Unexpected Ground-State Structure and Mechanical Properties of Ir2Zr Intermetallic Compound

1
College of Physics and Optoelectronics Technology, Baoji University of Arts and Sciences, Baoji 721016, China
2
College of Optoelectronic Technology, Chengdu University of Information Technology, Chengdu 610225, China
*
Author to whom correspondence should be addressed.
Materials 2018, 11(1), 103; https://doi.org/10.3390/ma11010103
Submission received: 12 December 2017
/
Revised: 1 January 2018
/
Accepted: 9 January 2018
/
Published: 10 January 2018
(This article belongs to the Special Issue Density Functional Theory (DFT) Calculation of Materials Properties)
Abstract
:Using an unbiased structure searching method, a new orthorhombic Cmmm structure consisting of ZrIr12 polyhedron building blocks is predicted to be the thermodynamic ground-state of stoichiometric intermetallic Ir2Zr in Ir-Zr systems. The formation enthalpy of the Cmmm structure is considerably lower than that of the previously synthesized Cu2Mg-type phase, by ~107 meV/atom, as demonstrated by the calculation of formation enthalpy. Meanwhile, the phonon dispersion calculations further confirmed the dynamical stability of Cmmm phase under ambient conditions. The mechanical properties, including elastic stability, rigidity, and incompressibility, as well as the elastic anisotropy of Cmmm-Ir2Zr intermetallic, have thus been fully determined. It is found that the predicted Cmmm phase exhibits nearly elastic isotropic and great resistance to shear deformations within the (100) crystal plane. Evidence of atomic bonding related to the structural stability for Ir2Zr were manifested by calculations of the electronic structures.
1. Introduction
The platinum-group metals (PGMs)—osmium, iridium, platinum, ruthenium, rhodium, and palladium—are immensely important in numerous technologies, but the experimental and computational data on their binary alloys still contain many gaps. In contrast to other transition metals [1,2,3], interest in PGMs is driven by their essential role in a wide variety of industrial applications, which is at odds with their high cost. The primary application of PGMs is in catalysis, where they are core ingredients in the chemical, petroleum, and automotive industries. Recently, the Ir-Zr binary alloys have become a topic that is currently attracting considerable interest for their potential applications in aeronautics and electronics applications. Like Ni-based alloys, Ir-Zr alloys have a two-phase face-centered cubic (fcc)/L12 structure, with the L12 (Ir3Zr) precipitates coherently embedded in the fcc Ir matrix, and such an alloy with fcc/L12 interfaces has been found to have higher strength than the single fcc or L12 intermetallic [4,5,6,7,8]. Moreover, it has been demonstrated that, for a given alloy, some unexpected stoichiometric intermetallics would appear in its microstructure during different heating and cooling processes, which would have a great effect on the properties of the alloys [9,10,11]. For instance, it has been reported that the phase transformation of IrZr significantly contributes to the great high-temperature shape memory effect for Ir-Zr alloys [10,11]. Hence, an insight into the intrinsic properties of the intermetallics, such as their mechanical properties, could be of great practical significance for the design of composite materials.
The Ir-Zr binary system has been reviewed by Okamoto [12], where the phase diagram was based primarily on the high-temperature X-ray and thermal analyses conducted by Eremenko et al. [13]. Six known stoichiometric intermetallics—Ir3Zr, Ir2Zr, IrZr, Ir3Zr5, IrZr2, and IrZr3—have been determined, in which Ir3Zr and IrZr melted congruently, and the others were formed by peritectic or peritectoid reactions. The follow-up studies on their structures, mechanical, thermodynamic, and dislocation properties have evoked significant interest in their potential applications. It is known that crystal structures are the key for understanding the mechanical properties of materials [14,15,16,17,18]. Thus, the crystal structures of these six intermetallic compounds have been extensively studied. Except for IrZr and Ir2Zr, consensus has been reached that the intermetallic phases Ir3Zr, Ir3Zr5, IrZr2, and IrZr3 adopt cubic Cu3Au-type (, Z = 1), hexagonal Mn5Si3-type (P63/mcm, Z = 2), tetragonal Al2Cu-type (I4/mcm, Z = 4), and tetragonal V3S-type (, Z = 8) structures, respectively. For IrZr, different room-temperature structures, including TiNi-type, FeB-type, and CrB-type, have been proposed, while a new orthorhombic Cmcm structure was experimentally characterized to be the most stable phase for IrZr [15], and was confirmed by the theoretical work in [10,16,18]. However, the crystal structure of Ir2Zr is the subject of continuing debate. Ir2Zr was proposed to have the C15 (Cu2Mg-type) structure [13], while recently, it was theoretically suggested to be unstable through formation enthalpy calculations [18]. Compared to IrZr, experimental and theoretical investigation of the crystal structures of Ir2Zr have rarely been undertaken, and there is a lack of confirmed reports on the existence of ground-state structure. Therefore, the peculiarity and the absence of characterized stable structures of Ir2Zr prompted our endeavor to investigate its structural stability under ambient conditions. Furthermore, the explorations of ground-state structures and related mechanical properties would provide more insights on other Ir-based intermetallic compounds. In order to address these points, we here performed extensive structure searches to explore the potential energetically stable Ir2Zr phase at ambient pressure using the newly developed Crystal structure AnaLYsis by Particle Swarm Optimization package (CALYPSO) [19,20], unbiased by any known information. This method has been successfully applied to extensive structures that have been confirmed by independent experiments [21,22,23]. Indeed, an orthorhombic Cmmm structure is uncovered to be the best ground-state candidate for Ir2Zr, and the crystal structures, mechanical behaviors, and electronic structures of this new phase were then fully investigated in comparison with the proposed Cu2Mg-type phase.
