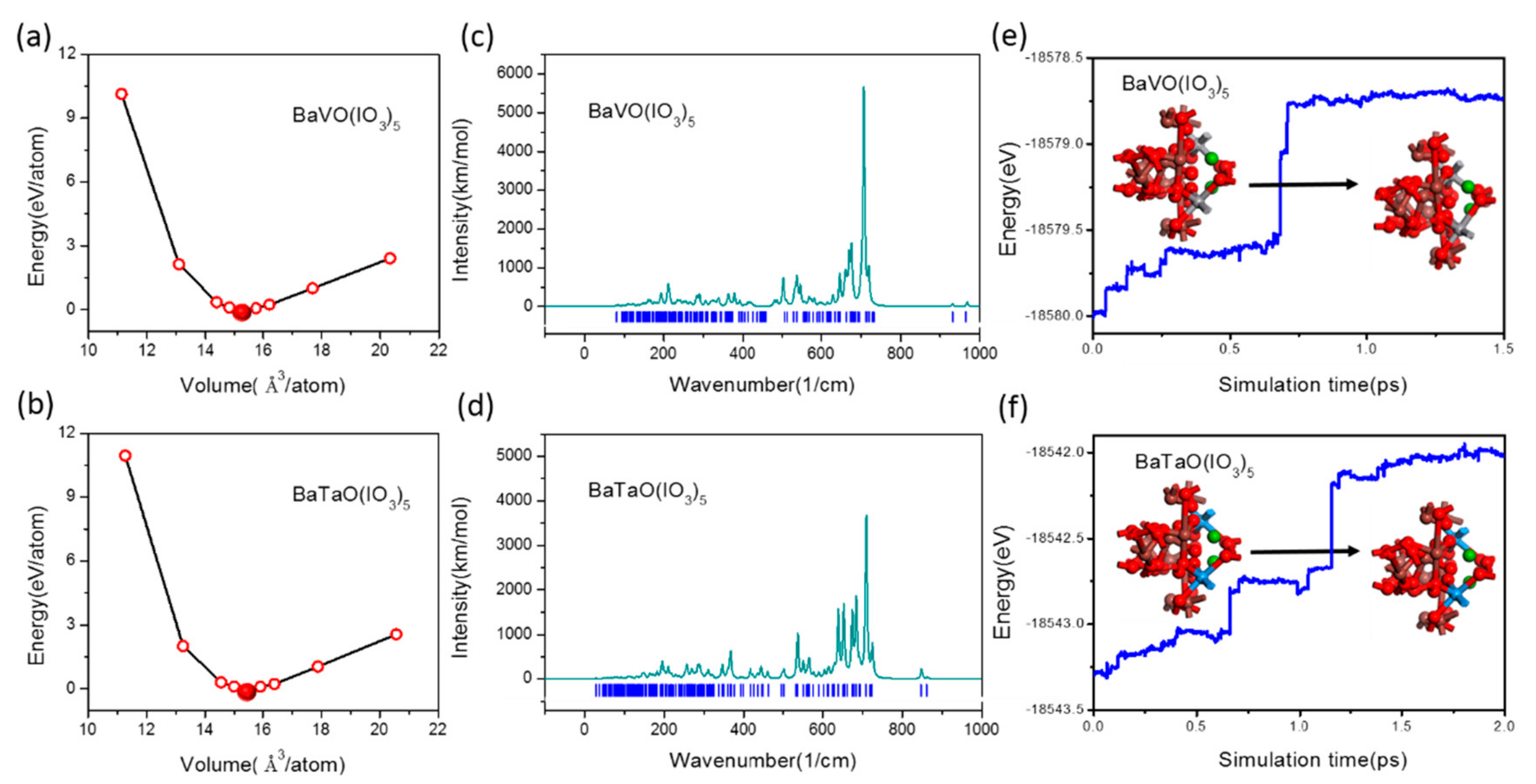
3.1. The Kinetic Stability and Thermodynamic Stability of BaMO(IO3)5 (M = V, Ta)
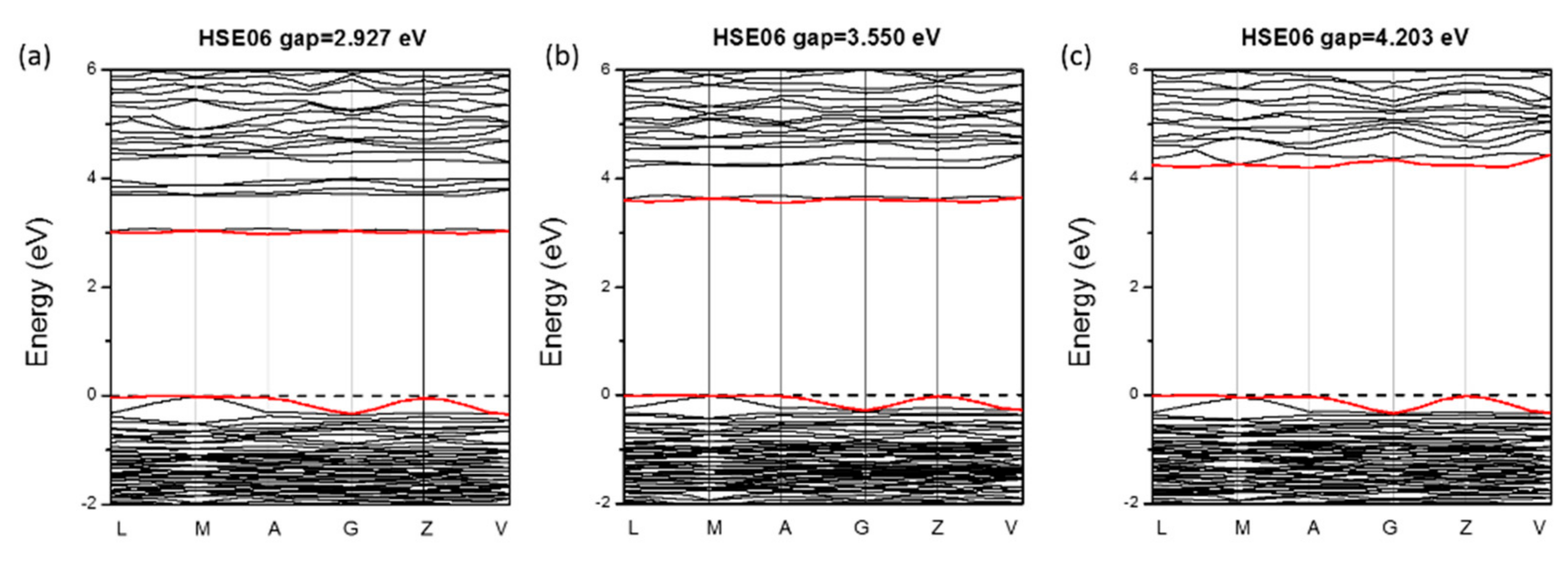
3.2. Reliability of Our Predictions on the NLO Properties
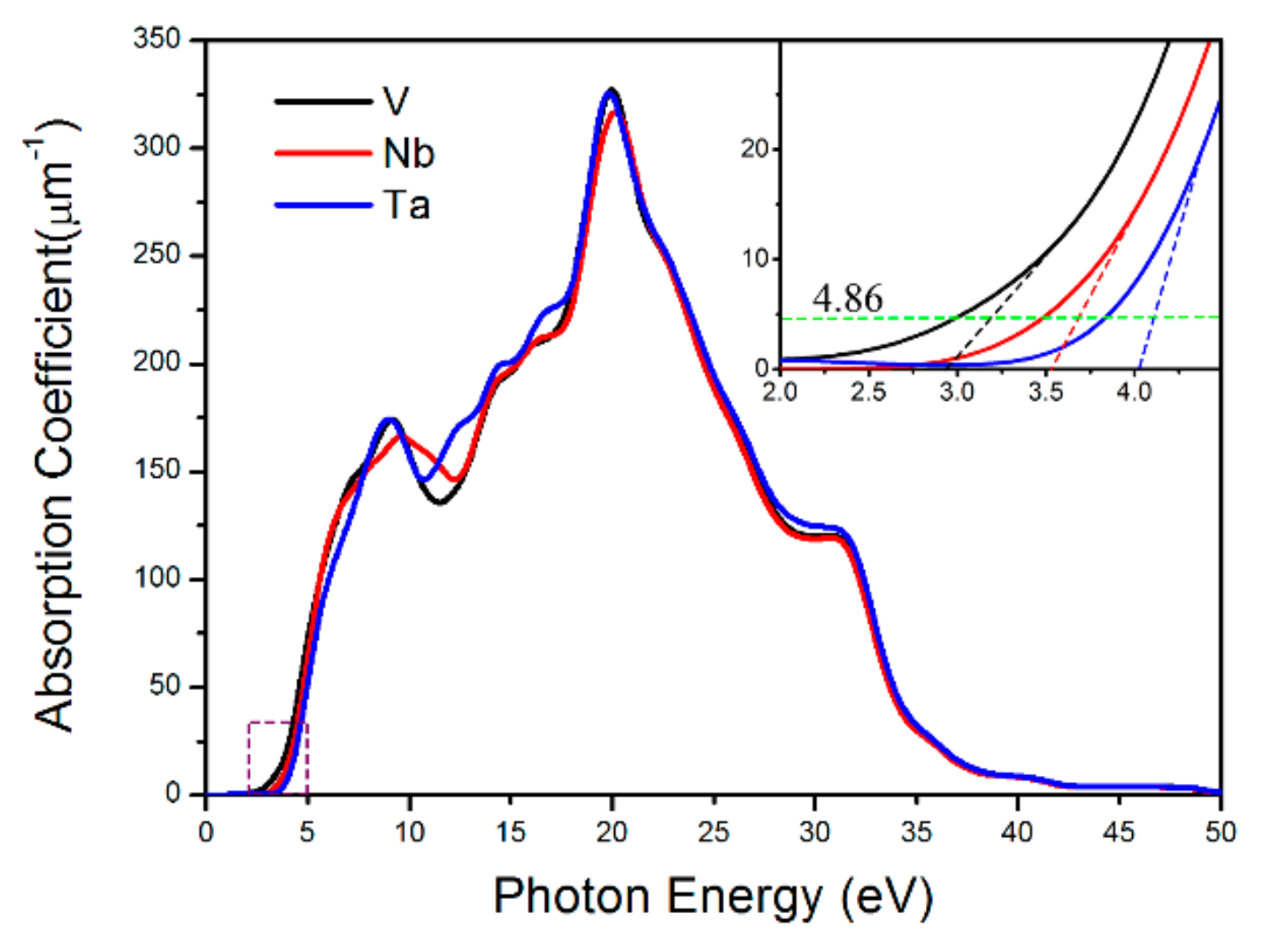
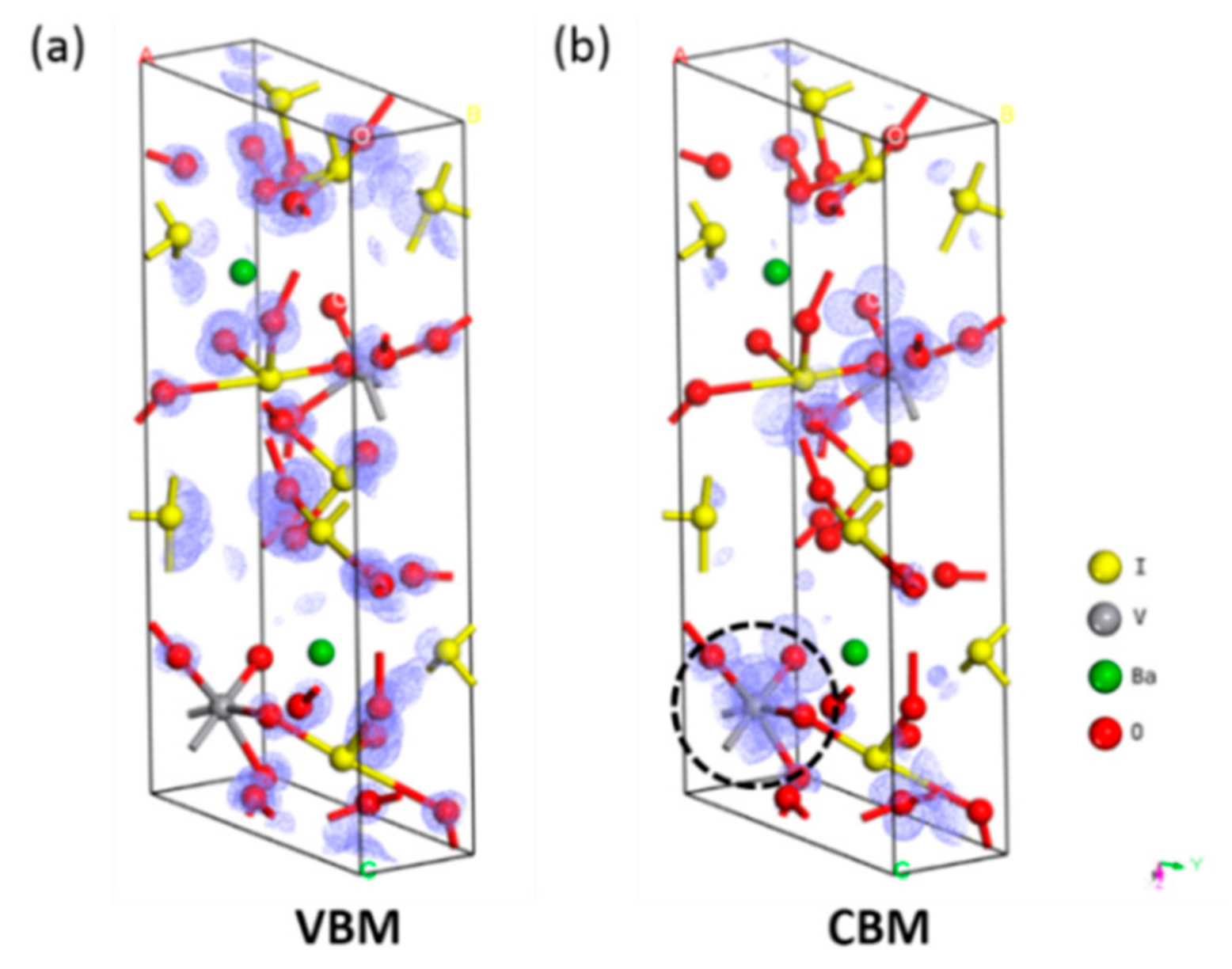
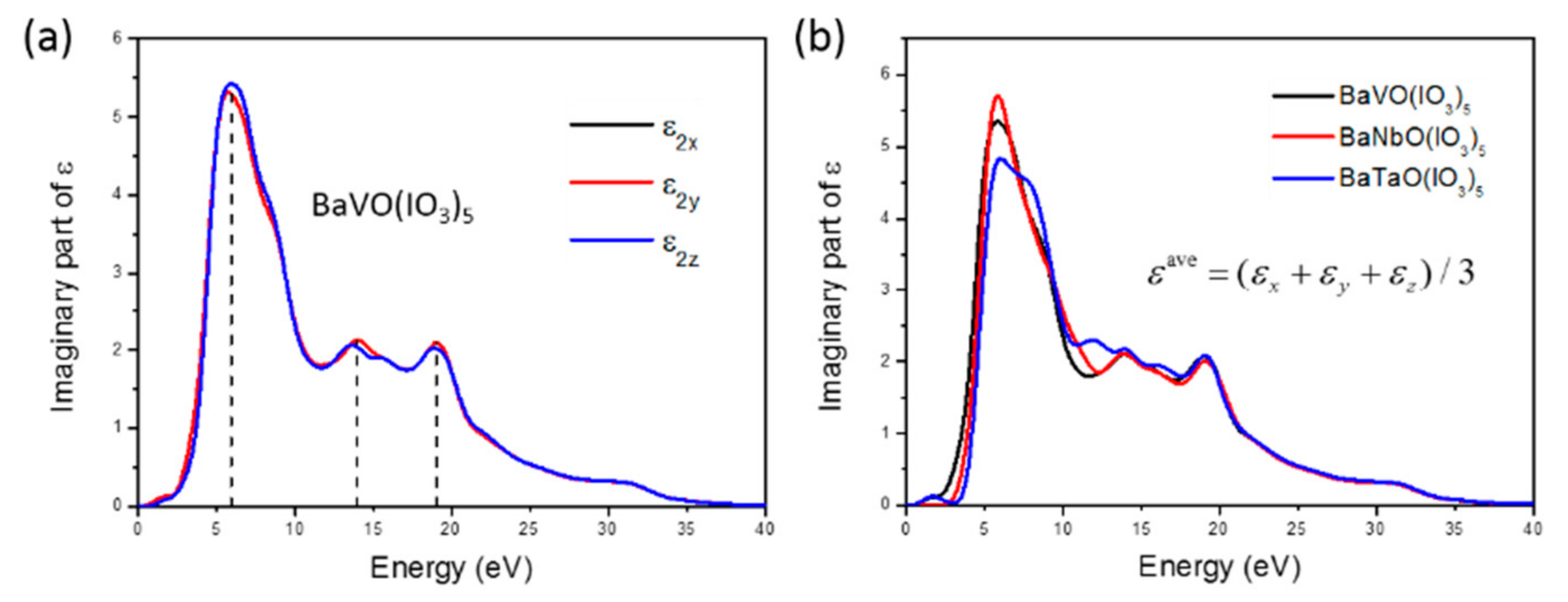
3.3. NLO Properties of BaVO(IO3)5 and BaTaO(IO3)5
3.4. Effect of Element Substitution on the NLO Performance
- (1)
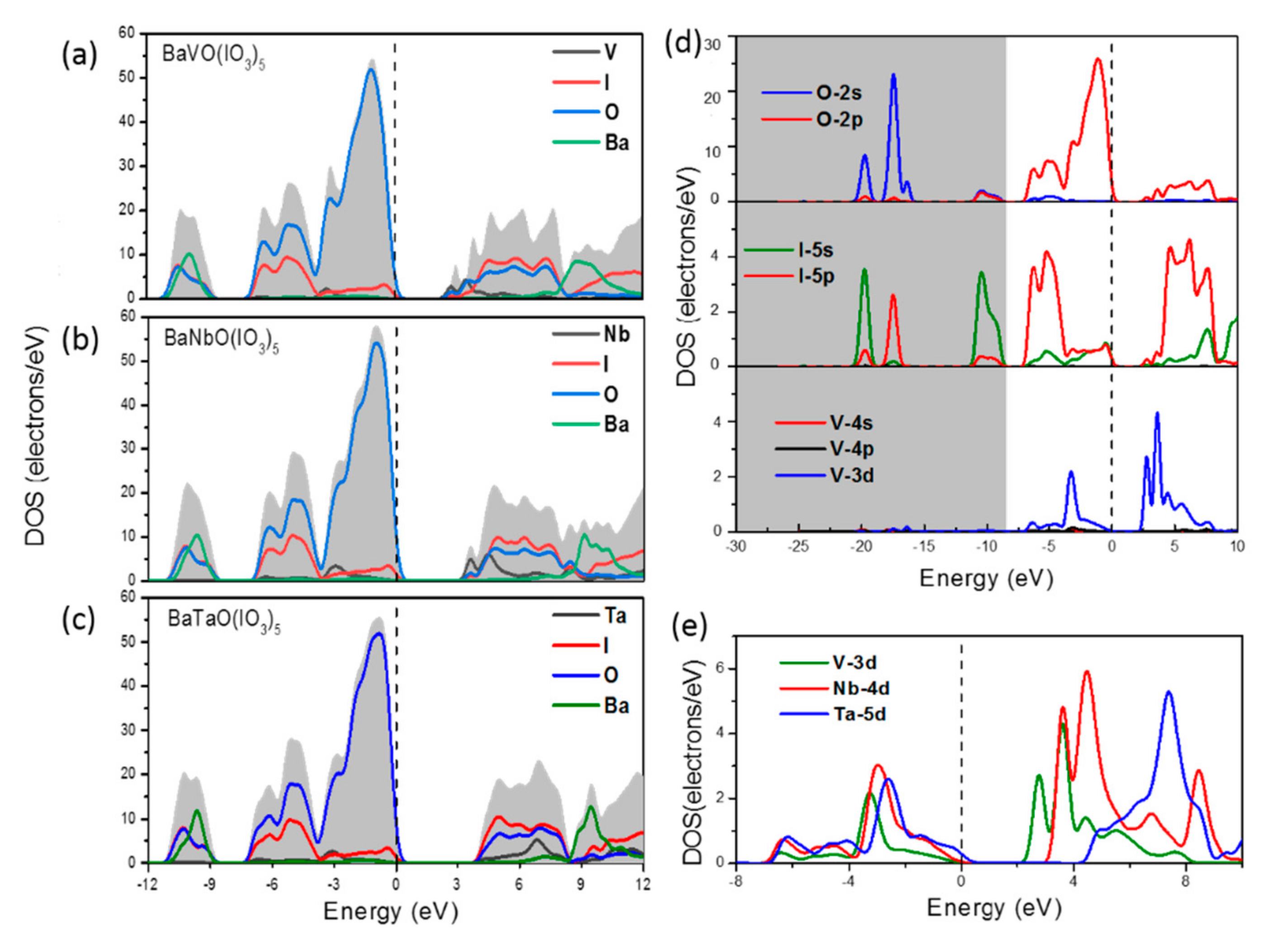
- There is an obvious sharp peak at the bottom of the conduction band, and from Figure 6d,e we can see that this peak results from the contributions of V-3d and Nb-4d;
- (2)
- In the conduction band region, the DOS of I and O are very comparable, which exhibits the full hybrid interaction in IO3−; at the same time, their energy range (I and O) is intertwined with that of the transition metal V and Nb.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, G.G.; Lu, G.W.; Yu, Y.H.; Zhang, W.S.; Zhao, K. Calculation for linear and nonlinear optical properties of LBO crystals. Chin. J. Lasers 2010, 37, 1342–1346. [Google Scholar] [CrossRef]
- Han, S.Y.; Lu, G.W.; Zhang, J.; Xia, H.R.; Wang, C.L. Calculations for the linear and nonlinear optical coefficients of UREA crystals. J. Synth. Cryst. 2006, 35, 1346–1350. [Google Scholar]
- Chen, X.; Liu, H.; Wu, Q.; Jiang, X.; Meng, X.; Lin, Z.; Qin, J. ABi2(IO3)2F5 (A = K, Rb and Cs): Combination of halide and oxide anionic units to create large SHG response with wide bandgap. Angew. Chem. Int. Ed. 2017, 56, 9492–9496. [Google Scholar]
- Yang, Z.; Pan, S.; Yu, H.; Lee, M.H. Electronic structure and optical properties of the nonlinear optical crystal Pb4O(BO3)2 by first-principles calculations. J. Solid State Chem. 2013, 198, 77–80. [Google Scholar] [CrossRef]
