Removal of Hexavalent Chromium in Portland Cement Using Ground Granulated Blast-Furnace Slag Powder

 , and
, and
Abstract
:1. Introduction
2. Experimental Methods
2.1. Material and Sample Descriptions
2.2. Accelerated Carbonation and Tank Leaching Test
2.3. Phase Identification
2.4. Scanning Transmission X-ray Microscopy (STXM)
3. Results and Discussion
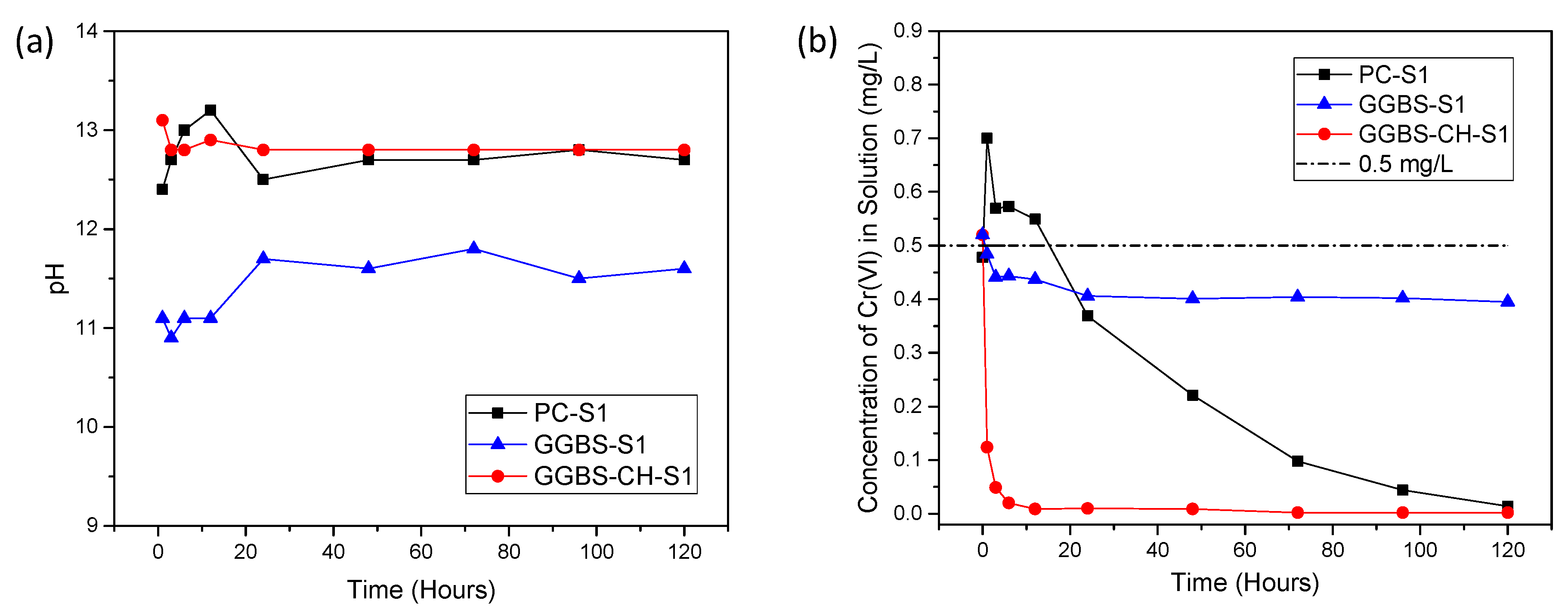
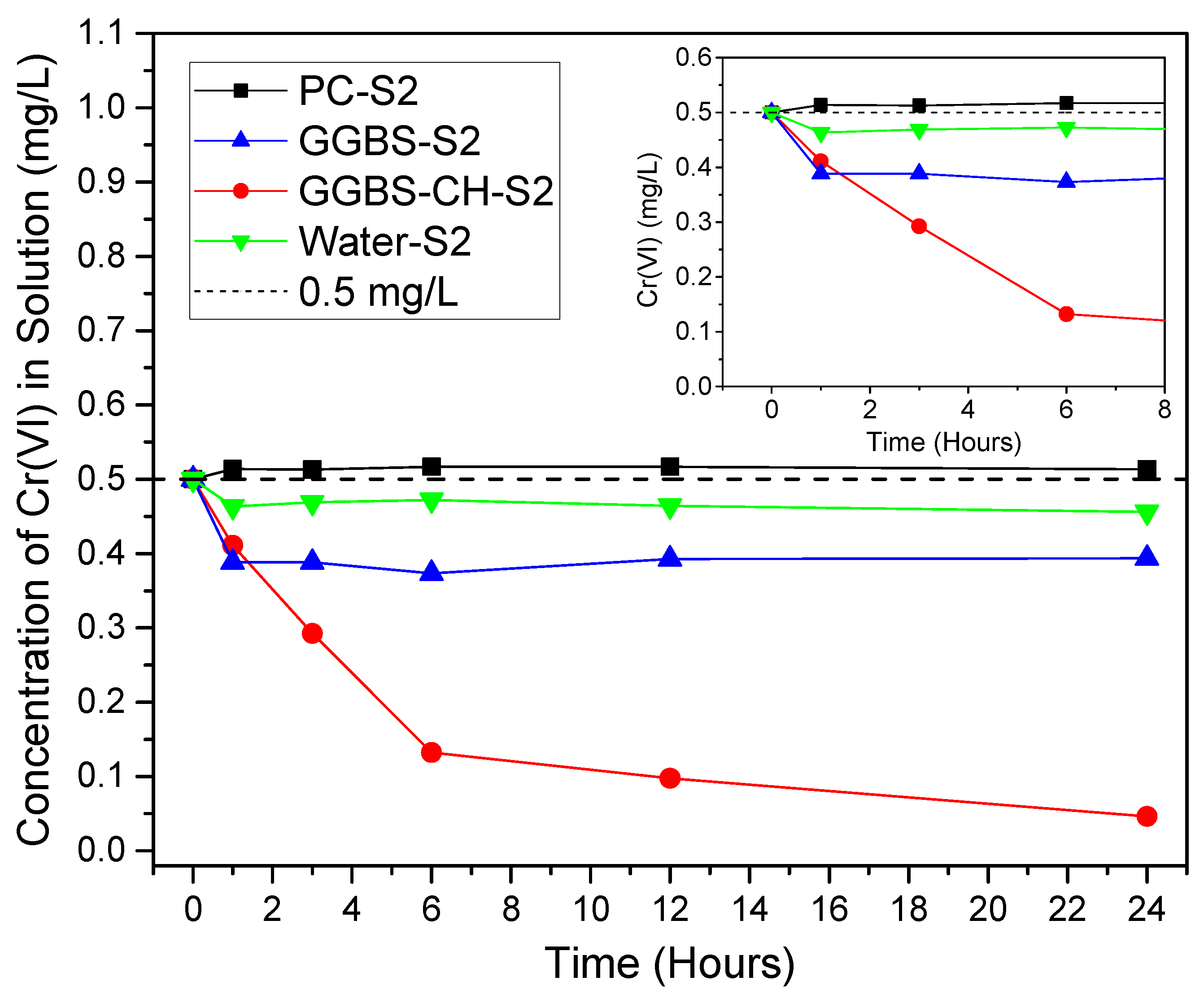
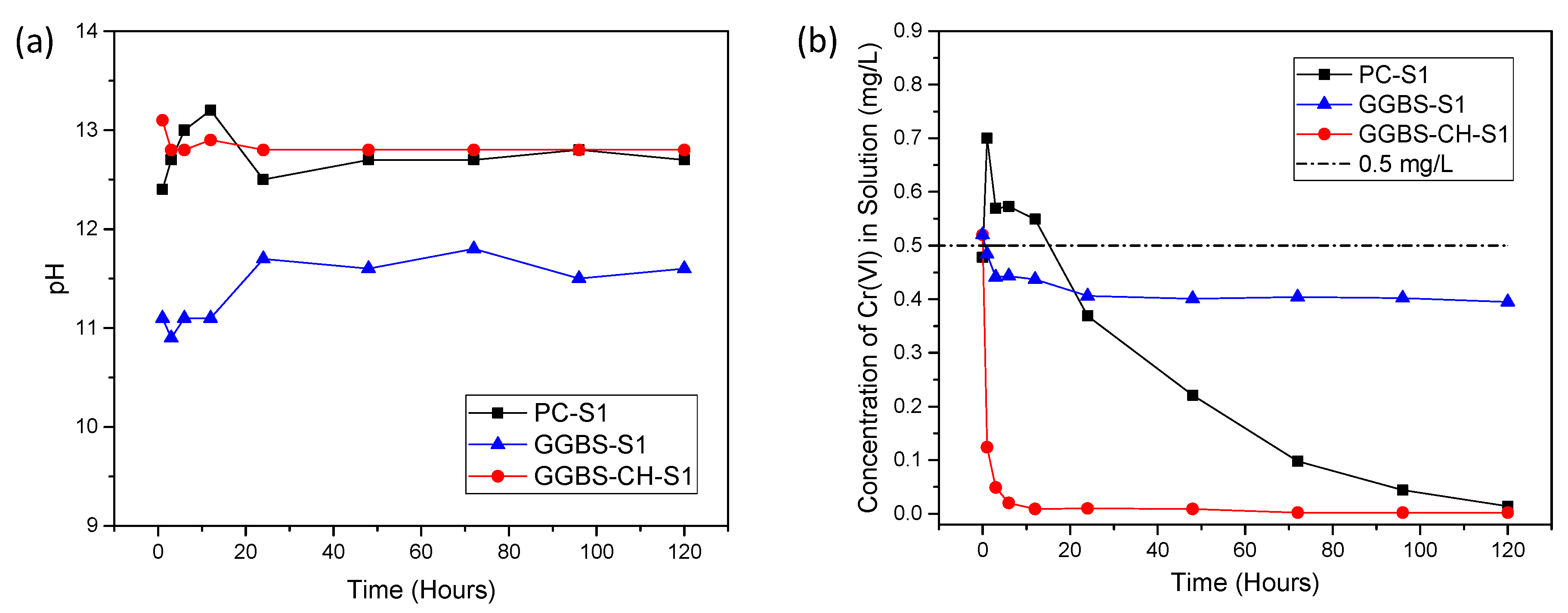
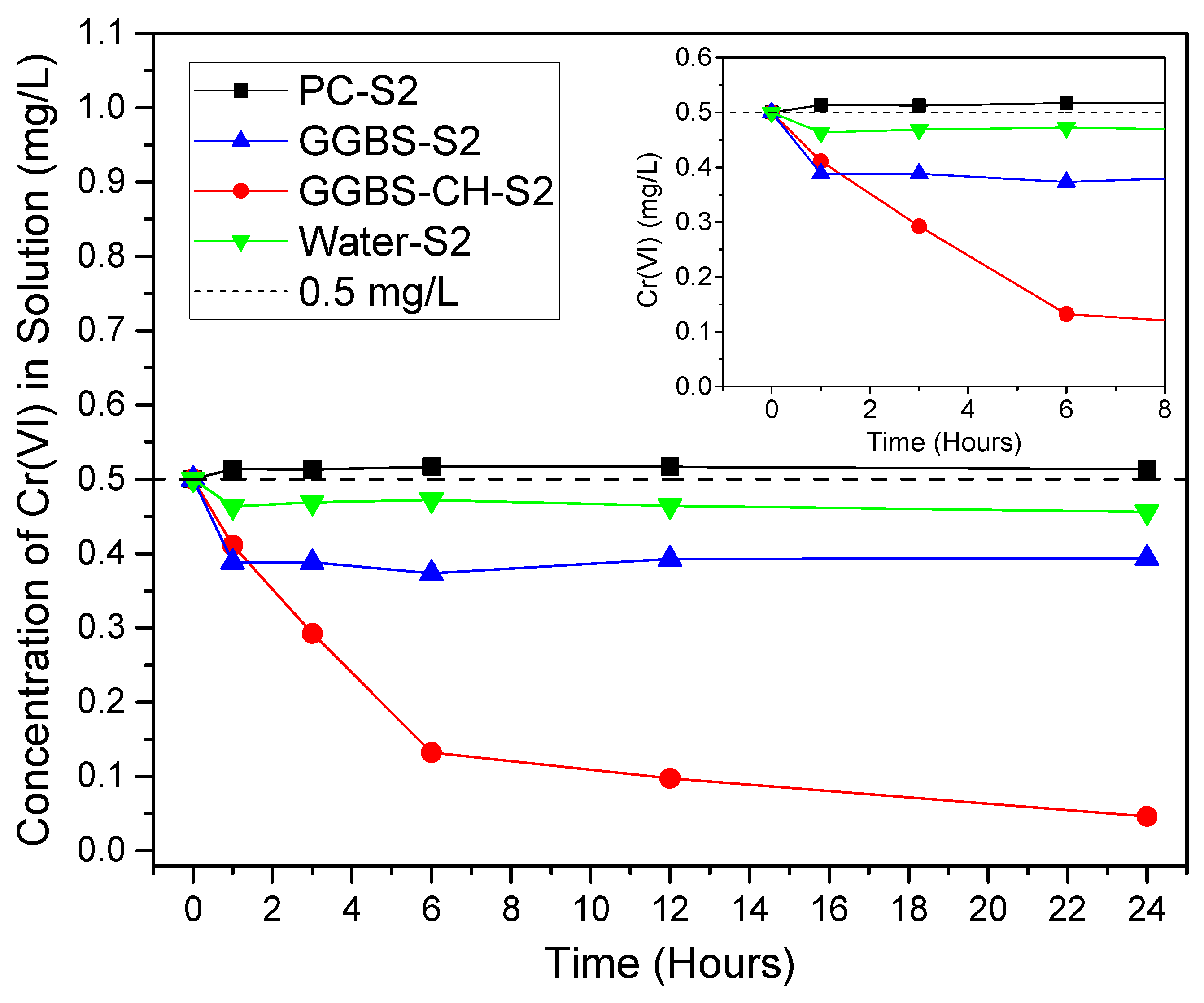
3.1. Cr(VI) Removal Using PC and GGBS-PC Samples
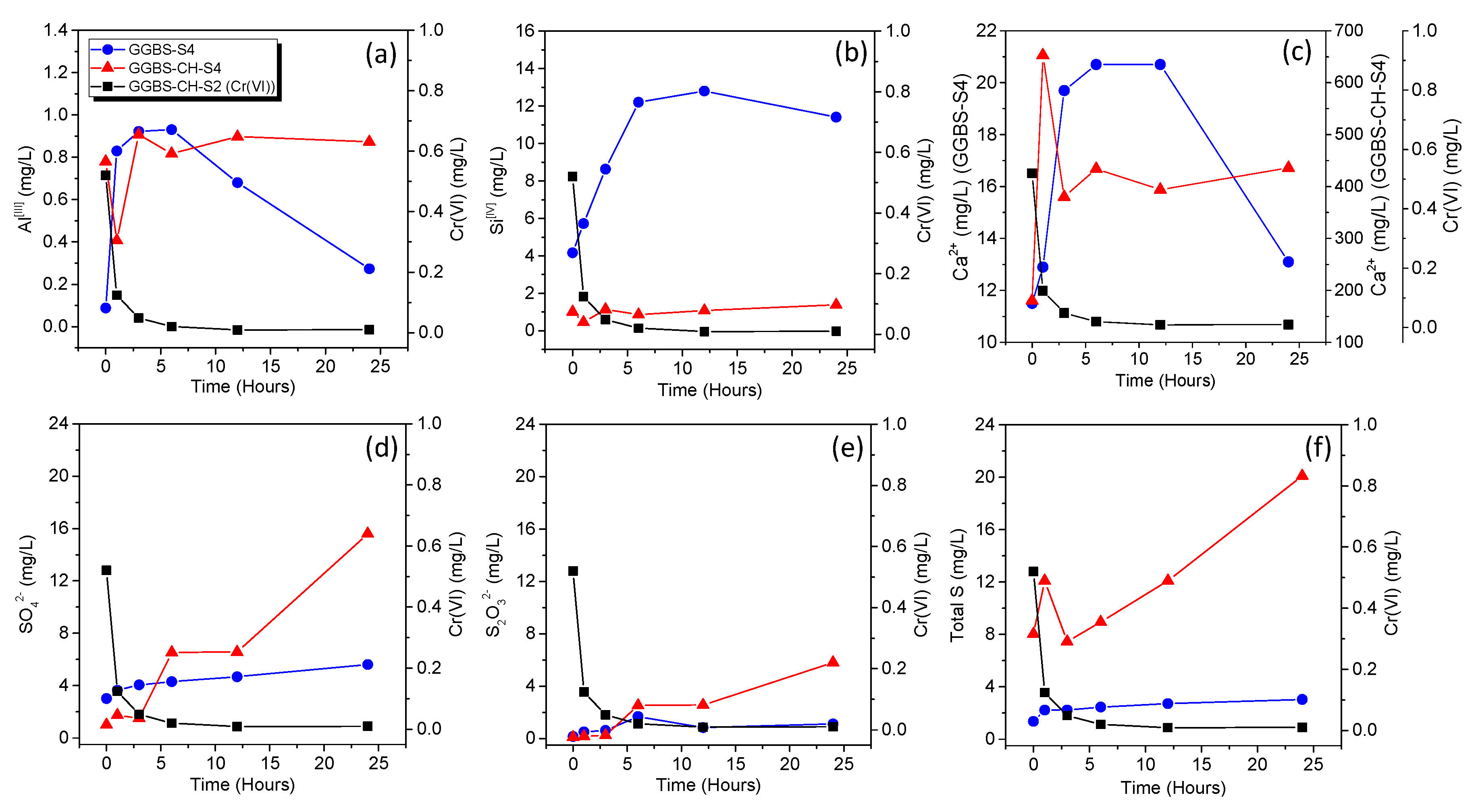
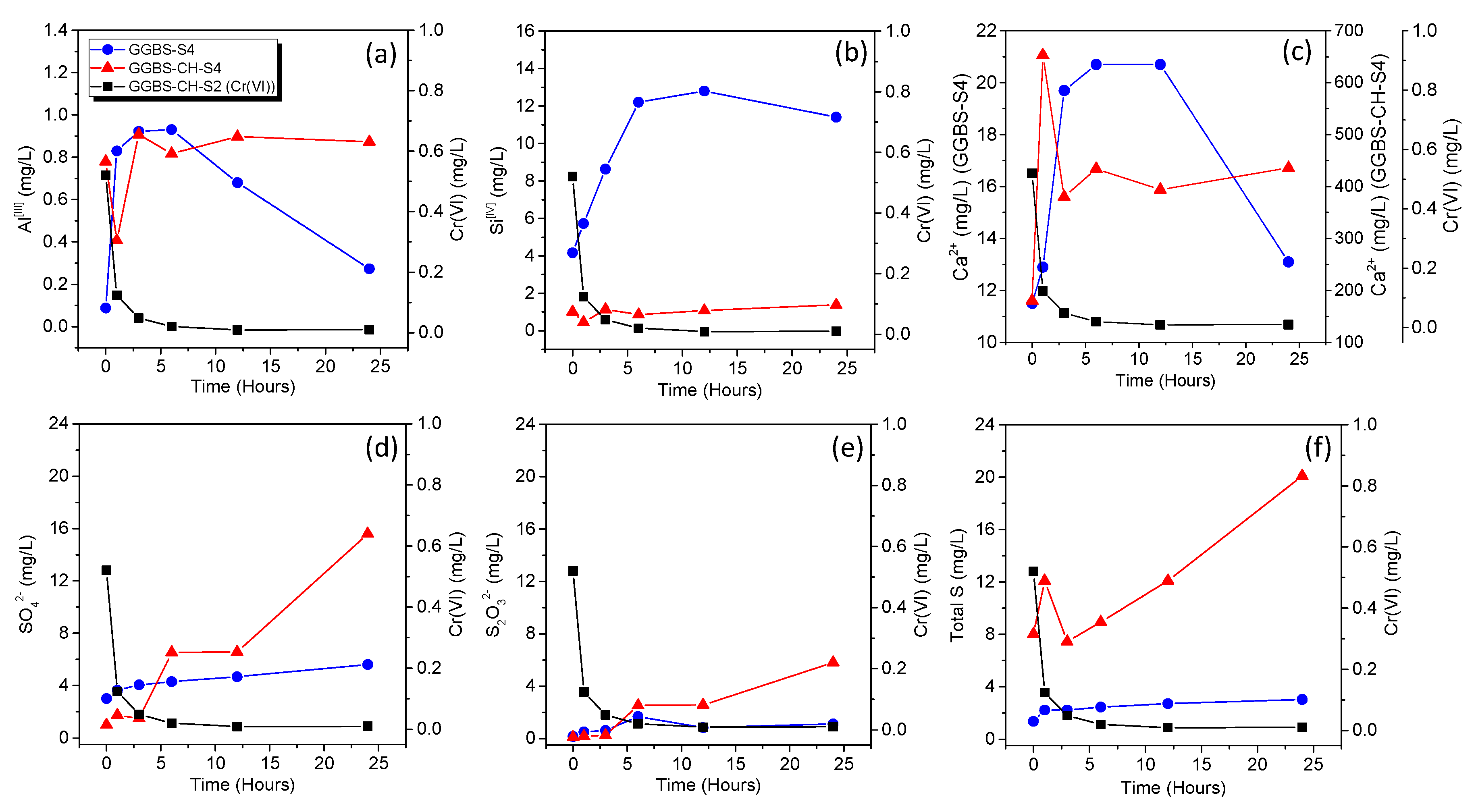
3.2. Cr(VI) Reduction via Dissolved Ions from GGBS
3.3. Cr(VI) Adsorption onto GGBS-PC Samples
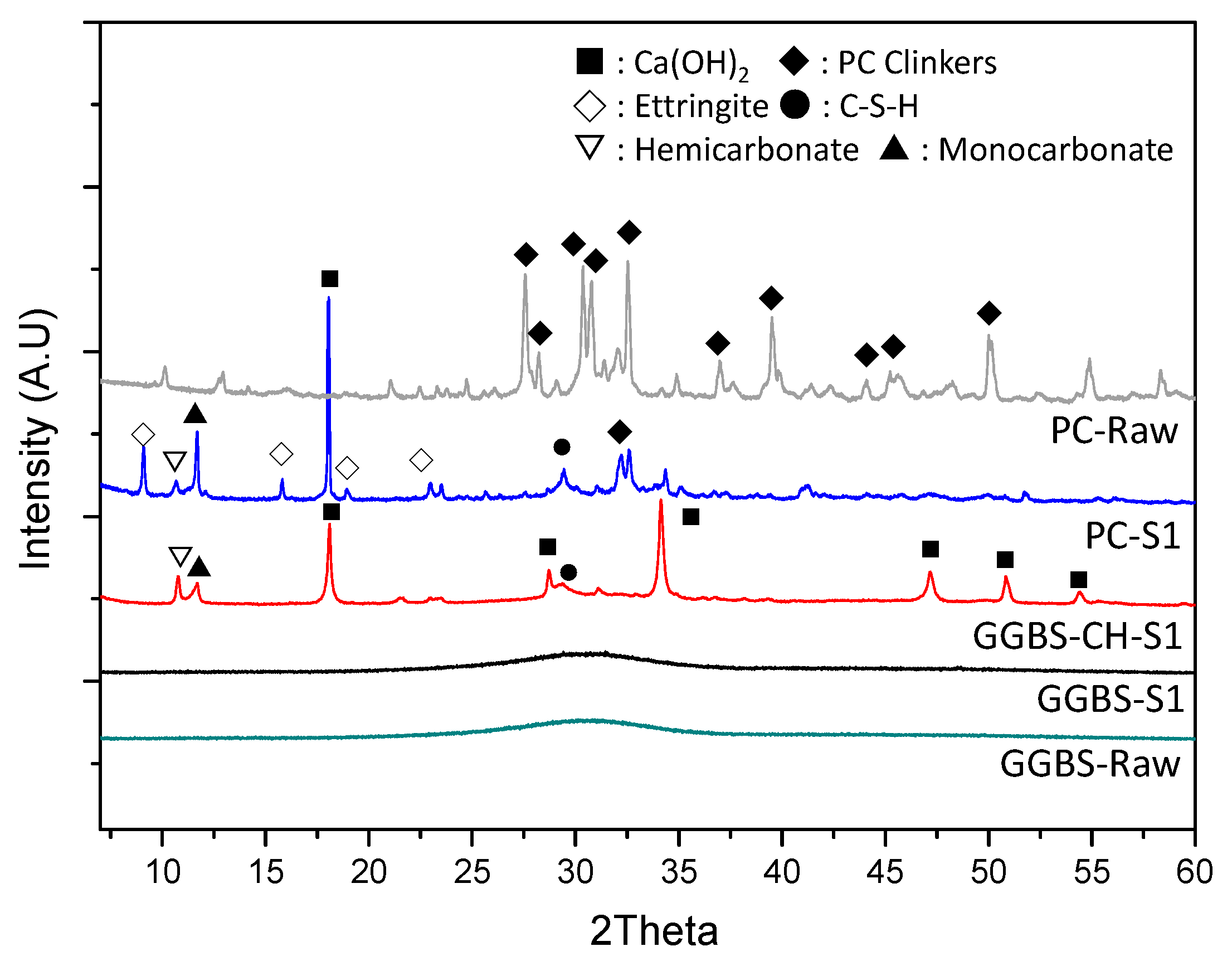
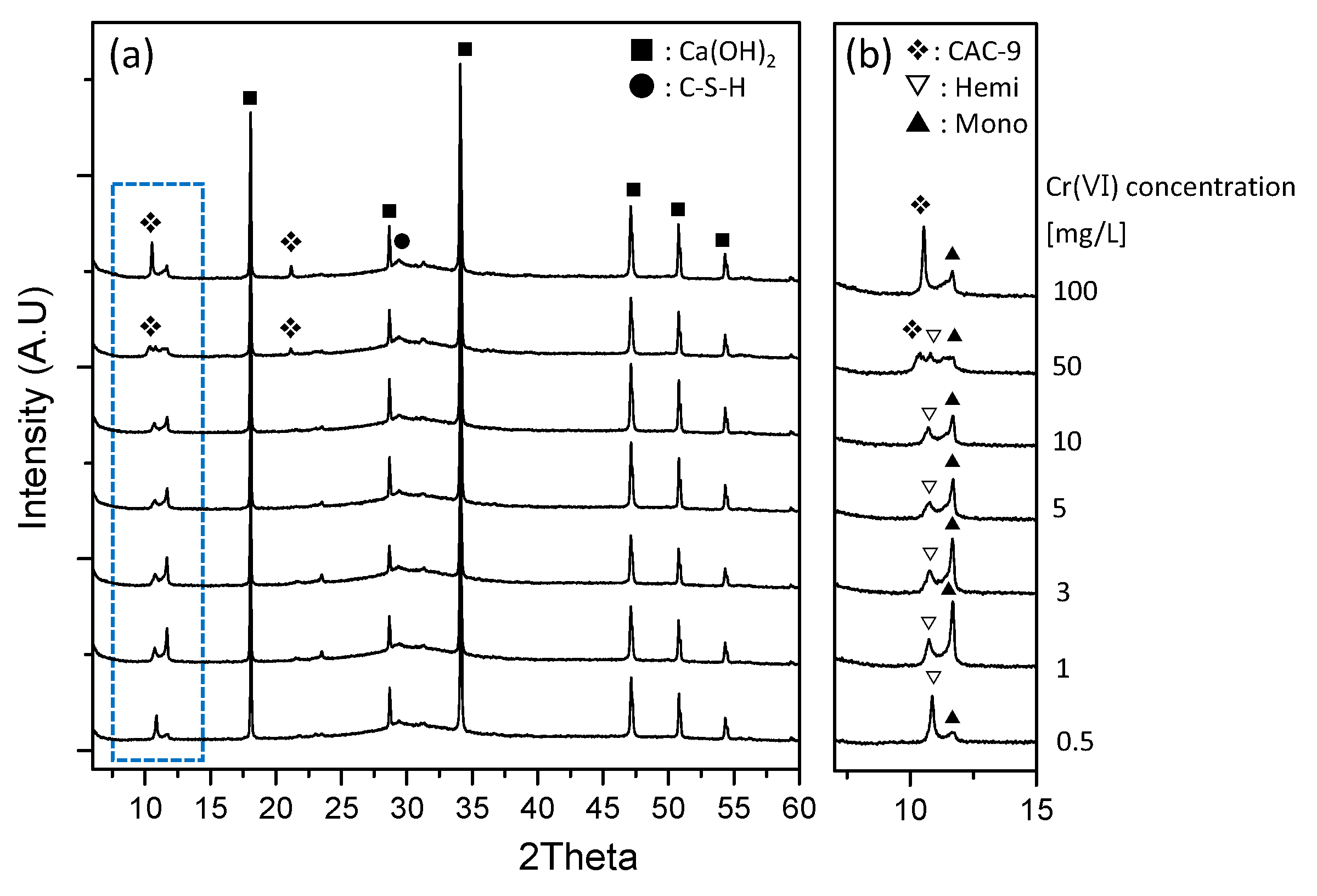
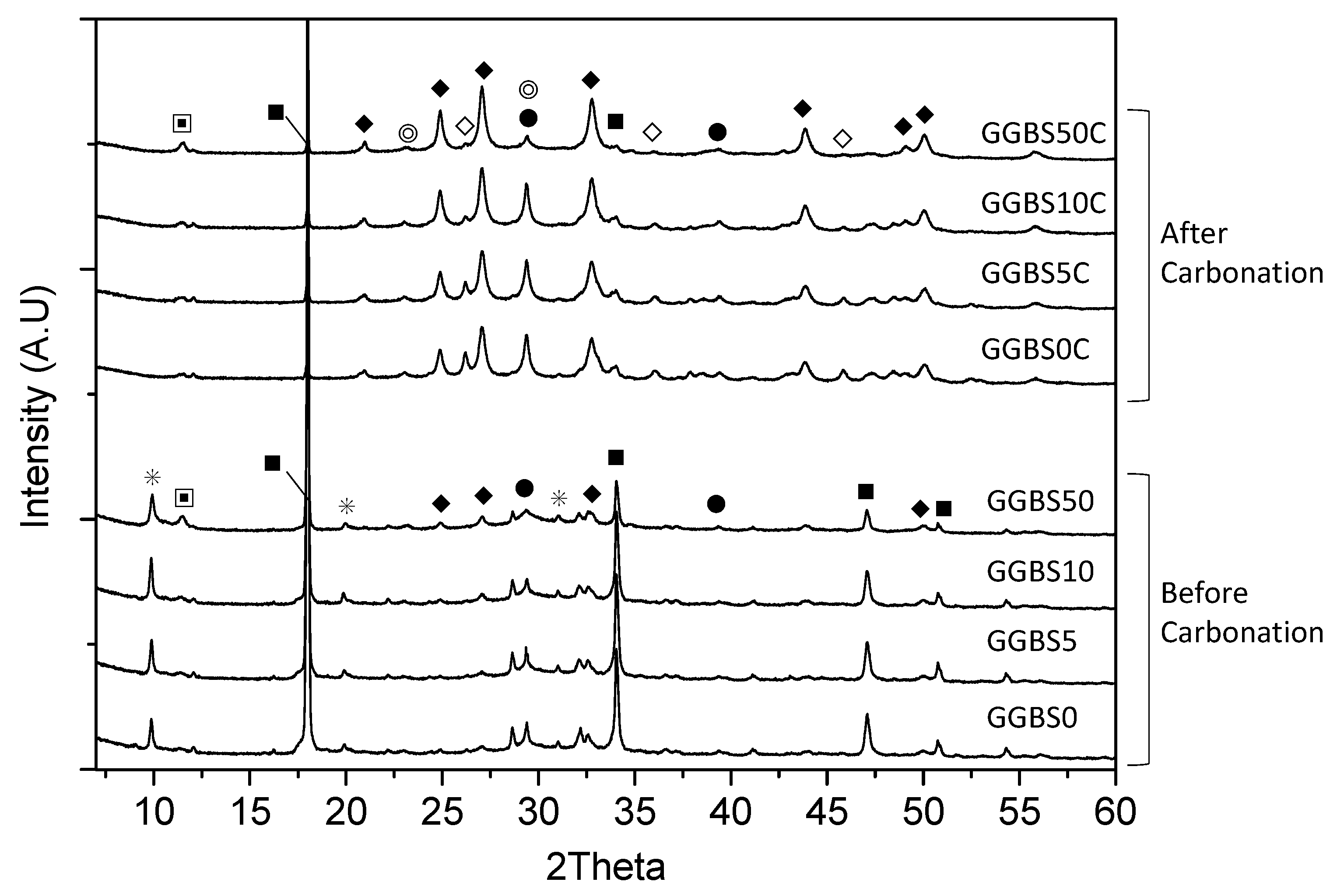