2. Computational Methods
The crystal structure searches for Ir2Zr were performed based on a global minimization of energy surfaces merging first-principle total-energy calculations as implemented in CALYPSO code [19,20], which was designed to predict stable or metastable crystal structures requiring only chemical compositions of a given compound at given external conditions (e.g., pressure). Here, using the CALYPSO code in combination with Vienna ab initio simulation package (VASP) [24], variable cell structure searches for Ir2Zr containing 1–6 formula units (f.u.) in the simulation cell were systematically performed at ambient pressure. During the structure searches, the 60% of the structures of each generation with the lowest enthalpies were selected to generate the structures for the next generation by Particle Swarm Optimization (PSO) operation, and the other structures in new generation were randomly generated to increase the structural diversity. The following local structural relaxations and electronic calculations were performed using the VASP code, in which the generalized-gradient approximation proposed by Perdew-Burke-Ernzerhof exchange-correlation functional [25,26] was used for the full optimization of all crystal structures. The electron and core interactions were included by using the frozen-core all-electron projector augmented wave potential of the metal atoms including d electrons as valence states [27]. The cutoff energy of 600 eV for the plane-wave expansions and dense k-point with grid density of 0.03 × 2π Å−1 (Monkhorst-Pack scheme) [28] were used in the Brillouin zone integration. The total energy and stress calculations were performed by using the tetrahedron method with Blöch corrections and Gaussian smearing method, respectively. The structural relaxation was performed using the conjugate gradient method until total energy is converged to within 10−5 eV and the force on each atom is less than 0.01 eV∙Å−1. The phonon spectra of the Cmmm structure was calculated by the finite displacement method, which is based on first-principles calculations of total energy, Hellman-Feynman forces, and the dynamical matrix as implemented in the PHONOPY package [29]. The independent single crystal elastic constants were determined from evaluation of stress tensor generated small strain (stress-strain approach) [30], and the polycrystalline elastic moduli including bulk modulus, shear modulus and Young’s modulus, as well as Poisson’s ratio, were thus estimated by the Voigt-Reuss-Hill approximation [31].
3. Results and Discussion
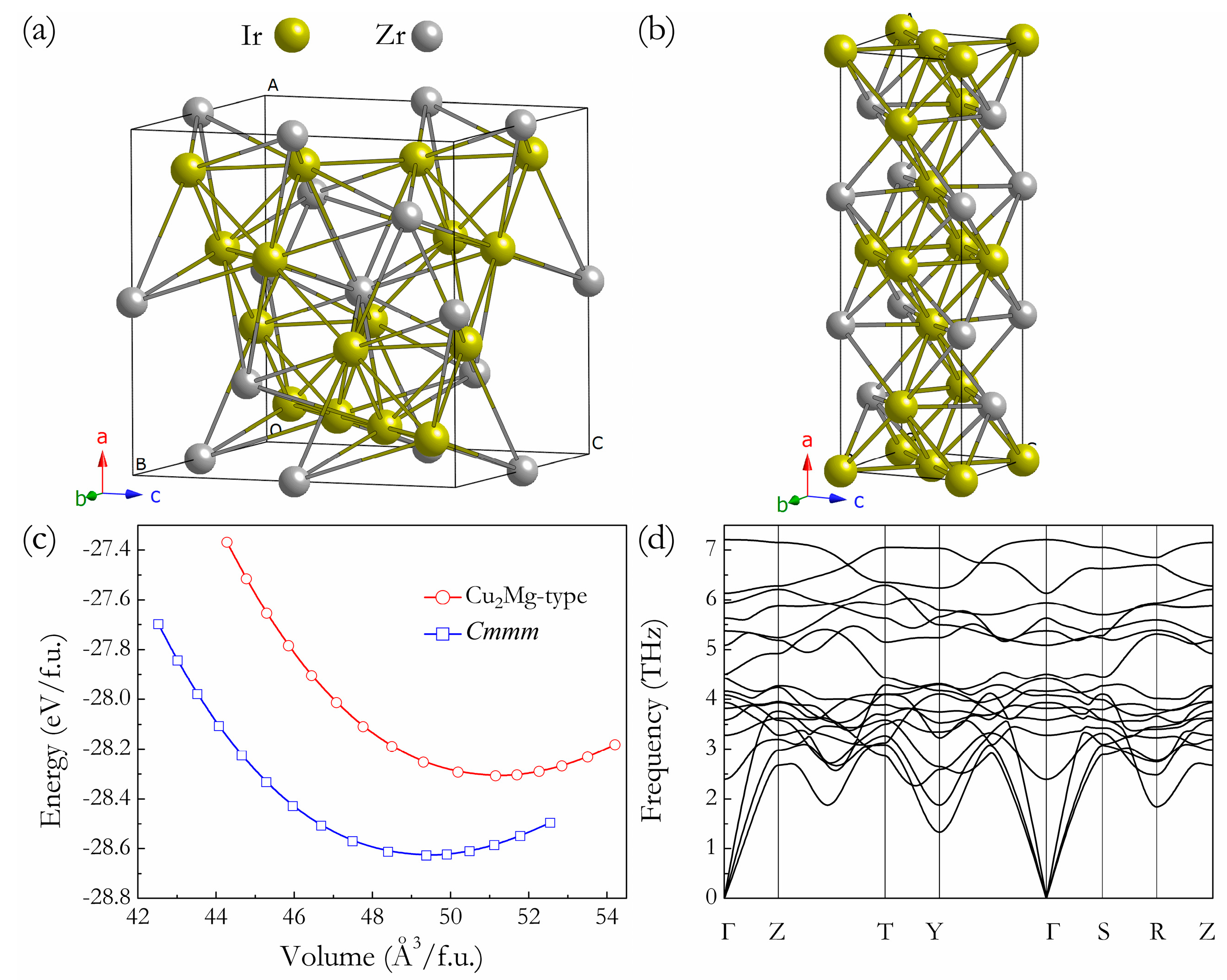
Under ambient conditions, the structure searches with the only input being the chemical composition of Ir:Zr = 2:1 predicted the most stable structure to be the orthorhombic Cmmm structure containing four f.u. per unit cell, as presented in Figure 1b, together with the previous experimental Cu2Mg-type phase Figure 1a. Compared to the building block (ZrIr10) in the Cu2Mg-type phase, each Zr atom is surrounded by twelve Ir atoms in the Cmmm structure, resulting in a different polyhedron building block. By the full relaxations of both lattice constants and internal atomic coordinations, the equilibrium structural parameters of Cmmm structure are calculated to be a = 12.477 Å, b = 4.012 Å, and c = 3.946 Å, with four inequivalent atoms Zr, Ir1, Ir2, and Ir3 occupying 4g (0.653, 0, 0), 2a (0, 0, 0), 2c (0.5, 0, 0.5), and 4g (0.836, 0, 0.5) positions, respectively. Figure 1c presents the dependence of the total energy on the f.u. volume for Cmmm and Cu2Mg-type phases; it can be clearly seen that, for Ir2Zr, the predicted Cmmm structure is energetically far more stable than Cu2Mg-type structure, and this further confirms our structural prediction. Moreover, by fitting the third-order Birch-Murnaghan equation of state (EOS) [32] based on the calculated E-V data (Figure 1c), the zero-pressure bulk modulus (B0) and its pressure derivatives (B0′) of Cmmm phase is determined to be 235 GPa and 4.691, respectively. The dynamical stability of a crystalline structure requires the eigen frequencies of its lattice vibrations be real for all wave vectors in the whole Brillouin zone. As shown in Figure 1d, no imaginary phonon frequency was detected in the whole Brillouin zone for the predicted Cmmm phase, indicating its dynamical stability at ambient pressure.