- Zhang, J.; Kang, L.; Lin, T.H.; Jiang, X.; Gong, P.; Lee, M.H.; Lin, Z. The mechanism for the nonlinear optical properties in La9Na3B8O27, La2Na3B3O9 and La2CaB10O19: ab initio studies. J. Phys. Condens. Mat. 2015, 27, 485501. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yao, J.; Lin, Z.; Wang, X.; He, R.; Yao, W.; Zhai, N.; Chen, C. NaSr3Be3B3O9F4: A promising deep-ultraviolet nonlinear optical material resulting from the cooperative alignment of the [Be3B3O12F]10− anionic group. Angew. Chem. Int. Ed. 2011, 50, 9141–9144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lee, M.H.; Yang, Z.; Jing, Q.; Pan, S.; Zhang, M.; Wu, H.; Su, X.; Li, C.S. Simulated pressure-induced blue-shift of phase-matching region and nonlinear optical mechanism for K3B6O10X (X = Cl, Br). Appl. Phys. Lett. 2015, 106, 031906. [Google Scholar]
- Ok, K.M.; Halasyamani, P.S.; Casanova, D.; Llunell, M.; Pere Alemany, A.; Alvarez, S. Distortions in octahedrally coordinated d0 transition metal oxides: A continuous symmetry measures approach. Chem. Mater. 2006, 18, 3176–3183. [Google Scholar] [CrossRef]
- Kang, L.; Zhou, M.; Yao, J.; Lin, Z.; Wu, Y.; Chen, C. Metal thiophosphates with good mid-infrared nonlinear optical performances: A First-principles prediction and analysis. J. Am. Chem. Soc. 2015, 137, 13049–13059. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.L.; Mao, J.G. Recent advances on second-order NLO materials based on metal iodates. Coord. Chem. Rev. 2015, 288, 1–17. [Google Scholar] [CrossRef]
- Chang, H.Y.; Kim, S.H.; Halasyamani, P.S.; Ok, K.M. Alignment of lone pairs in a new polar material: Synthesis, characterization, and functional properties of Li2Ti(IO3)6. J. Am. Chem. Soc. 2016, 40, 2426–2427. [Google Scholar]