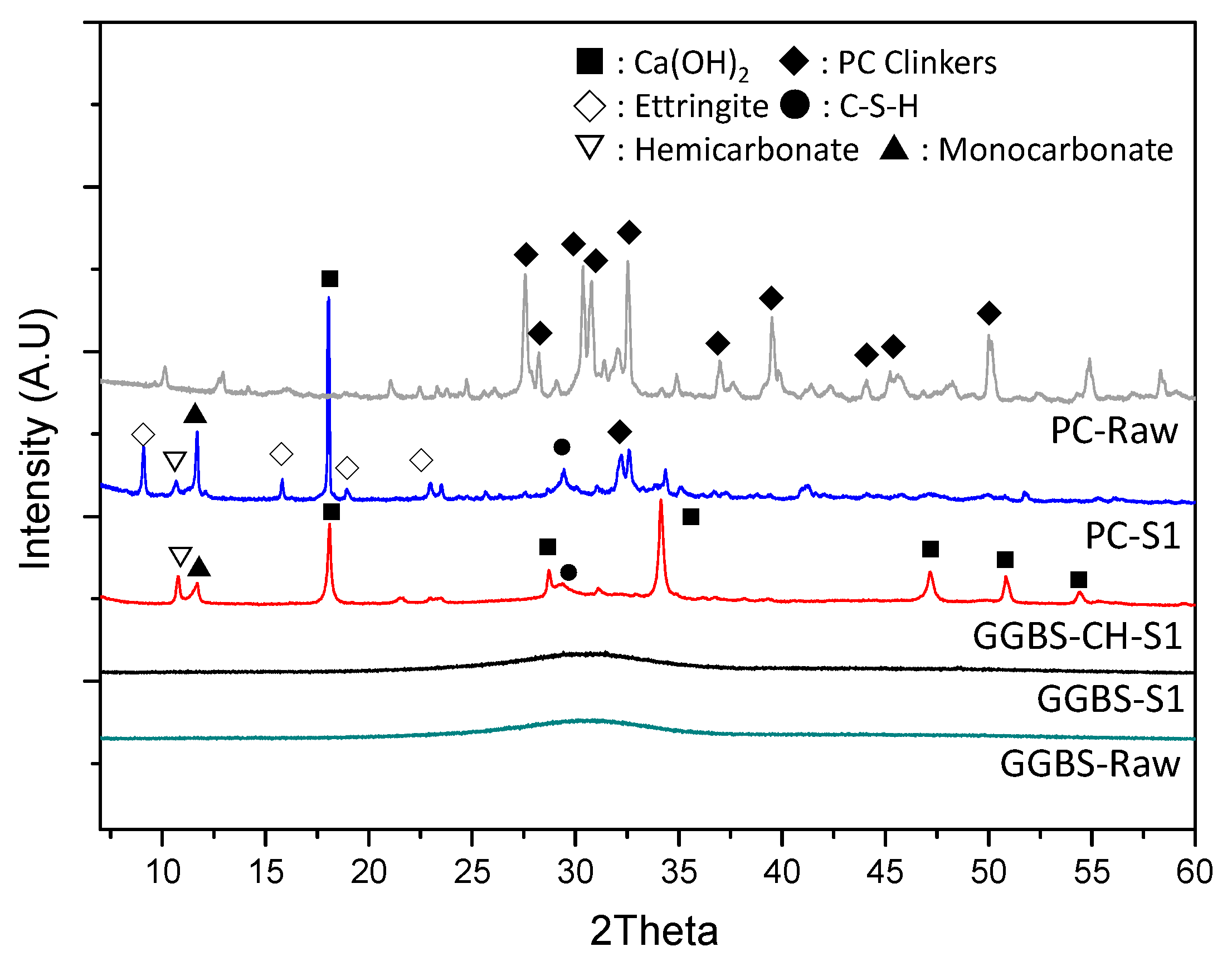
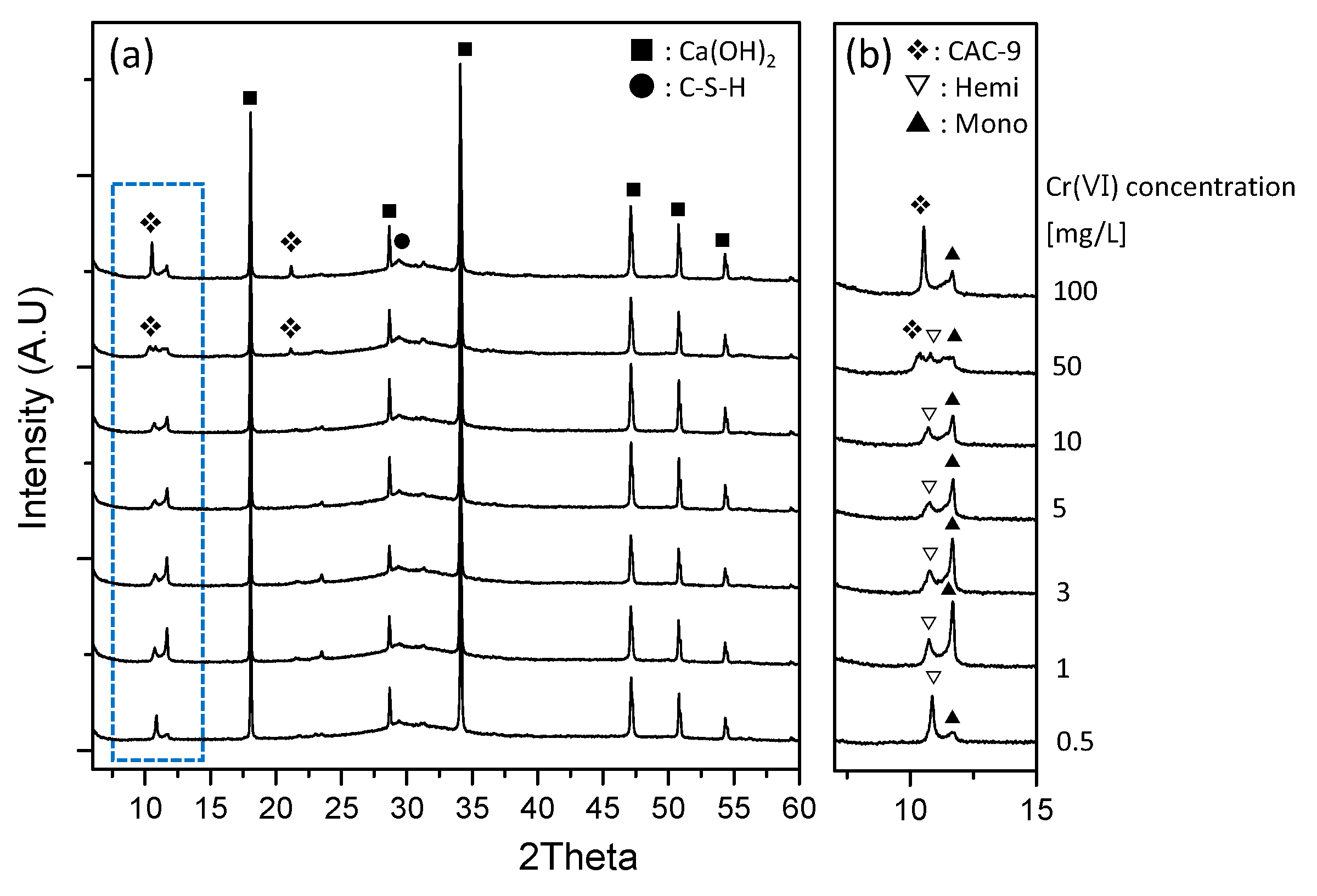
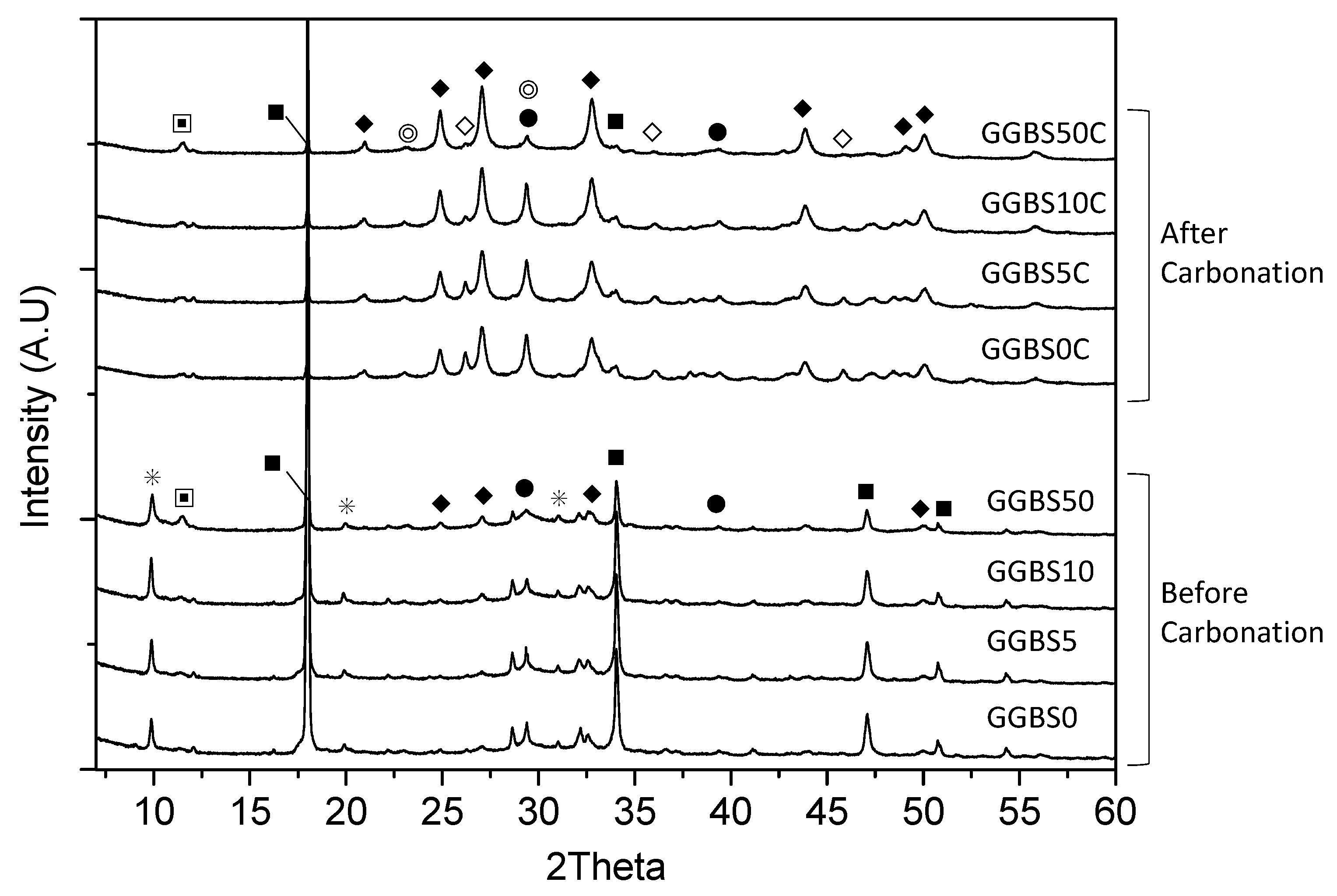
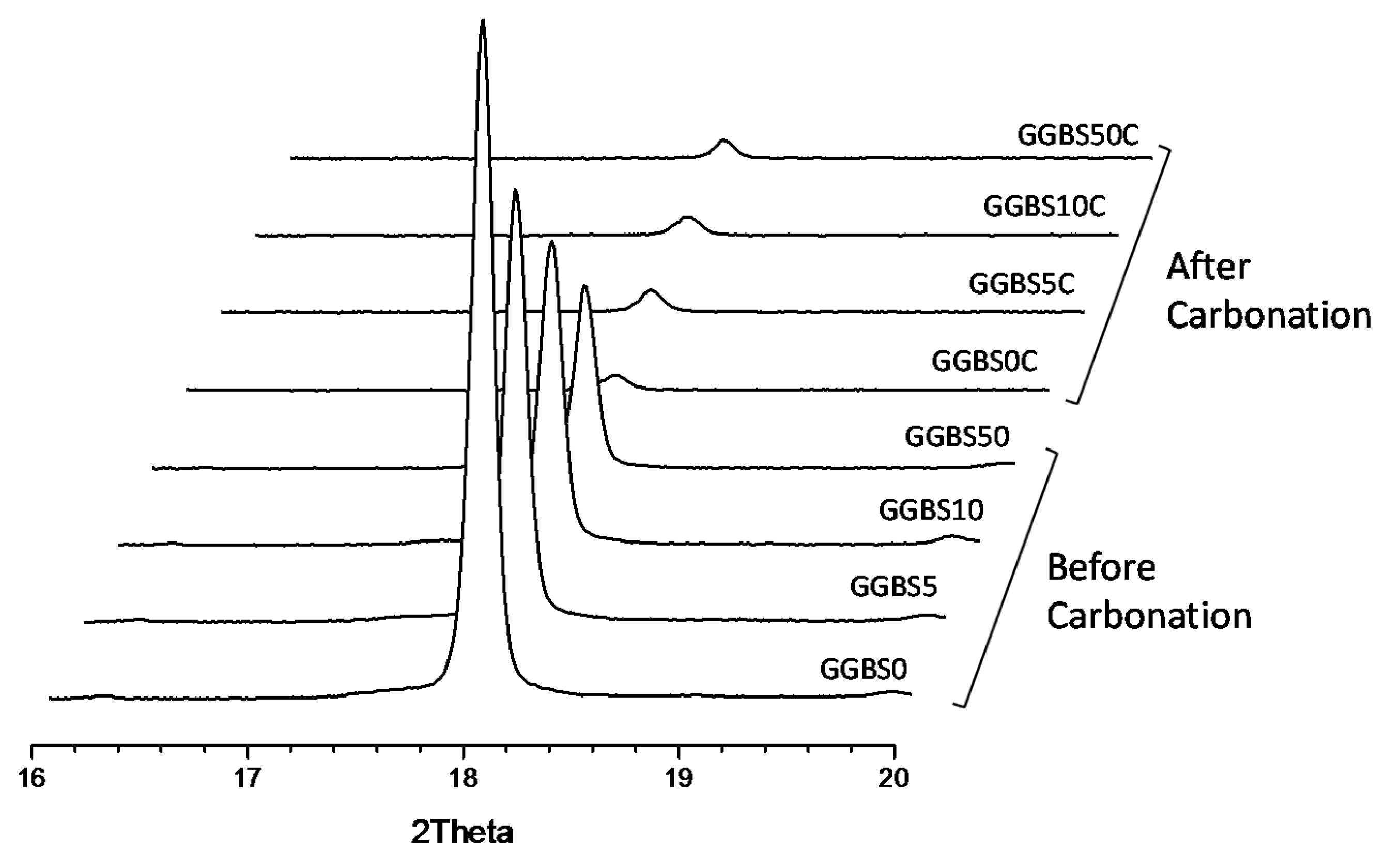
3.4. XRD Characterization of the Reacted Phases
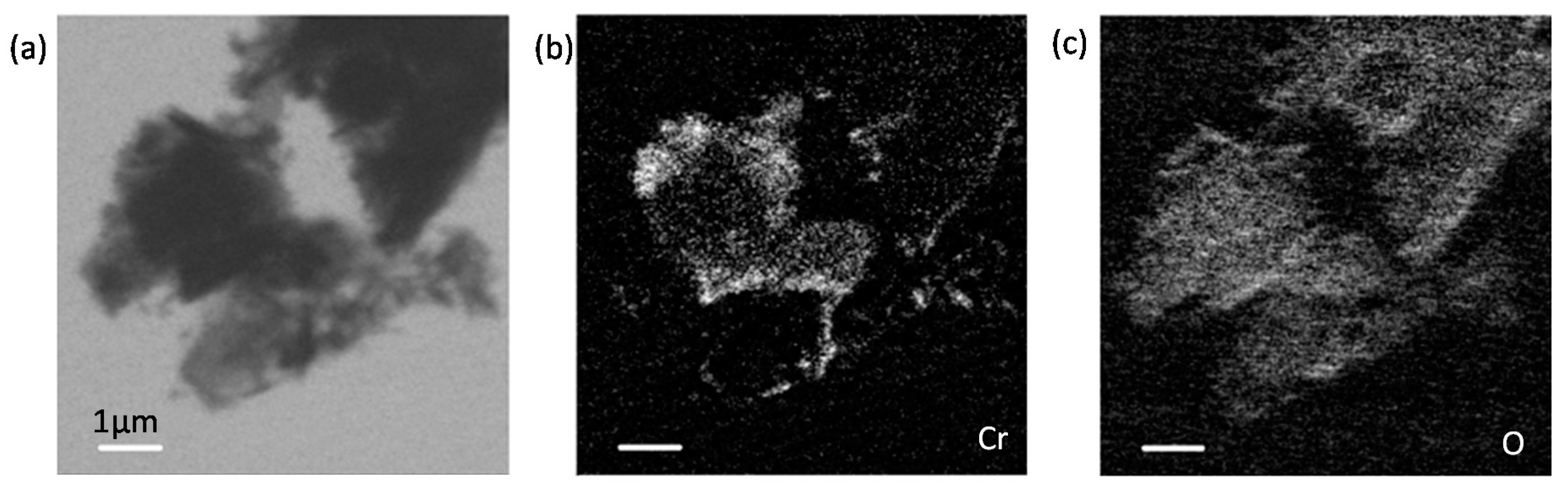
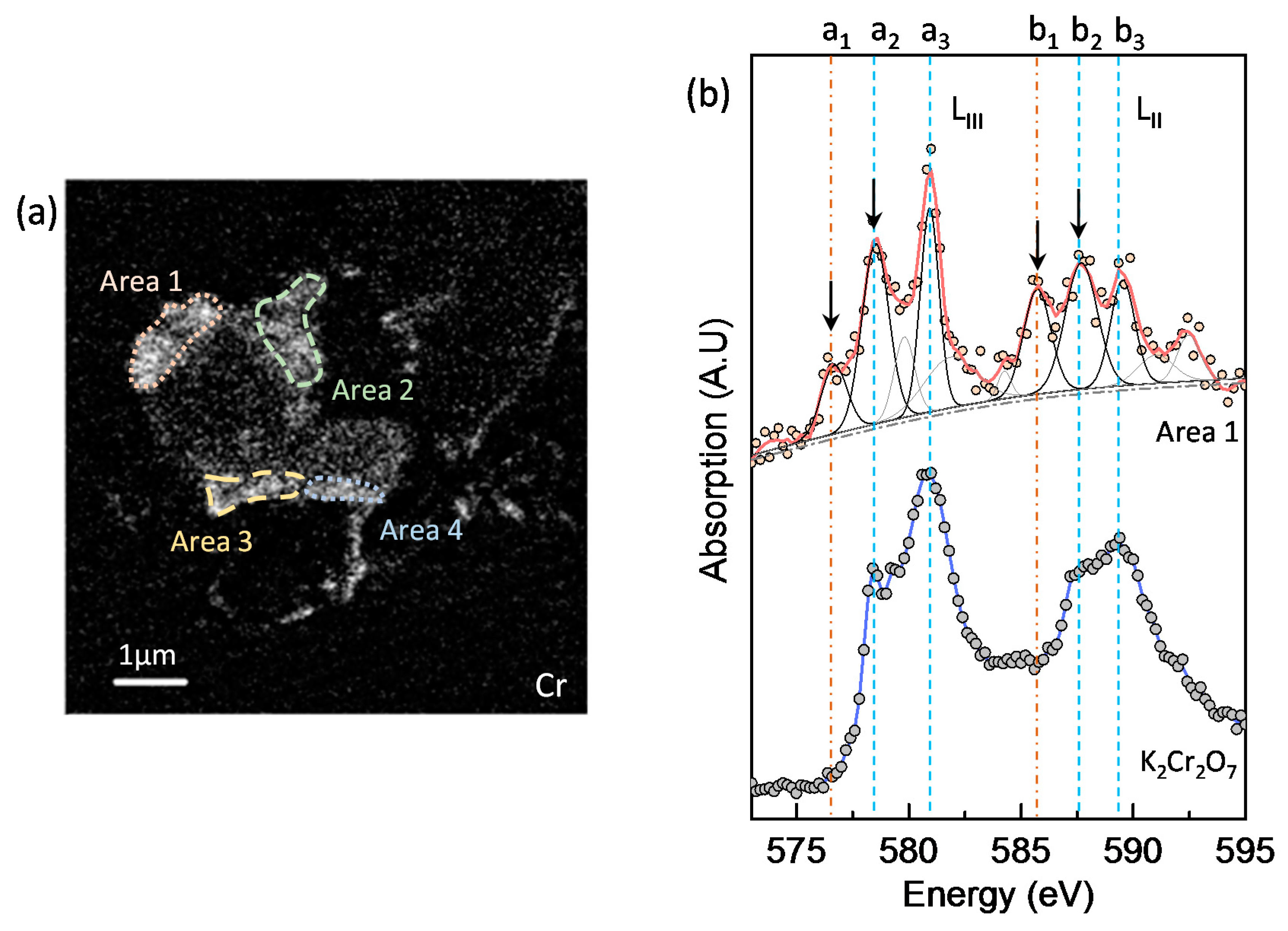
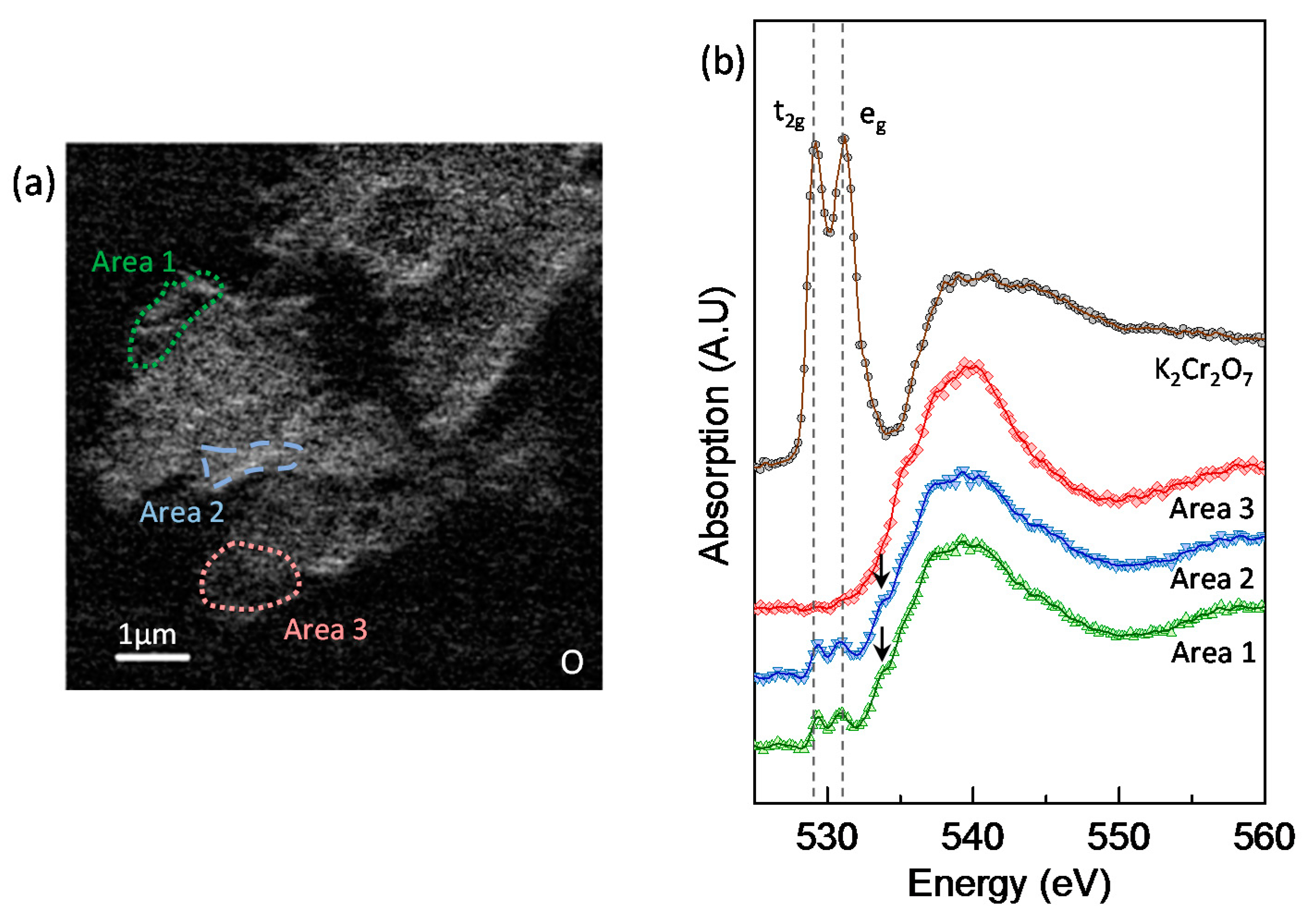
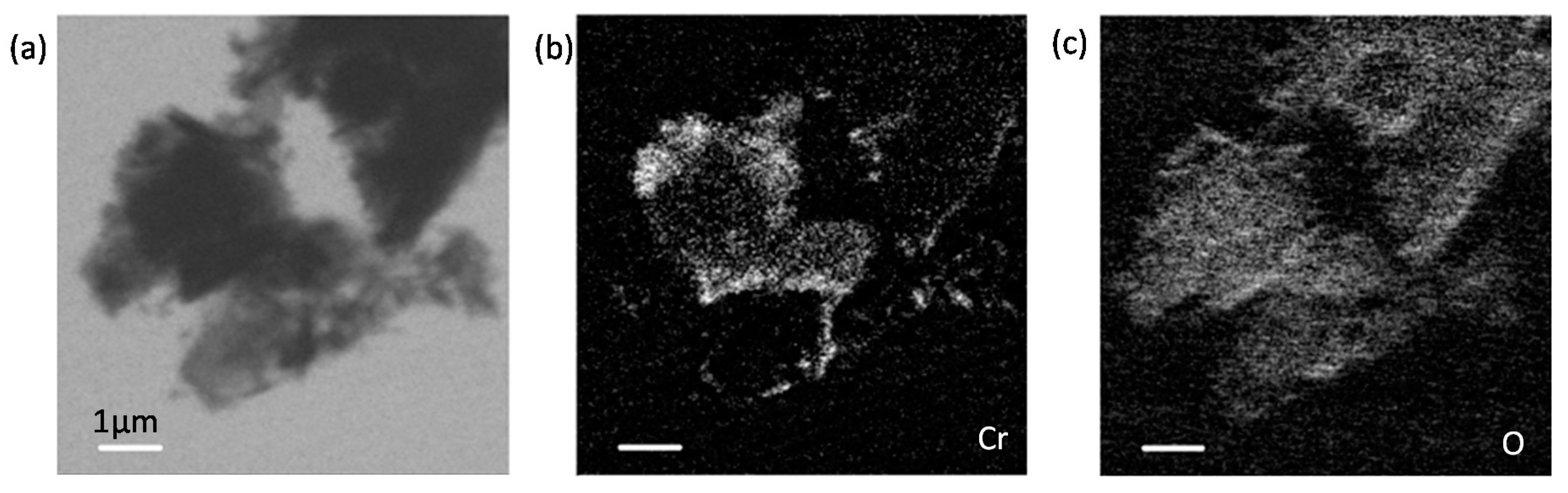
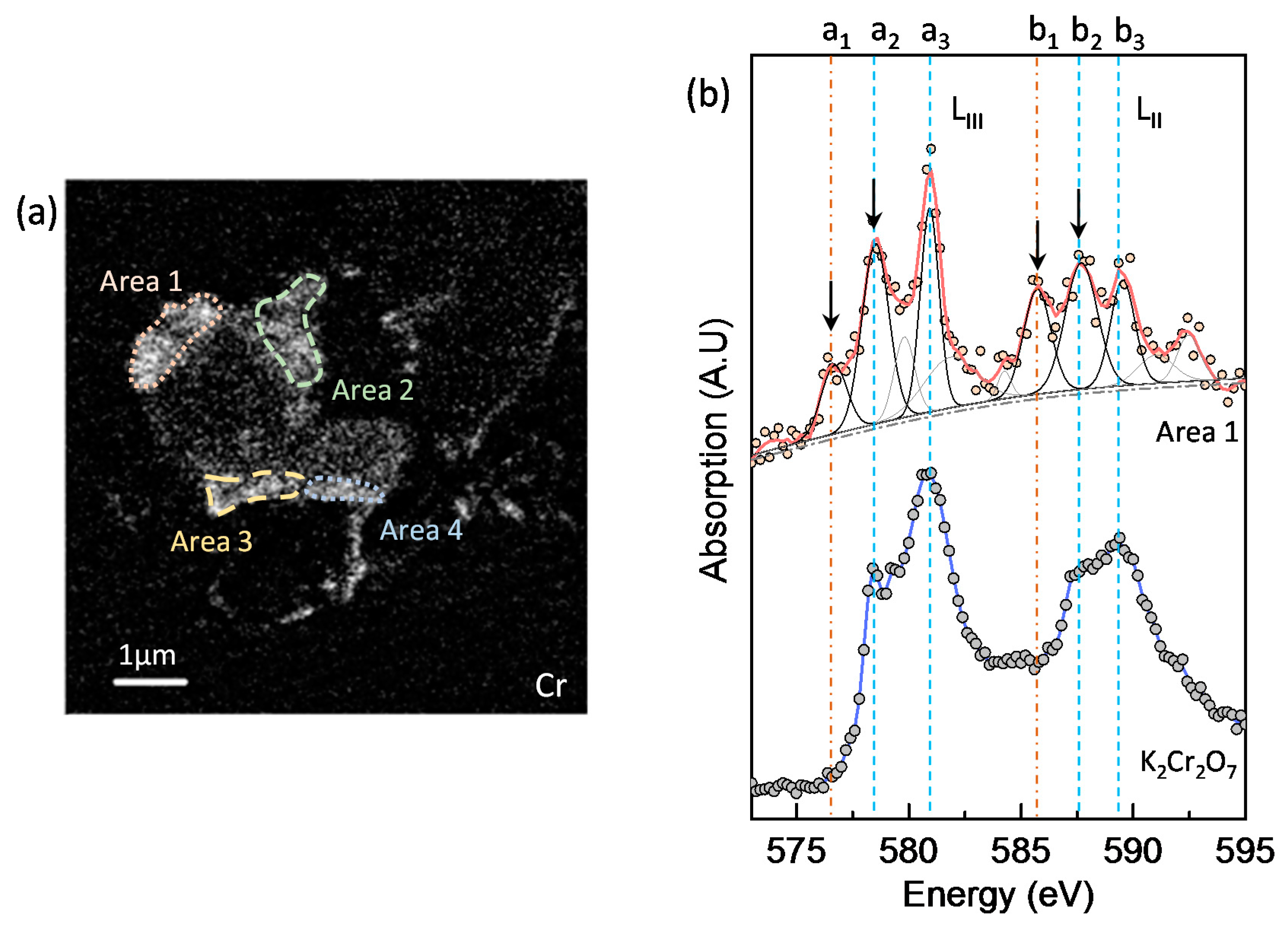
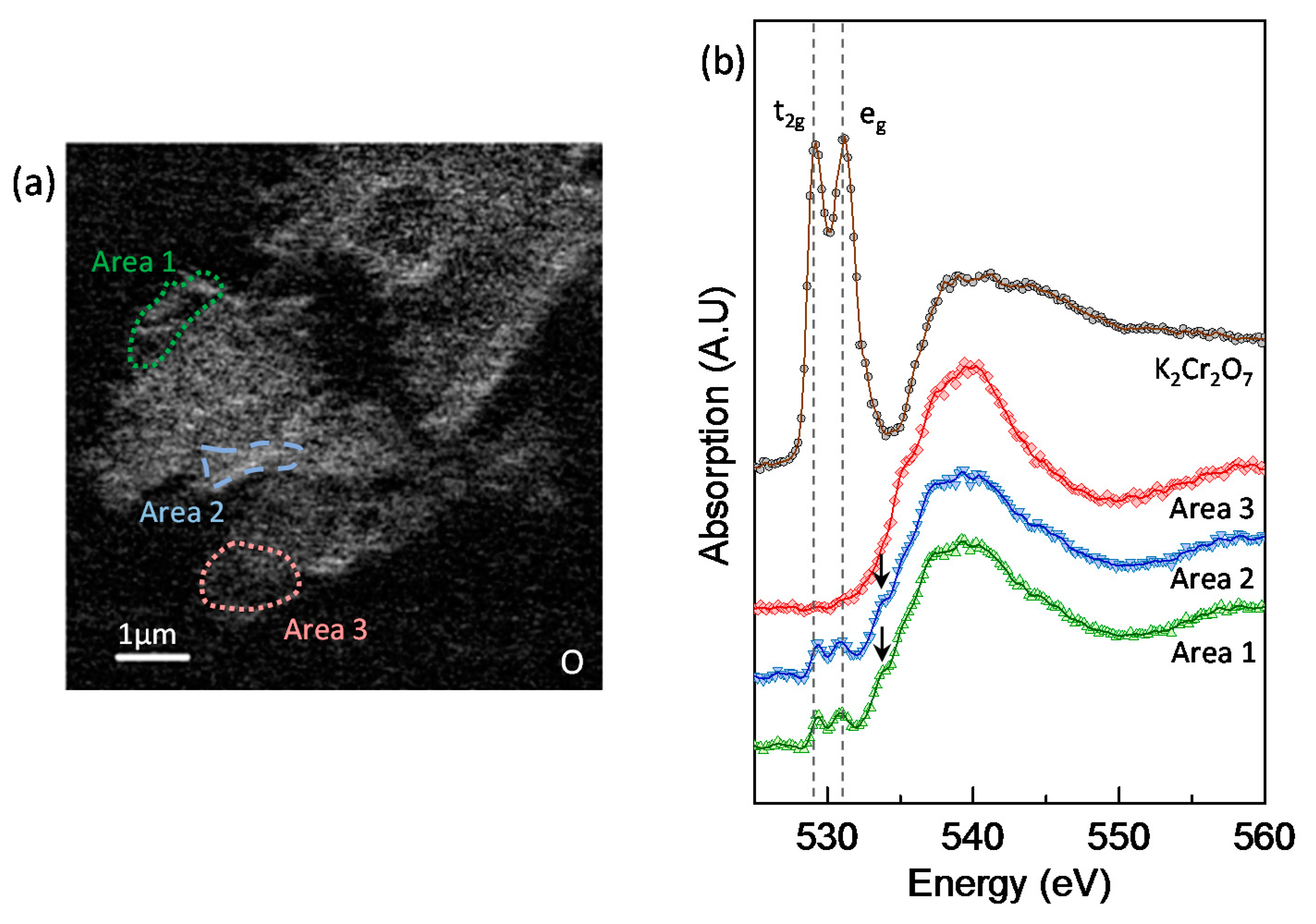
3.5. Chromium Speciation via STXM
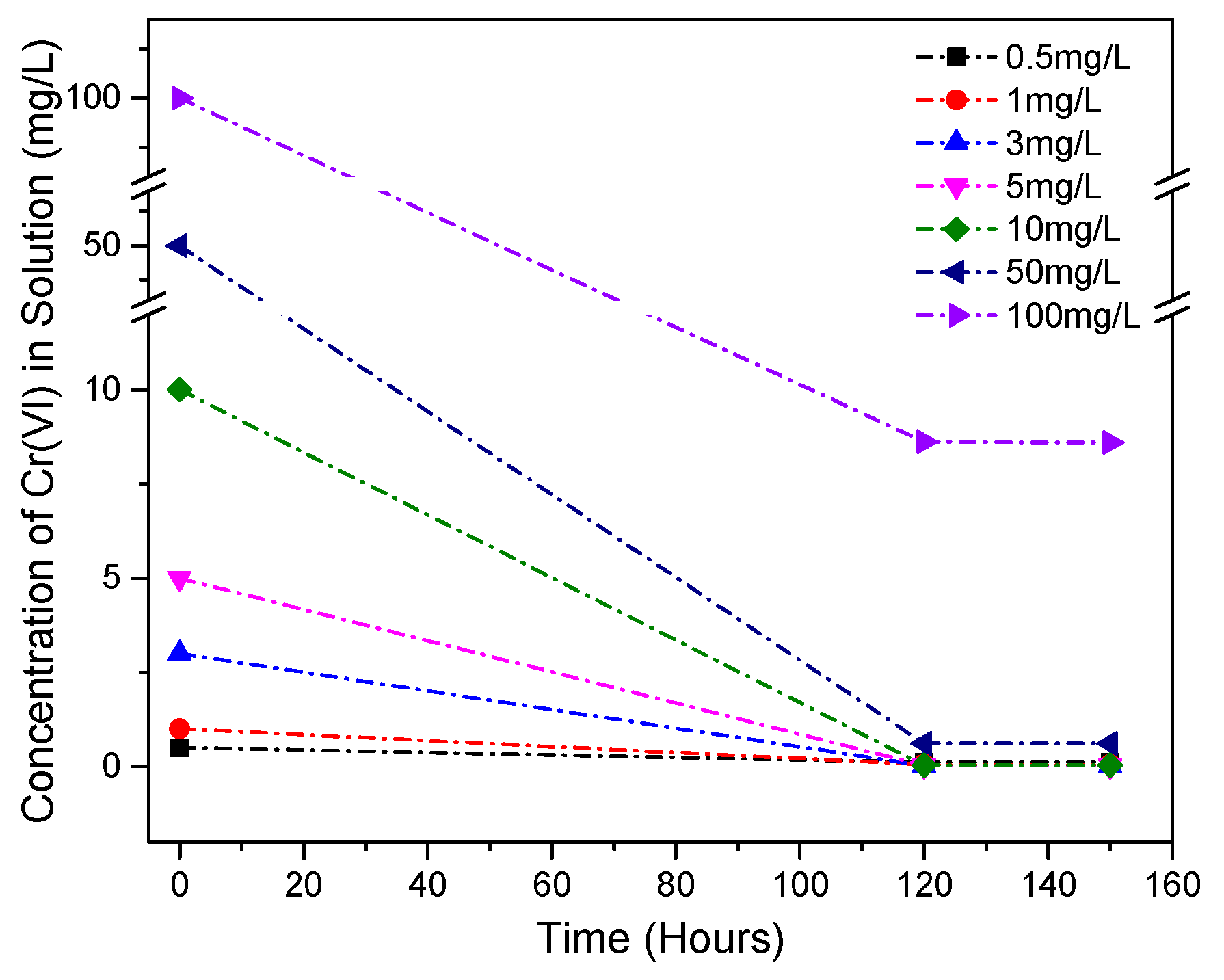
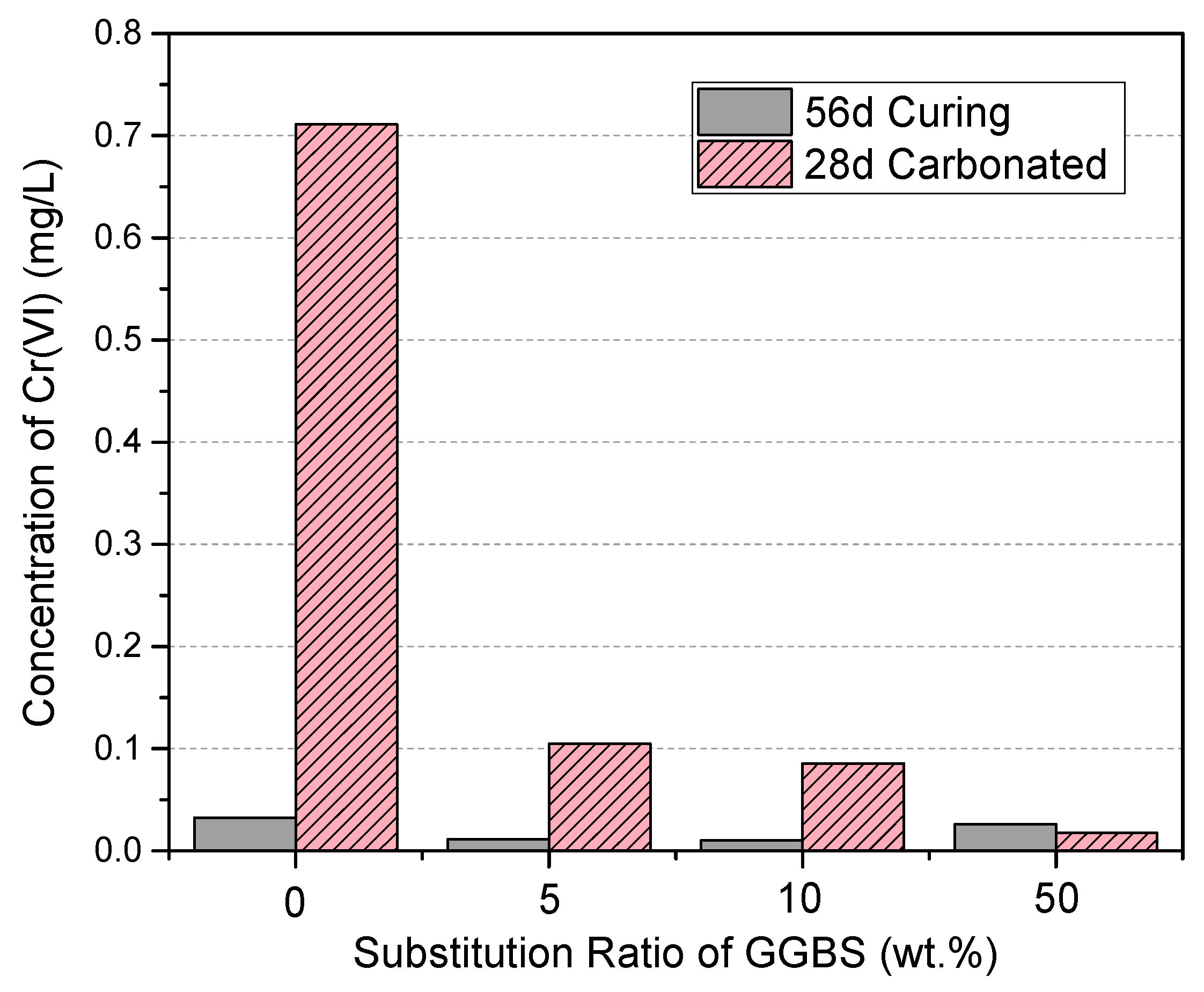
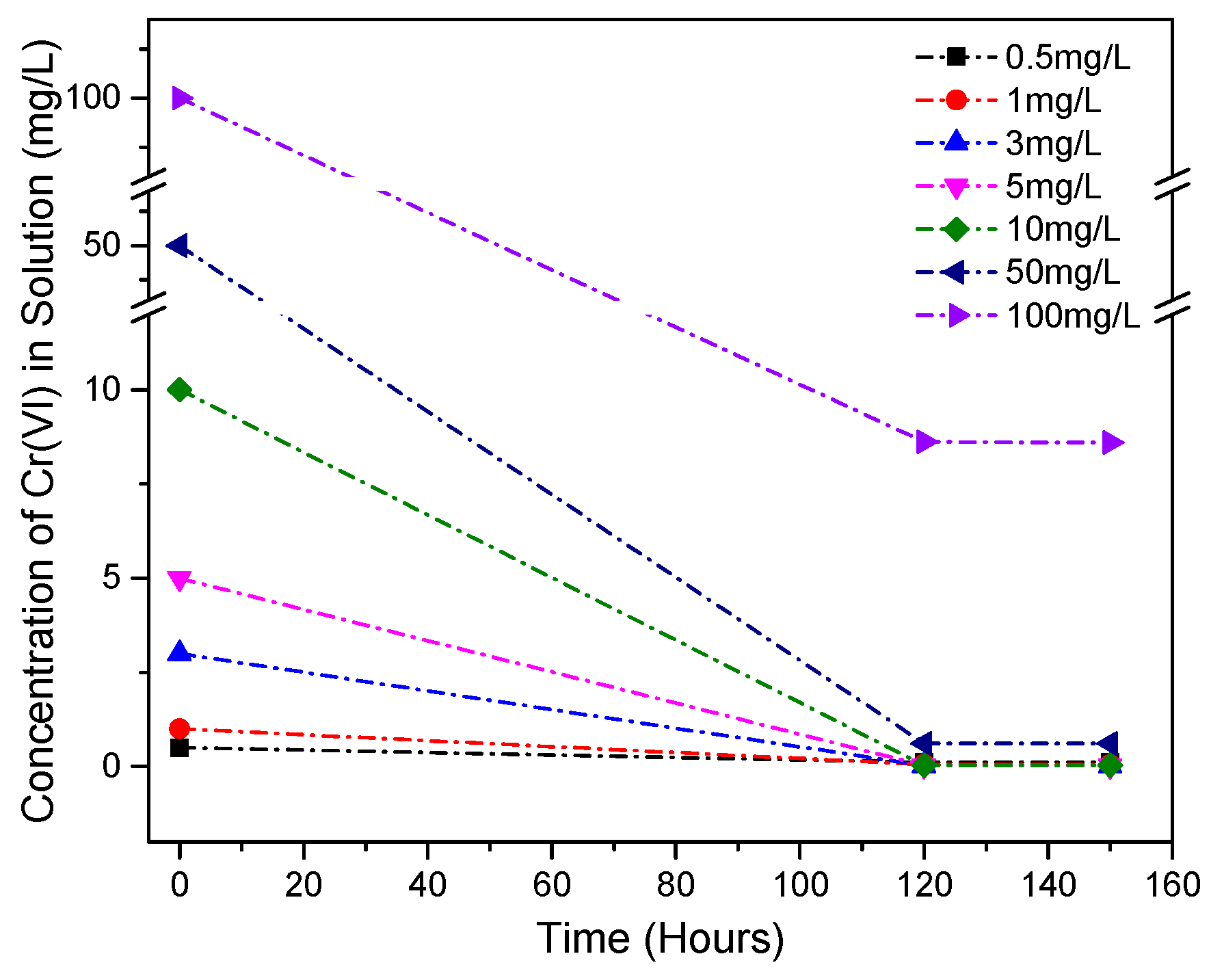
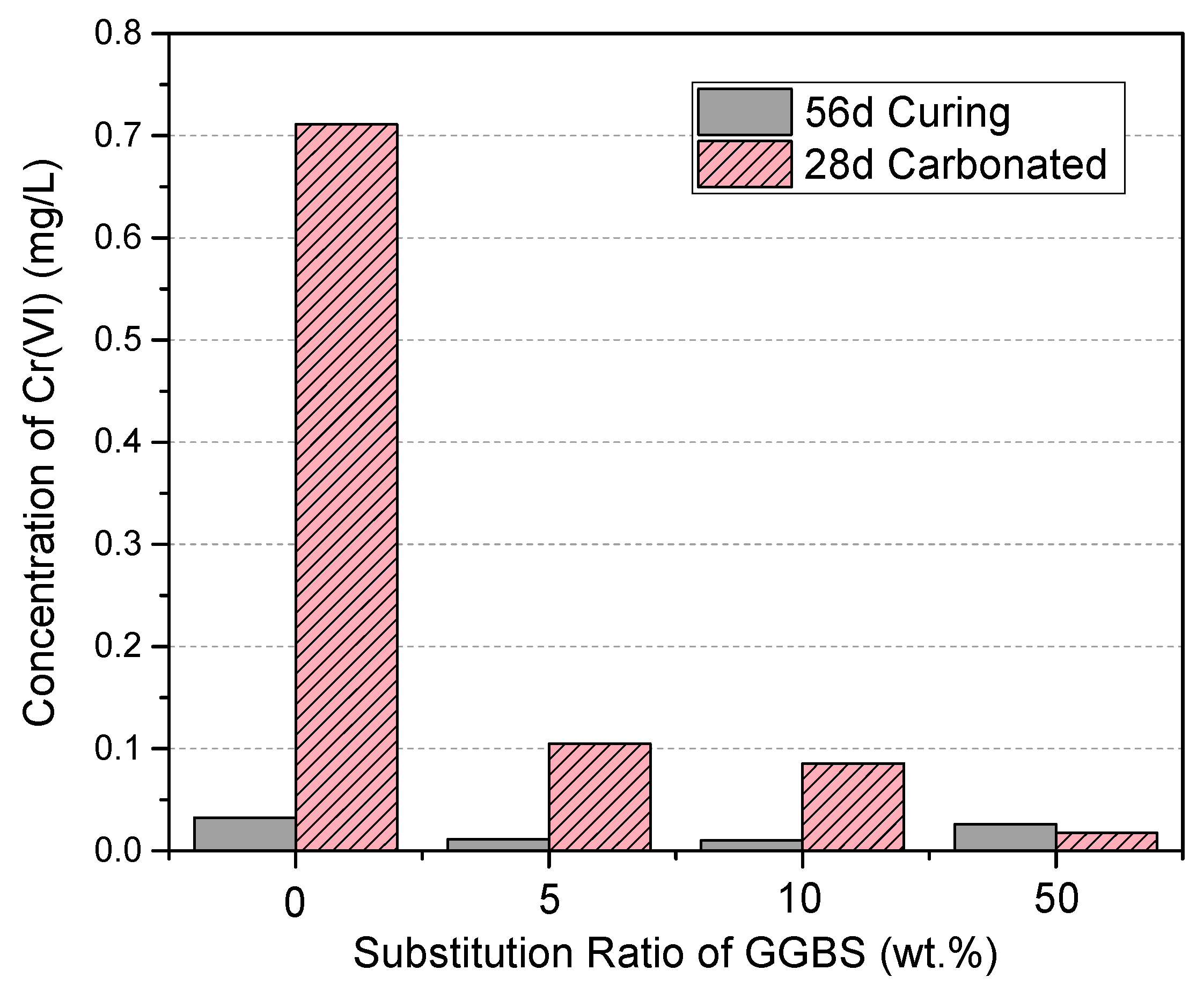
3.6. Cr(VI) Dissolution and the Effects of Accelerated Carbonation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Frías, M.; de Rojas, M.I.S.; García, N.; Luxán, M.P. Contribution of toxic elements: Hexavalent chromium in materials used in the manufacture of cement. Cem. Concr. Res. 1994, 24, 533–541. [Google Scholar] [CrossRef]