The formation enthalpy vs. composition plot, called a convex hull, is the set of lines connecting the lowest energy structures, and any structure whose formation enthalpy lies on the convex hull is deemed stable and synthesizable in principle [33]. In the Ir-Zr system, the formation enthalpy of each IrxZry intermetallic with respect to the fcc-Ir and α-Zr separate phases is quantified by the formula. Based on the calculated formation enthalpy for each IrxZry intermetallic compound, we reconstructed the convex hull line for the Ir-Zr system, as shown in Figure 2. One can see that the formation enthalpy of Ir-Zr intermetallic decreases when x < 0.5, while the formation enthalpy increases when x > 0.5. The Cmcm-IrZr is the most stable intermetallic compound among all the studied intermetallic compounds, which is in excellent agreement with the previous works [18]. The predicted Cmmm structure for Ir2Zr matches evidently with the convex hull curve between the experimental L12-Ir3Zr and Cmcm-IrZr phases, indicating the possible synthesis of this composition in the real experiment. However, the previously proposed Cu2Mg-type phase for Ir2Zr is located above the convex hull line and possesses a higher formation enthalpy, at 107 meV/atom, than that of the Cmmm phase, suggesting that the Cu2Mg-type phase for Ir2Zr is indeed metastable. In addition, we suppose that the synthetic conditions of Cmmm-Ir2Zr may be similar to those observed for the formation of Ir3Zr5 for their similar values for formation enthalpies presented in the convex hull line.
As a new intermetallic phase in the Ir-Zr system, the mechanical properties of Cmmm-Ir2Zr are important for high-temperature applications. Table 1 and Table 2 present the calculated results on the mechanical parameters of Cmmm-Ir2Zr and Cu2Mg-Ir2Zr, including the single crystal elastic constants (Cij), polycrystalline elastic modulus, and hardness, which are determined from the calculated Cij by applying a set of given strains. Under the stress-strain approach [30], for a given set of strains ε = (ε1, ε2, ε3, ε4, ε5, ε6) (where ε1, ε2, and ε3 are the normal strains and others are the shear strains) imposed on a crystal, correspondingly, one set of stresses σ = (σ1, σ2, σ3, σ4, σ5, σ6) can be determined on the deformed lattice in terms of first-principles calculations; herein, the elastic stiffness constant matrix C links ε and σ by σ = ε C. Based on the obtained elastic constants, bulk modulus B and shear modulus G are evaluated by using the Voigt-Reuss-Hill approximation [31], and the Young’s modulus E and Poisson’s ratio v are derived from the equations of E = 9BG = (3B + G) and v = (3B − 2G) = (6B + 2G), respectively. The empirical model of Hv = 2(k2G)0.585 − 3 (k = G/B) proposed by Chen et al. [34] employed here to estimate the hardness of Ir2Zr. As listed in Table 1, the three independent Cij calculated for the cubic Cu2Mg-Ir2Zr are in excellent agreement with the previous theoretical results [18], confirming the reliability of the present results and the accuracy of the elastic constant calculations. The mechanical stability of the predicted Cmmm phase satisfies the Born-Huang criterion for an orthorhombic crystal [35]: (C11 > 0, C44 > 0, C55 > 0, C66 > 0, C11C22 > C122, C11C22C33 + 2C12C13C23 − C11C232 − C22C132 − C33C122 > 0), thus suggesting that the Cmmm phase is mechanically stable under ambient conditions. In addition, the calculated results for the Cmmm phase showed that the Cij possess the trend C11 ≈ C22 ≈ C33, suggesting that it is nearly the same uniaxial compression resistance along the three main crystal directions. Polycrystalline elastic modulus, another important parameter, also contains information regarding the hardness of a material with respect to various types of deformation. Bulk modulus B measures the resistance of a material to volume change and provides an estimate of its response to a hydrostatic pressure, shear modulus G describes the resistance of a material to shape change, and Young’s modulus E measures the resistance against uniaxial tension. From Table 2, firstly, it should be noted that the derived Hill bulk modulus B of Cmmm phase (239 GPa) agrees well with that obtained directly from the fitting of the Birch-Murnaghan EOS (B0 = 235 GPa), which further demonstrates the accuracy of our elastic constant calculations. Secondly, the calculated values of bulk modulus B, shear modulus G, Young’s modulus E, and hardness for Cmmm phase are close to those of Cu2Mg-type phase, indicating their similar polycrystalline mechanical behaviors. Thirdly, according to the Pugh criterion [36], the intrinsically brittle nature of both Cmmm and Cu2Mg-type structures for Ir2Zr can be revealed by its B/G ratio of 2.038 and 1.920, which is larger than the critical value of 1.75.