- Sun, C.F.; Hu, C.L.; Xu, X.; Ling, J.B.; Hu, T.; Kong, F.; Long, X.F.; Mao, J.G. BaNbO(IO3)5: A new polar material with a very large SHG response. J. Am. Chem. Soc. 2009, 131, 9486–9487. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Ramo, D.; Lin, Z.; Bristowe, P.; Qin, J.; Chen, C. First principles selection and design of mid-IR nonlinear optical halide crystals. J. Mater. Chem. C 2013, 1, 7363–7370. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-Principles Simulation: Ideas, Illustrations and the CASTEP Code. J. Phys. Condens. Mat. 2002, 14, 2717. [Google Scholar] [CrossRef]
- Wu, Q.; Meng, X.; Zhong, C.; Chen, X.; Qin, J. Rb2CdBr2I2: A new IR nonlinear optical material with a large laser damage threshold. J. Am. Chem. Soc. 2014, 136, 5683–5686. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Hu, C.; Mutailipu, M.; Sun, Y.; Wu, K.; Zhang, M.; Pan, S. Oxyhalides: Prospecting ore for optical functional materials with large laser damage thresholds. J. Mater. Chem. C 2018, 6, 2435–2442. [Google Scholar] [CrossRef]
- Mao, F.F.; Hu, C.L.; Chen, J.; Mao, J.G. A series of mixed-metal germanium iodates as second-order nonlinear optical materials. Chem. Mater. 2018, 30, 2443–2452. [Google Scholar] [CrossRef]
- Chen, C.; Lin, Z.; Wang, Z. The development of new borate-based UV nonlinear optical crystals. Appl. Phys. B Lasers Opt. 2005, 80, 1–25. [Google Scholar] [CrossRef]
- Halasyamani, P.S. Asymmetric cation coordination in oxide materials: Influence of lone-pair cations on the intra-octahedral distortion in d0 transition metals. Cheminform 2004, 35, 73–85. [Google Scholar] [CrossRef]
- Bersuker, I.B. The Jahn-Teller Effect. J. Lumin. 1984, 31, 29–36. [Google Scholar]