- Klemm, W.A. Hexavalent Chromium in Portland-Cement. Cem. Concr. Aggreg. 1994, 16, 43–47. [Google Scholar]
- Macphee, D.E.; Glasser, F.P. Immobilization Science of Cement Systems. MRS Bull. 1993, 18, 66–71. [Google Scholar] [CrossRef]
- Costa, M. Potential hazards of hexavalent chromate in our drinking water. Toxicol. Appl. Pharmacol. 2003, 188, 1–5. [Google Scholar] [CrossRef]
- Kjuus, H.; Lenvik, K.; Kjærheim, K.; Austad, J. Epidemiological Assessment of the Occurrence of Allergic Dermatitis in Workers in the Construction Industry Related to the Content of Cr (VI) in Cement; National Institute of Occupational Health: Ahmedabad, India, 2003. [Google Scholar]
- Dresel, P.E.; Qafoku, N.; McKinley, J.P.; Fruchter, J.S.; Ainsworth, C.C.; Liu, C.; Ilton, E.; Phillips, J. Geochemical Characterization of Chromate Contamination in the 100 Area Vadose Zone at the Hanford Site; The US Department of Energy: Washington, DC, USA, 2008.
- Chen, Q.Y.; Tyrer, M.; Hills, C.D.; Yang, X.M.; Carey, P. Immobilisation of heavy metal in cement-based solidification/stabilisation: A review. Waste Manag. 2009, 29, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Benard, A.; Susini, J.; Borschneck, D.; Hazemann, J.L.; Cheylan, P.; Vichot, A.; Bottero, J.Y. First insights of Cr speciation in leached Portland cement using X-ray spectromicroscopy. Environ. Sci. Technol. 2003, 37, 4864–4870. [Google Scholar] [CrossRef] [PubMed]
- Malviya, R.; Chaudhary, R. Factors affecting hazardous waste solidification/stabilization: A review. J. Hazard. Mater. 