The elastic anisotropic property, which is strongly related to the mechanical strength of solid materials, has important implications in engineering applications, such as microcracks, anisotropic plastic deformation, elastic durability, etc. Compared to the reported results of other known intermetallic compounds in the Ir-Zr system, the studies on the elastic anisotropic behaviors of this new Cmmm phase are of great importance for its technical applications in high-temperature environments. As outlined by Panda et al. [37] and He et al. [38], executing the appropriate coordinate system transformations for the compliances allows the determination of the variation of bulk modulus B, Young’s moduli E, and shear modulus G with crystallographic direction, [uvw], for a given crystallographic plane, (hkl), containing these directions, (i.e., B[uvw], E[uvw], and G(hkl)[uvw]). For orthorhombic Cmmm phase, the bulk modulus B and Young’s modulus E can be expressed as:
where α, β, and γ are the direction cosines of [uvw] direction, and s11, s22, etc. are the elastic compliance constants given by Ney [39]. The shear modulus G on the (hkl) shear plane with shear stress applied along [uvw] direction is given by:
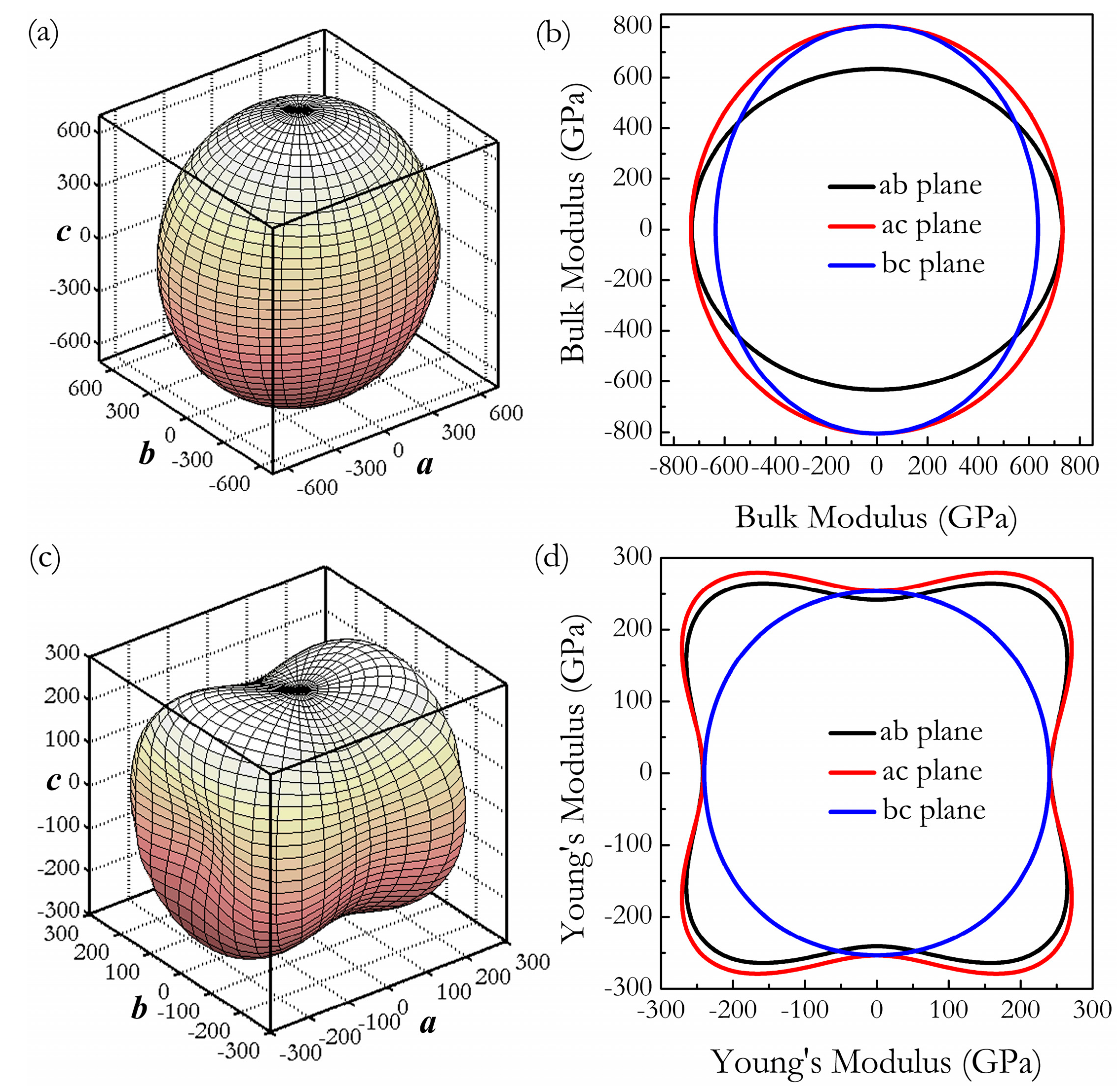
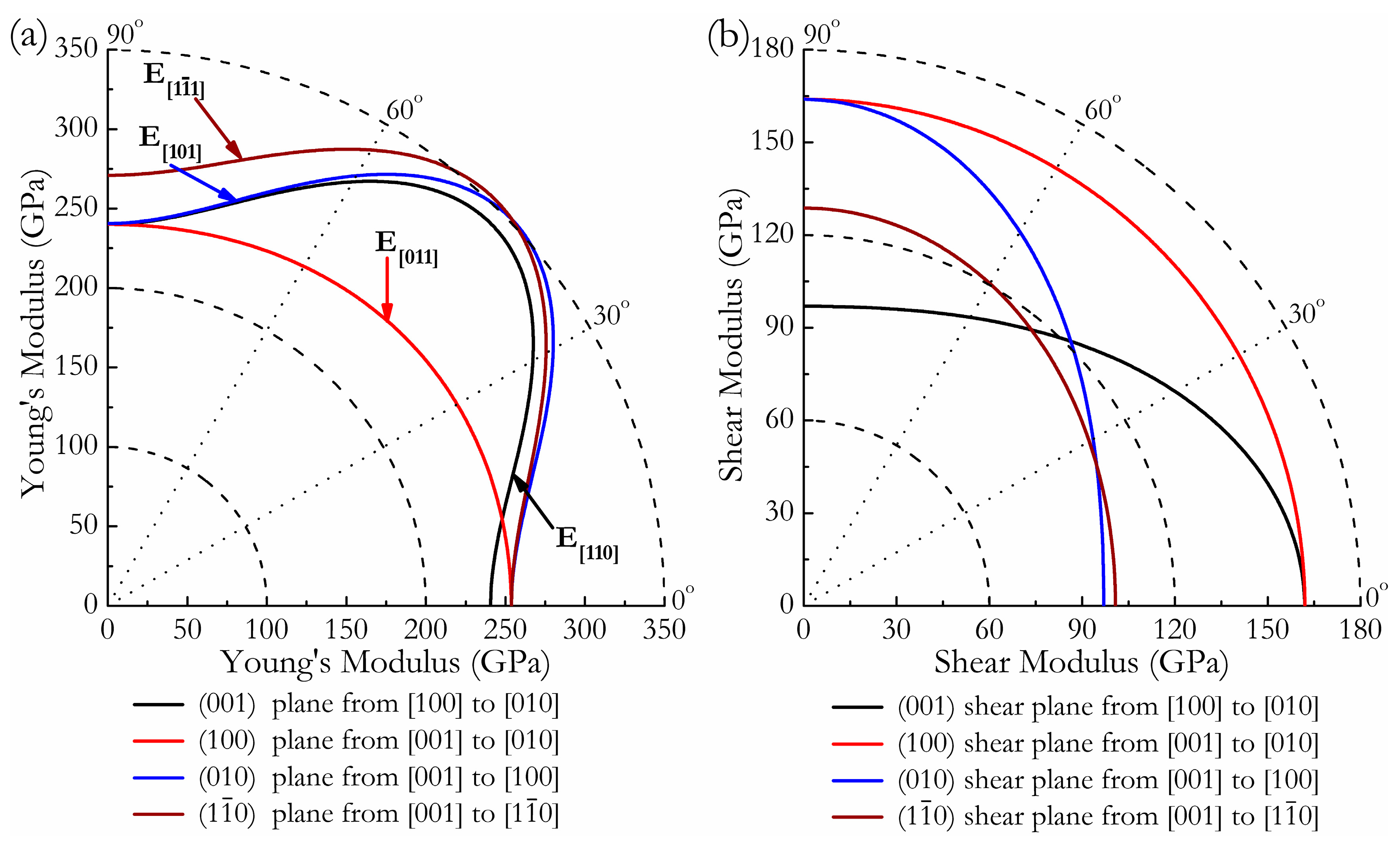
where α1, β1, γ1, α2, β2, γ2 are the direction cosines of the [uvw] and [HKL] directions in the coordinate systems, where the [HKL] denotes the vector normal to the (hkl) shear plane. The three-dimensional (3D) surface representations showing the variation of the bulk modulus B and Young’s modulus E are plotted in Figure 3a,c, and the distance from the origin of the system of coordinates to this surface is equal to the B or E in a given direction. The plane projections (ab, ac, and bc planes) of the directional dependences of the bulk modulus B and Young’s modulus E are given in Figure 3b,d for comparison. It can be seen that Cmmm-Ir2Zr exhibits a highly pronounced elastic anisotropy, as its 3D picture shows a large deviation from the spherical shape, which qualifies an isotropic medium. From Figure 3b,d, the distributions of bulk modulus B and Young’s modulus E within the crystal plane bc display the largest and the smallest elastic anisotropy behaviors, respectively. In more detail, the changes of Young’s moduli along different