- Bersuker, I.B. Modern aspects of the Jahn-Teller effect theory and applications to molecular problems. Chem. Rev. 2001, 101, 1067–1114. [Google Scholar] [CrossRef] [PubMed]
- Halasyamani, P.S.; Poeppelmeier, K.R. Noncentrosymmetric Oxides. Chem. Mater. 1998, 10, 2753–2769. [Google Scholar] [CrossRef]
- Zhang, J.J.; Zhang, Z.H.; Tao, X.T. Research advances of novel nonlinear optical crystals based on second-order Jahn-Teller effects(SOJT). J. Shandong Univ. 2011, 46, 99–120. [Google Scholar]
- Ok, K.M.; Halasyamani, P.S. New d0 transition metal iodates: Synthesis, structure, and characterization of BaTi(IO3)6, LaTiO(IO3)5, Ba2VO2(IO3)4‚(IO3), K2MoO2(IO3)4, and BaMoO2(IO3)4‚H2O. Inorg. Chem. 2005, 44, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.P.; Hu, C.L.; Xu, X.; Sun, C.F.; Zhang, J.H.; Mao, J.G. NaVO2(IO3)2(H2O): A unique layered material produces a very strong shg response. Chem. Mater. 2010, 22, 1545–1550. [Google Scholar] [CrossRef]
- Milman, V.; Winkler, B.; White, J.A.; Pickard, C.J.; Payne, M.C.; Akhmatskaya, E.V.; Nobes, R.H. Electronic structure, properties, and phase stability of inorganic crystals: A pseudopotential plane-wave study. Int. J. Quantum Chem. 2015, 77, 895–910. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Morris, A.J.; Nicholls, R.J.; Pickard, C.J.; Yates, J.R. OptaDOS: A tool for obtaining density of states, core-level and optical spectra from electronic structure codes. Comput. Phys. Commun. 2014, 185, 1477–1485. [Google Scholar] [CrossRef]
- Geng, L.; Li, Q.; Meng, C.Y.; Dai, K.; Lu, H.Y.; Lin, C.S.; Cheng, W.D. BaBi(SeO3)2Cl: A new polar material showing high second-harmonic generation efficiency enhanced by constructive alignment of chloride ions. J. Mater. Chem. C 2015, 3, 12290–12296. [Google Scholar] [CrossRef]