2006, 137, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Pera, J.; Thevenin, G.; Chabannet, M. Design of a novel system allowing the selection of an adequate binder for solidification/stabilization of wastes. Cem. Concr. Res. 1997, 27, 1533–1542. [Google Scholar] [CrossRef]
- Glasser, F.P. Fundamental aspects of cement solidification and stabilisation. J. Hazard. Mater. 1997, 52, 151–170. [Google Scholar] [CrossRef]
- Zamorani, E.; Sheikh, I.A.; Serrini, G. Physical properties measurements and leaching behavior of chromium compounds solidified in a cement matrix. Nucl. Chem. Waste Manag. 1988, 8, 239–245. [Google Scholar] [CrossRef]
- Poellmann, H.; Auer, S.; Kuzel, H.J.; Wenda, R. Solid solution of ettringites. Cem. Concr. Res. 1993, 23, 422–430. [Google Scholar] [CrossRef]
- Gougar, M.L.D.; Scheetz, B.E.; Roy, D.M. Ettringite and C-S-H Portland cement phases for waste ion immobilization: A review. Waste Manag. 1996, 16, 295–303. [Google Scholar] [CrossRef]
- Wieczorek-Ciurowa, K.; Fela, K.; Kozak, A.J. Chromium(III)-ettringite formation and its thermal stability. J. Therm. Anal. Calorim. 2001, 65, 655–660. [Google Scholar] [CrossRef]
- Chrysochoou, M.; Dermatas, D. Evaluation of ettringite and hydrocalumite formation for heavy metal immobilization: Literature review and experimental study. J. Hazard. Mater. 2006, 136, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Perkins, R.B.; Palmer, C.D. Solubility of Ca-6[Al(OH)6]2(CrO4)3 center dot 26H2O, the chromate analog of ettringite; 5–75 °C. Appl. Geochem. 2000, 15, 1203–1218. [Google Scholar] [CrossRef]
- Macias, A.; Kindness, A.; Glasser, F.P. Impact of carbon dioxide on the immobilization potential of cemented wastes: Chromium. Cem. Concr. Res. 1997, 27, 215–225. [Google Scholar] [CrossRef]
- Wazne, M.; Jagupilla, S.C.; Moon, D.H.; Christodoulatos, C.; Koutsospyros, A. Leaching Mechanisms of Cr(VI) from Chromite Ore Processing Residue. J. Environ. Qual. 2008, 37, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Richardson, I.G.; Groves, G.W. Microstructure and microanalysis of hardened cement pastes involving ground granulated blast-furnace slag. J. Mater. Sci. 1992, 27, 6204–6212. [Google Scholar] [CrossRef]
- Kilcoyne, A.L.D.; Tyliszczak, T.; Steele, W.F.; Fakra, S.; Hitchcock, P.; Franck, K.; Anderson, E.; Harteneck, B.; Rightor, E.G.; Mitchell, G.E.; et al. Interferometer-controlled scanning transmission X-ray microscopes at the Advanced Light Source. J. Synchrotron Radiat. 2003, 10, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, A.P. aXis 2000—Analysis of X-ray Images and Spectra. Available online: http://unicorn.mcmaster.ca/aXis2000.html (accessed on 21 December 2017).