crystal directions within four specific planes (001), (100), (010), and are presented in Figure 4a. For example, the variation of Young’s modulus E in the (100) plane for the quadrant of directions [uvw] between [001] (θ = 0°) and [010] (θ = 90°), the Cmmm phase displays the smallest elastic anisotropy behavior (as shown in Figure 3d) with a maximum of 254 GPa and a minimum of 240 GPa. The selected directional Young’s moduli along the five principal crystallographic directions are denoted as in Figure 4a, and it can be seen that the calculated values for E decrease in the following order: E[100] ≈ E[010] < E[011] < E[001] < E[110] < E[110] < E[101] < < . Similarly, Figure 4b presents the orientation dependence of the shear modulus along arbitrary shear directions within these four shear planes. Compared to the other three shear planes, the calculations show that the shear modulus for a shear deformation within the (100) plane is not only nearly isotropic [Gmax = G[010] = 164 GPa and Gmin = G[001] = 162 GPa] but also has greater resistance to shear deformations.
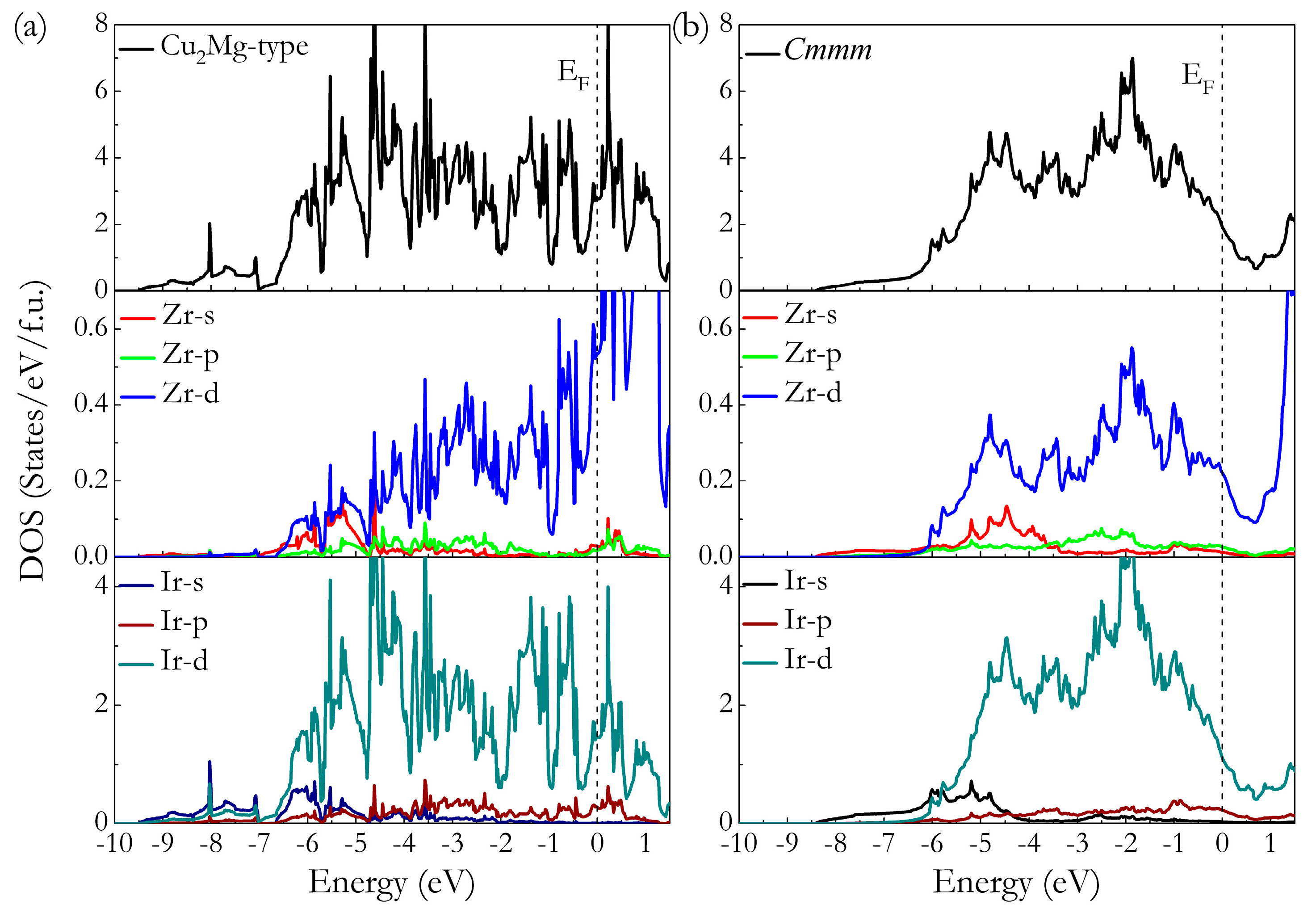
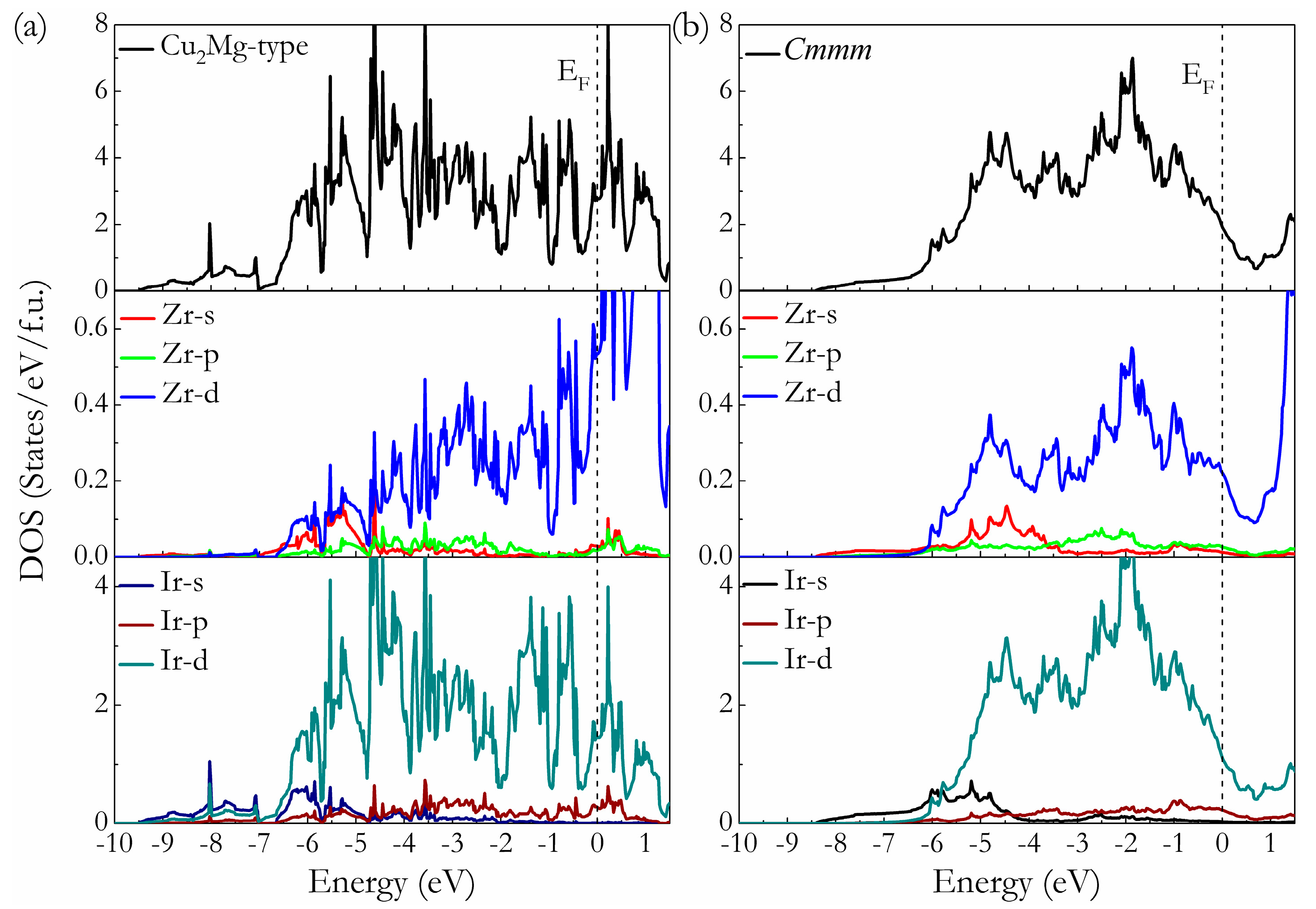
To understand the bonding mechanism of intermetallic Ir2Zr on a fundamental level, the total and partial density of state (t-DOS and p-DOS) of Cu2Mg-type and Cmmm phases at ambient pressure are calculated and presented in Figure 5, where the vertical dashed line is the Fermi level (EF). It is clear that both structures are metal for the finite values of t-DOS at the EF. The typical feature of the t-DOS for these two compounds is the presence of so-called “pseudogap” (a sharp valley around the EF), a borderline between the bonding and antibonding orbital [40]. It has been suggested that the location of EF in the DOS profiles can reflect the structural stability of the compounds. For the Cu2Mg-type phase, the EF lies to the right of the pseudogap (i.e., within the antibonding states), suggesting their metastable nature, while for Cmmm phase, the EF locates at the left of the pseudogap (i.e., within the bonding states) with relative lower electronic density of state N(EF) values, revealing its structural stability. These findings support the previous results acquired from the formation enthalpy calculations. The feature of p-DOSs for both phases indicates that there is