- Milman, V.; Refson, K.; Clark, S.J.; Pickard, C.J.; Yates, J.R.; Gao, S.P.; Hasnip, P.J.; Probert, M.I.J.; Perlov, A.; Segall, M.D. Electron and vibrational spectroscopies using DFT, plane waves and pseudopotentials: CASTEP implementation. J. Mol. Struct. Theochem. 2010, 954, 22–35. [Google Scholar] [CrossRef]
- Zhang, W.L.; Cheng, W.D.; Zhang, H.; Geng, L.; Lin, C.S.; He, Z.Z. A strong second-harmonic generation material Cd4BiO(BO3)3 originating from 3-Chromophore asymmetric structures. Cheminform 2010, 132, 1508–1509. [Google Scholar] [CrossRef]
- Boyd, R.W. Nonlinear Optics; Elsevier: New York, NY USA, 1992. [Google Scholar]
- Kurtz, S.K.; Perry, T.T. A powder technique for the evaluation of nonlinear optical materials. J. Appl. Phys. 1968, 39, 3798–3813. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.L.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.S.; Qteish, A.; Payne, M.C.; Heine, V.V. Optimized and transferable nonlocal separable ab initio pseudopotentials. Phys. Rev. B Condens. Mat. 1993, 47, 4174. [Google Scholar] [CrossRef]
- Jiang, X.; Kang, L.; Luo, S.; Gong, P.; Lee, M.H.; Lin, Z. Development of nonlinear optical materials promoted by density functional theory simulations. Int. J. Mod. Phys. B 2014, 28, 1430018. [Google Scholar] [CrossRef]
- Lin, Z.S.; Kang, L.; Zheng, T.; He, R.; Huang, H.; Chen, C.T. Strategy for the optical property studies in ultraviolet nonlinear optical crystals from density functional theory. Comput. Mater. Sci. 2012, 60, 99–104. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Lin, Z.S.; Qin, J.G.; Chen, C.T. Two novel nonlinear optical carbonates in the deep-ultraviolet region: KBeCO3F and RbAlCO3F2. Sci. Rep. 2013, 3, 1366. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Luo, S.Y.; Peng, G.; Ye, N.; Wu, Y.C.; Chen, C.T.; Lin, Z.S. First-Principles Design of a deep-ultraviolet nonlinear-optical crystal from KBe2BO3F2 to NH4Be2BO3F2. Inorg. Chem. 2015, 54, 10533–10535. [Google Scholar] [CrossRef] [PubMed]