- Bae, S.; Taylor, R.; Hernández-Cruz, D.; Yoon, S.; Kilcoyne, D.; Monteiro, P.J.M. Soft X-ray Spectromicroscopic Investigation of Synthetic C-S-H and C3S Hydration Products. J. Am. Ceram. Soc. 2015, 98, 2914–2920. [Google Scholar] [CrossRef]
- Bae, S.; Taylor, R.; Shapiro, D.; Denes, P.; Joseph, J.; Celestre, R.; Marchesini, S.; Padmore, H.; Tyliszczak, T.; Warwick, T.; et al. Soft X-ray Ptychographic Imaging and Morphological Quantification of Calcium Silicate Hydrates (C-S-H). J. Am. Ceram. Soc. 2015, 98, 4090–4095. [Google Scholar] [CrossRef]
- Bae, S.; Taylor, R.; Kilcoyne, D.; Moon, J.; Monteiro, P.J.M. Effects of Incorporating High-Volume Fly Ash into Tricalcium Silicate on the Degree of Silicate Polymerization and Aluminum Substitution for Silicon in Calcium Silicate Hydrate. Materials 2017, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Kanematsu, M.; Hernandez-Cruz, D.; Moon, J.; Kilcoyne, D.; Monteiro, P.J.M. In Situ Soft X-ray Spectromicroscopy of Early Tricalcium Silicate Hydration. Materials 2016, 9, 976. [Google Scholar] [CrossRef] [PubMed]
- Rashad, A.M.; Bai, Y.; Basheer, P.A.M.; Milestone, N.B.; Collier, N.C. Hydration and properties of sodium sulfate activated slag. Cem. Concr. Compos. 2013, 37, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Jimenez, A.; Puertas, F. Effect of activator mix on the hydration and strength behaviour of alkali-activated slag cements. Adv. Cem. Res. 2003, 15, 129–136. [Google Scholar] [CrossRef]
- Chang, J.J. A study on the setting characteristics of sodium silicate-activated slag pastes. Cem. Concr. Res. 2003, 33, 1005–1011. [Google Scholar] [CrossRef]
- Song, S.; Sohn, D.; Jennings, H.M.; Mason, T.O. Hydration of alkali-activated ground granulated blast furnace slag. J. Mater. Sci. 2000, 35, 249–257. [Google Scholar] [CrossRef]
- Roy, A. Sulfur speciation in granulated blast furnace slag: An X-ray absorption spectroscopic investigation. Cem. Concr. Res. 2009, 39, 659–663. [Google Scholar] [CrossRef]
- Grolimund, D.; Trainor, T.P.; Fitts, J.P.; Kendelewicz, T.; Liu, P.; Chambers, S.A.; Brown, G.E. Identification of Cr species at the aqueous solution-hematite interface after Cr(VI)-Cr(III) reduction using GI-XAFS and Cr L-edge NEXAFS. J. Synchrotron Radiat. 1999, 6, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Naftel, S.J.; Sham, T.K.; Yiu, Y.M.; Yates, B.W. Calcium L-edge XANES study of some calcium compounds. J. Synchrotron Radiat. 2001, 8, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, J. NEXAFS Spectroscopy; Springer: Berlin, Germany, 1992. [Google Scholar]
- Doyle, C.S.; Kendelewicz, T.; Bostick, B.C.; Brown, G.E. Soft X-ray spectroscopic studies of the reaction of fractured pyrite surfaces with Cr(Vl)-containing aqueous solutions. Geochim. Cosmochim. Acta 2004, 68, 4287–4299. [Google Scholar] [CrossRef]
- Shen, C.; Chen, H.; Wu, S.; Wen, Y.; Li, L.; Jiang, Z.; Li, M.; Liu, W. Highly efficient detoxification of Cr(VI) by chitosan-Fe(III) complex: Process and mechanism studies. J. Hazard. Mater. 2013, 244–245, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Kucheyev, S.O.; van Buuren, T.; Baumann, T.F.; Satcher, J.H.; Willey, T.M.; Meulenberg, R.W.; Felter, T.E.; Poco, J.F.; Gammon, S.A.; Terminello, L.J. Electronic structure of titania aerogels from soft X-ray absorption spectroscopy. Phys. Rev. B 2004, 69, 245102. [Google Scholar] [CrossRef]
- Andersson, K.; Allard, B.; Bengtsson, M.; Magnusson, B. Chemical-Composition of Cement Pore Solutions. Cem. Concr. Res. 1989, 19, 327–332. [Google Scholar] [CrossRef]
- Rimmelé, G.; Barlet-Gouédard, V.; Porcherie, O.; Goffé, B.; Brunet, F. Heterogeneous porosity distribution in Portland cement exposed to CO2-rich fluids. Cem. Concr. Res. 2008, 38, 1038–1048. [Google Scholar] [CrossRef]
- Johannesson, B.; Utgenannt, P. Microstructural changes caused by carbonation of cement mortar. Cem. Concr. Res. 2001, 31, 925–931. [Google Scholar] [CrossRef]
- Puertas, F.; Palacios, M.; Vazquez, T. Carbonation process of alkali-activated slag mortars. J. Mater. Sci. 2006, 41, 3071–3082. [Google Scholar] [CrossRef]
- Slegers, P.A.; Rouxhet, P.G. Carbonation of the hydration products of tricalcium silicate. Cem. Concr. Res. 1976, 6, 381–388. [Google Scholar] [CrossRef]
- Morandeau, A.; Thiery, M.; Dangla, P. Investigation of the carbonation mechanism of CH and C-S-H in terms of kinetics, microstructure changes and moisture properties. Cem. Concr. Res. 2014, 56, 153–170. [Google Scholar] [CrossRef] [Green Version]
- Glasser, F.; Matschei, T. Interactions between Portland cement and carbon dioxide. In Proceedings of the ICCC Conference, Montreal, QC, Canada, 8–13 July 2007. [Google Scholar]
- Thiery, M.; Villain, G.; Dangla, P.; Platret, G. Investigation of the carbonation front shape on cementitious materials: Effects of the chemical kinetics. Cem. Concr. Res. 2007, 37, 1047–1058. [Google Scholar] [CrossRef]
- Borges, P.H.R.; Costa, J.O.; Milestone, N.B.; Lynsdale, C.J.; Streatfield, R.E. Carbonation of CH and C-S-H in composite cement pastes containing high amounts of BFS. Cem. Concr. Res. 2010, 40, 284–292. [Google Scholar] [CrossRef]
- Richardson, I.G.; Brough, A.R.; Groves, G.W.; Dobson, C.M. The Characterization of Hardened Alkali-Activated Blast-Furnace Slag Pastes and the Nature of the Calcium Silicate Hydrate (C-S-H) Phase. Cem. Concr. Res. 1994, 24, 813–829. [Google Scholar] [CrossRef]
- Wang, S.D.; Scrivener, K.L. Hydration Products of Alkali-Activated Slag Cement. Cem. Concr. Res. 1995, 25, 561–571. [Google Scholar] [CrossRef]
- Suda, Y.; Saeki, T.; Saito, T. Relation between Chemical Composition and Physical Properties of C-S-H Generated from Cementitious Materials. J. Adv. Concr. Technol. 2015, 13, 275–290. [Google Scholar] [CrossRef]
- Beaudoin, J.J.; Feldman, R.F. The significance of helium diffusion measurements in studying the removal of structural water in inorganic hydrated systems. Cem. Concr. Res. 1978, 8, 223–231. [Google Scholar] [CrossRef]
- Black, L.; Breen, C.; Yarwood, J.; Garbev, K.; Stemmermann, P.; Gasharova, B. Structural features of C-S-H(I) and its carbonation in air—A Raman spectroscopic study. Part II: Carbonated phases. J. Am. Ceram. Soc. 2007, 90, 908–917. [Google Scholar] [CrossRef]
- Richardson, I.G.; Groves, G.W. The structure of the calcium silicate hydrate phases present in hardened pastes of white Portland cement/blast-furnace slag blends. J. Mater. Sci. 1997, 32, 4793–4802. [Google Scholar] [CrossRef]
- Ayub, T.; Khan, S.U.; Memon, F.A. Mechanical Characteristics of Hardened Concrete with Different Mineral Admixtures: A Review. Sci. World J. 2014, 2014, 875085. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Chemical Compositions (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SiO2 | Al2O3 | Fe2O3 | CaO | MgO | SO3 | Na2O | K2O | TiO2 | P2O5 | MnO | Total | |
| PC | 20.94 | 5.45 | 2.83 | 64.96 | 1.5 | 2.05 | 0.32 | 0.48 | 0.27 | 0.31 | 0.08 | 99.22 |
| GGBS | 33.03 | 14.82 | 0.42 | 41.44 | 6.51 | 2.07 | 0.27 | 0.27 | 0.53 | 0.04 | 0.18 | 99.9 |
| Sample | Mix Proportions | Measurements | ||||
|---|---|---|---|---|---|---|
| Distilled Water (L) | GGBS (g) | CH (g) | PC (g) | Cr(VI) Concentration (mg/L) | ||
| PC-S1 | 2 | - | - | 40 | 0.5 | UV-vis, XRD, pH |
| GGBS-S1 | 40 | - | - | UV-vis, XRD, pH | ||
| GGBS-CH-S1 | 40 | 20 | - | UV-vis, XRD, pH, Soft X-ray absorption microscopy | ||
| PC-S2 | 2.4 | - | - | 48 | 0.5 | UV-vis |
| GGBS-S2 | 48 | - | - | |||
| GGBS-CH-S2 | 48 | 24 | - | |||
| GGBS-CH-S3 | 2 | 40 | 20 | - | 0.5~100 | UV-vis, XRD |
| GGBS-S4 | 2 | 40 | - | - | - | Ion chromatography |
| GGBS-CH-S4 | 2 | 40 | 20 | - | - | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, S.; Hikaru, F.; Kanematsu, M.; Yoshizawa, C.; Noguchi, T.; Yu, Y.; Ha, J. Removal of Hexavalent Chromium in Portland Cement Using Ground Granulated Blast-Furnace Slag Powder. Materials 2018, 11, 11. https://doi.org/10.3390/ma11010011
Bae S, Hikaru F, Kanematsu M, Yoshizawa C, Noguchi T, Yu Y, Ha J. Removal of Hexavalent Chromium in Portland Cement Using Ground Granulated Blast-Furnace Slag Powder. Materials. 2018; 11(1):11. https://doi.org/10.3390/ma11010011
Chicago/Turabian StyleBae, Sungchul, Fumino Hikaru, Manabu Kanematsu, Chiaki Yoshizawa, Takafumi Noguchi, Youngsang Yu, and Juyoung Ha. 2018. "Removal of Hexavalent Chromium in Portland Cement Using Ground Granulated Blast-Furnace Slag Powder" Materials 11, no. 1: 11. https://doi.org/10.3390/ma11010011