strong interaction between Ir and Zr atoms through d-d hybridization, which can be seen from the hybridization peaks of Ir-d and Zr-d orbitals plotted in Figure 5a,b, signifying the existence of directional covalent-like bonding in Ir2Zr intermetallic compound. A careful comparison further reveals that the p-DOS of the Ir and Zr atoms in Cmmm-Ir2Zr are more localized than those in Cu2Mg-Ir2Zr, resulting a small bandwidth of p-DOS and a lower N(EF). It is known that for the most stable structure, there is enough room to accommodate all its valence electrons into bonding states, so as to bring the EF to a valley position separating bonding and antibonding states (pseudogap) favorable for structural stability. Therefore, the formation of Cmmm intermetallic phase is energetically more favorable than the previously proposed Cu2Mg-type for Ir2Zr.
4. Conclusions
In summary, we have extensively explored the ground-state structures of stoichiometric Ir2Zr intermetallic compound by using the recently developed particle swarm optimization algorithm in crystal structure prediction. A new orthorhombic Cmmm structure was proposed as the best candidate at ground state that is energetically more favorable than the previous experimental Cu2Mg-type structure, as demonstrated by the total energy and formation enthalpy calculations. Then, the mechanical and electronic properties of this new predicted Cmmm phase were fully investigated in comparisons with Cu2Mg-type phase. The predicted Cmmm phase exhibits nearly elastic isotropic behavior and great resistance to shear deformations within (100) crystal plane according to the directional elastic moduli calculations. Evidence of atomic bonding related to the structural stability for Ir2Zr was obtained by calculation of the electronic structures.
Acknowledgments
This work was supported by the National Undergraduate Innovative Training Program (No. 201610721004), the Natural Science New Star of Science and Technologies Research Plan in Shaanxi Province of China (Grant No. 2017KJXX-53), the Natural Science Basic Research plan in Shaanxi Province of China (Grant No. 2016JM1016), and the Baoji University of Arts and Sciences Key Research (Grant No. ZK16068). The all authors thank the computing facilities at High Performance Computing Center of Baoji University of Arts and Sciences.