- Garza, A.J.; Scuseria, G.E. Predicting band gaps with hybrid density functionals. J. Phys. Chem. Lett. 2016, 7, 4165–4170. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef] [Green Version]
- Maggard, P.A.; Nault, T.S.; Stern, C.L.; Poeppelmeier, K.R. Alignment of acentric MoO3F33− anions in a polar material: (Ag3MoO3F3)(Ag3MoO4)Cl. J. Solid State Chem. 2003, 175, 27–33. [Google Scholar] [CrossRef]
- Izumi, H.K.; Kirsch, J.E.; Stern, C.L.; Poeppelmeier, K.R. Examining the out-of-center distortion in the [NbOF5]2− anion. Inorg. Chem. 2005, 44, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.F.; Hu, C.L.; Xu, X.; Yang, B.P.; Mao, J.G. Explorations of new second-order nonlinear optical materials in the potassium vanadyl iodate system. J. Am. Chem. Soc. 2011, 133, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.C.; Kim, S.H.; Kang, M.O.; Halasyamani, P.S. New polar oxides: Synthesis, characterization, calculations, and structure−property relationships in RbSe2V3O12 and TlSe2V3O12. Chem. Mater. 2009, 21, 1654–1662. [Google Scholar]
- Sun, C.F.; Hu, T.; Xu, X.; Mao, J.G. Syntheses, crystal structures, and properties of three new lanthanum(III) vanadium iodates. Dalton T. 2010, 39, 7960–7967. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, T.; Hong, Y.C.; Baek, J.; Halasyamani, P.S. Two new noncentrosymmetric polar oxides: Synthesis, characterization, second-harmonic generating, and pyroelectric measurements on TlSeVO5 and TlTeVO5. Cheminform 2010, 38, 4710–4715. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BaNbO(IO3)5 | Our Results | Reference | ||