Author Contributions
Meiguang Zhang and Ke Cheng conceived and designed the projects; Meiguang Zhang, Rui Cao, Meijie Zhao and Juan Du performed the calculations; Meiguang Zhang Rui Cao, Meijie Du and Juan Du prepared the manuscript; Ke Cheng revised the paper; all authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kuznetsova, T.; Zubar, T.; Chizhik, S.; Gilewicz, A.; Lupicka, O.; Warcholinski, B. Surface microstructure of Mo(C)N coatings investigated by AFM. J. Mater. Eng. Perform. 2016, 25, 5450–5459. [Google Scholar] [CrossRef]
- Warcholinski, B.; Gilewicz, A.; Lupicka, O.; Kuprin, A.S.; Tolmachova, G.N.; Ovcharenko, V.D.; Kolodiy, I.V.; Sawczak, M.; Kochmanska, A.E.; Kochmanski, P.; et al. Structure of CrON coatings formed in vacuum arc plasma fluxes. Surf. Coat. Technol. 2017, 309, 920–930. [Google Scholar] [CrossRef]
- Warcholinski, B.; Gilewicz, A.; Kuznetsova, T.A.; Zubar, T.I.; Chizhik, S.A.; Abetkovskaia, S.O.; Lapitskaya, V.A. Mechanical properties of Mo(C)N coatings deposited using cathodic arc evaporation. Surf. Coat. Technol. 2017, 319, 117–128. [Google Scholar] [CrossRef]
- Yamabe-Mitarai, Y.; Ro, Y.; Maruko, T.; Harada, H. Microstructure dependence of strength of Ir-base refractory superalloys. Intermetallics 1999, 7, 49–58. [Google Scholar] [CrossRef]
- Ran, H.T.; Du, Z.M. Thermodynamic assessment of the Ir-Zr system. J. Alloys Compd. 2006, 413, 101–105. [Google Scholar] [CrossRef]
- Sha, J.B.; Yamabe-Mitarai, Y.; Harada, H. Microstructural evaluation and mechanical properties of Ir-Hf-Zr ternary alloys at room and high temperatures. Intermetallics 2006, 14, 1364–1369. [Google Scholar] [CrossRef]
- Yamabe-Mitarai, Y.; Gu, Y.; Harada, H.; Huang, C. Compressive creep properties of Ir-base refractory superalloys. Metall. Mater. Trans. A 2005, 36, 547–557. [Google Scholar] [CrossRef]
- Yamabe-Mitarai, Y.; Harada, H. Face centered cubic and L12 two-phase structure of Ir-Nb-Zr alloys. J. Alloys Compd. 2003, 361, 169–179. [Google Scholar] [CrossRef]
- Kim, W.Y.; Tanaka, H.; Kasama, A.; Hanada, S. Microstructure and room temperature fracture toughness of Nbss/Nb5Si3 in situ composites. Intermetallics 2001, 9, 827–834. [Google Scholar] [CrossRef]
- Chen, B.S.; Li, Y.Z.; Guan, X.Y.; Wang, C.; Wang, C.X.; Gao, Z.Y. First-principles study of structural, elastic and electronic properties of ZrIr alloy. Comput. Mater. Sci. 2015, 105, 66–70. [Google Scholar] [CrossRef]
- Kudryavtsev, Y.V.; Semenova, O.L. Shape memory effect in ZrIr and Zr-Ir-Co alloys. Powder Metall. Met. Ceram. 2011, 50, 471–478. [Google Scholar] [CrossRef]
- Okamoto, H. The Ir-Zr (iridium-zirconium) system. J. Phase Equilib. 1992, 13, 653–656. [Google Scholar] [CrossRef]
- Eremenko, V.N.; Semenova, E.L.; Shtepa, T.D. State diagram of the Zr-Ir system. Russ. Metall. 1980, 5, 210–213. [Google Scholar]
- Semenova, E.L.; Kudryavtsev, Y.V. Structural phase transformation and shape memory effect in ZrRh and ZrIr. J. Alloys Compd. 1994, 203, 165–168. [Google Scholar] [CrossRef]
- Stalick, J.K.; Waterstrat, R.M. The crystal structure of martensitic ZrIr and ZrRh. J. Alloys Compd. 2009, 477, 123–126. [Google Scholar] [CrossRef]
- Xing, W.; Chen, X.Q.; Li, D.Z.; Li, Y.Y.; Fu, C.L.; Meschel, S.V.; Ding, X. First-principles studies of structural stabilities and enthalpies of formation of refractory intermetallics: TM and TM3 (T = Ti, Zr, Hf; M = Ru, Rh, Pd, Os, Ir, Pt). Intermetallics 2012, 28, 16–24. [Google Scholar] [CrossRef]
- Hart, G.L.; Curtarolo, S.; Massalski, T.B.; Levy, O. Comprehensive search for new phases and compounds in binary alloy systems based on platinum-group metals, using a computational first-principles approach. Phys. Rev. X 2013, 3, 041035. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, B.; Zhan, Y. Ab initio investigation into the structure and properties of Ir-Zr intermetallics for high-temperature structural applications. Comput. Mater. Sci. 2017, 131, 146–159. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lv, J.; Zhu, L.; Ma, Y.M. Crystal structure prediction via particle swarm optimization. Phys. Rev. B 2010, 82, 094116. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lv, J.; Zhu, L.; Ma, Y.M. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef]
- Lv, J.; Wang, Y.C.; Zhu, L.; Ma, Y.M. Predicted novel high-pressure phases of lithium. Phys. Rev. Lett. 2011, 106, 015503. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, H.; Wang, Y.C.; Lv, J.; Ma, Y.M.; Cui, Q.L.; Ma, Y.M.; Zou, G.T. Substitutionally alloy of Bi and Te at high pressure. Phys. Rev. Lett. 2011, 106, 145501. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.W.; Hao, J.; Liu, H.Y.; Li, Y.L.; Ma, Y.M. The metallization and superconductivity of dense hydrogen sulfide. J. Chem. Phys. 2014, 140, 174712. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Milman, V.; Warren, M.C. Elasticity of hexagonal BeO. J. Phys. Condens. Matter 2001, 13, 241. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 1947, 71, 809. [Google Scholar] [CrossRef]
- Ghosh, G.; Van de Walle, A.; Asta, M. First-principles calculations of the structural and thermodynamic properties of bcc, fcc and hcp solid solutions in the Al-TM (TM = Ti, Zr and Hf) systems: A comparison of cluster expansion and supercell methods. Acta Mater. 2008, 56, 3202–3221. [Google Scholar] [CrossRef]
- Chen, X.Q.; Niu, H.Y.; Li, D.Z.; Li, Y.Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Born, M. On the stability of crystal lattices. Math. Proc. Camb. Philos. Soc. 1940, 36, 160–172. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Panda, K.B.; Ravi Chandran, K.S. Determination of elastic constants of titanium diboride (TiB2) from first principles using FLAPW implementation of the density functional theory. Comput. Mater. Sci. 2006, 35, 134–150. [Google Scholar] [CrossRef]
- He, Y.; Schwarz, R.B.; Migliori, A. Elastic constants of single crystal γ-TiAl. J. Mater. Res. 1995, 10, 1187–1195. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals: Their Representation by Tensors and Matrices; Oxford University Press: Oxford, UK, 1985. [Google Scholar]
- Vajeeston, P.; Ravindran, P.; Ravi, C.; Asokamani, R. Electronic structure, bonding, and ground-state properties of AlB2-type transition-metal diborides. Phys. Rev. B 2001, 63, 045115. [Google Scholar] [CrossRef]
Figure 1.