|---|---|---|---|---|
| Lattice Parameter | a (Å) | 7.95 | – | 7.93exp |
| b (Å) | 7.95 | – | 7.93exp | |
| c (Å) | 23.90 | – | 24.08exp | |
| α (°) | 136.09 | – | 136.46exp | |
| β (°) | 136.09 | – | 136.46exp | |
| γ (°) | 56.51 | – | 56.51exp | |
| Eg (eV) | 3.55 | 2.55cal | 3.64exp | |
| ε(0) | 4.51 | 4.50cal | – | |
| ∆n | 0.02 | 0.03cal | – | |
| d11 at 1064 nm (×10−9 esu) | 18.66 | 19.80cal | 15.40exp | |
| SHG (×10−9 esu) | d11 | d12 | d13 | d15 | d24 | d33 |
|---|---|---|---|---|---|---|
| BaVO(IO3)5 | 23.24 | 23.42 | 22.05 | 22.64 | 23.03 | 21.23 |
| BaNbO(IO3)5 | 18.66 | 18.39 | 17.97 | 18.31 | 18.18 | 17.60 |
| BaTaO(IO3)5 | 17.02 | 16.58 | 16.26 | 16.64 | 16.19 | 15.85 |
| Crystals | nx | ny | nz | ∆n |
| BaVO(IO3)5 | 2.22 | 2.22 | 2.26 | 0.04 |
| BaNbO(IO3)5 | 2.15 | 2.15 | 2.17 | 0.02 |
| BaTaO(IO3)5 | 2.12 | 2.12 | 2.14 | 0.02 |
| Parameters | BaVO(IO3)5 | BaNbO(IO3)5 | BaTaO(IO3)5 | |
|---|---|---|---|---|
| Bond lengths (Å) | M-O16 | 1.653 | 1.855 | 1.754 |
| M-O6 | 1.936 | 2.093 | 1.958 | |
| M-O7 | 1.866 | 2.063 | 1.924 | |
| M-O10 | 1.981 | 2.106 | 1.954 | |
| M-O13 | 1.891 | 2.069 | 1.924 | |
| M-O2 | 2.470 | 2.381 | 2.342 | |
| O-I1 | 1.833 | 1.844 | 1.840 | |
| Bond angles (°) | O16-M-O2 | 169.52 | 170.58 | 170.98 |
| O10-M-O7 | 156.25 | 156.44 | 156.33 | |
| O6-M-O13 | 159.25 | 165.49 | 165.42 | |
| Δd | 1.004 | 0.605 | 0.663 |
| Groups | Dipole Moment (D) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X-Component | Y-Component | Z-Component | Total Magnitude | |||||||||
| M = V | M = Nb | M = Ta | M = V | M = Nb | M = Ta | M = V | M = Nb | M = Ta | M = V | M = Nb | M = Ta | |
| I(1)O3 | 0.28 | −0.34 | −0.56 | 7.50 | 7.36 | 7.40 | −11.11 | −11.35 | −11.31 | 13.40 | 13.53 | 13.53 |
| I(2)O3 | −1.46 | −0.62 | −1.60 | −8.48 | −8.39 | −8.05 | −10.57 | −10.68 | −10.67 | 13.63 | 13.60 | 13.46 |
| I(3)O3 | 8.36 | 7.77 | 7.38 | −6.95 | −6.70 | −7.51 | −8.14 | −8.51 | −8.15 | 13.59 | 13.33 | 13.32 |
| I(4)O3 | −7.17 | −6.50 | −6.69 | 1.59 | 2.03 | 2.08 | −10.60 | −11.07 | −10.98 | 12.90 | 13.00 | 13.02 |
| I(5)O3 | 7.92 | 7.53 | 7.74 | −9.82 | −9.94 | −9.92 | −3.84 | −3.73 | −3.98 | 13.19 | 13.01 | 13.19 |
| MO6 | 7.32 | 6.37 | 5.92 | 7.73 | 6.44 | 5.79 | 20.65 | 13.98 | 11.68 | 23.23 | 16.66 | 14.32 |
| NDM | 15.26 | 14.21 | 12.19 | −8.43 | −9.20 | −10.20 | −23.62 | −31.37 | −33.41 | 89.93 | 83.12 | 80.84 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Cui, M.; Yan, H.; Yu, Y.; Li, M.; Li, X.; Chu, L.; Jiang, B.; Qin, M. Excellent Infrared Nonlinear Optical Crystals BaMO(IO3)5 (M = V, Ta) Predicted by First Principle Calculations. Materials 2018, 11, 1809. https://doi.org/10.3390/ma11101809
Li Y, Cui M, Yan H, Yu Y, Li M, Li X, Chu L, Jiang B, Qin M. Excellent Infrared Nonlinear Optical Crystals BaMO(IO3)5 (M = V, Ta) Predicted by First Principle Calculations. Materials. 2018; 11(10):1809. https://doi.org/10.3390/ma11101809
Chicago/Turabian StyleLi, Yingfeng, Mengqi Cui, Hejin Yan, Yangxin Yu, Meicheng Li, Xiang Li, Lihua Chu, Bing Jiang, and Mingde Qin. 2018. "Excellent Infrared Nonlinear Optical Crystals BaMO(IO3)5 (M = V, Ta) Predicted by First Principle Calculations" Materials 11, no. 10: 1809. https://doi.org/10.3390/ma11101809
APA StyleLi, Y., Cui, M., Yan, H., Yu, Y., Li, M., Li, X., Chu, L., Jiang, B., & Qin, M. (2018). Excellent Infrared Nonlinear Optical Crystals BaMO(IO3)5 (M = V, Ta) Predicted by First Principle Calculations. Materials, 11(10), 1809. https://doi.org/10.3390/ma11101809