Crystal structures of Cu2Mg-Ir2Zr (a) and Cmmm-Ir2Zr (b), the large and small spheres represent Ir and Zr atoms, respectively. The total energy vs. f.u. volume for Cu2Mg-type and Cmmm structures (c) and phonon dispersion curves of Cmmm-Ir2Zr at 0 GPa (d).
Figure 1.
Crystal structures of Cu2Mg-Ir2Zr (a) and Cmmm-Ir2Zr (b), the large and small spheres represent Ir and Zr atoms, respectively. The total energy vs. f.u. volume for Cu2Mg-type and Cmmm structures (c) and phonon dispersion curves of Cmmm-Ir2Zr at 0 GPa (d).
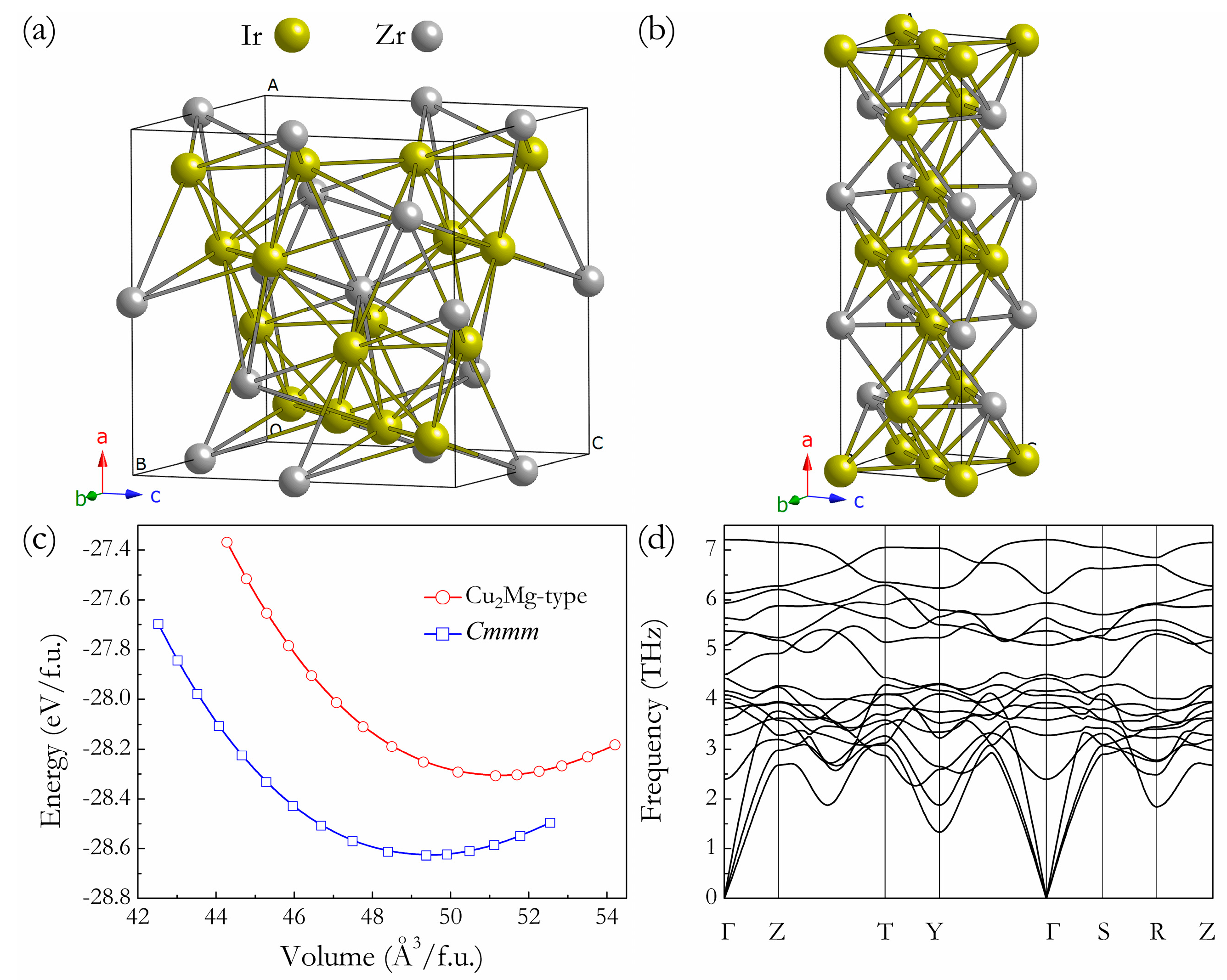
Figure 2.
The formation enthalpy vs. composition curves for stoichiometric intermetallics in Ir-Zr system with fcc-Ir and α-Zr as reference states. The solid line denotes the ground state convex hull.
Figure 2.
The formation enthalpy vs. composition curves for stoichiometric intermetallics in Ir-Zr system with fcc-Ir and α-Zr as reference states. The solid line denotes the ground state convex hull.

Figure 3.
Three-dimensional surface representations of bulk modulus B (a) and the Young’s modulus E (b) at 0 GPa, and the projections of bulk modulus B (c) and the Young’s modulus E (d) within ab, ac, and bc planes.
Figure 3.
Three-dimensional surface representations of bulk modulus B (a) and the Young’s modulus E (b) at 0 GPa, and the projections of bulk modulus B (c) and the Young’s modulus E (d) within ab, ac, and bc planes.

Figure 4.
Orientation dependences of the Young’s modulus E (a) and shear modulus G (b) for Cmmm-Ir2Zr.
Figure 4.
Orientation dependences of the Young’s modulus E (a) and shear modulus G (b) for Cmmm-Ir2Zr.
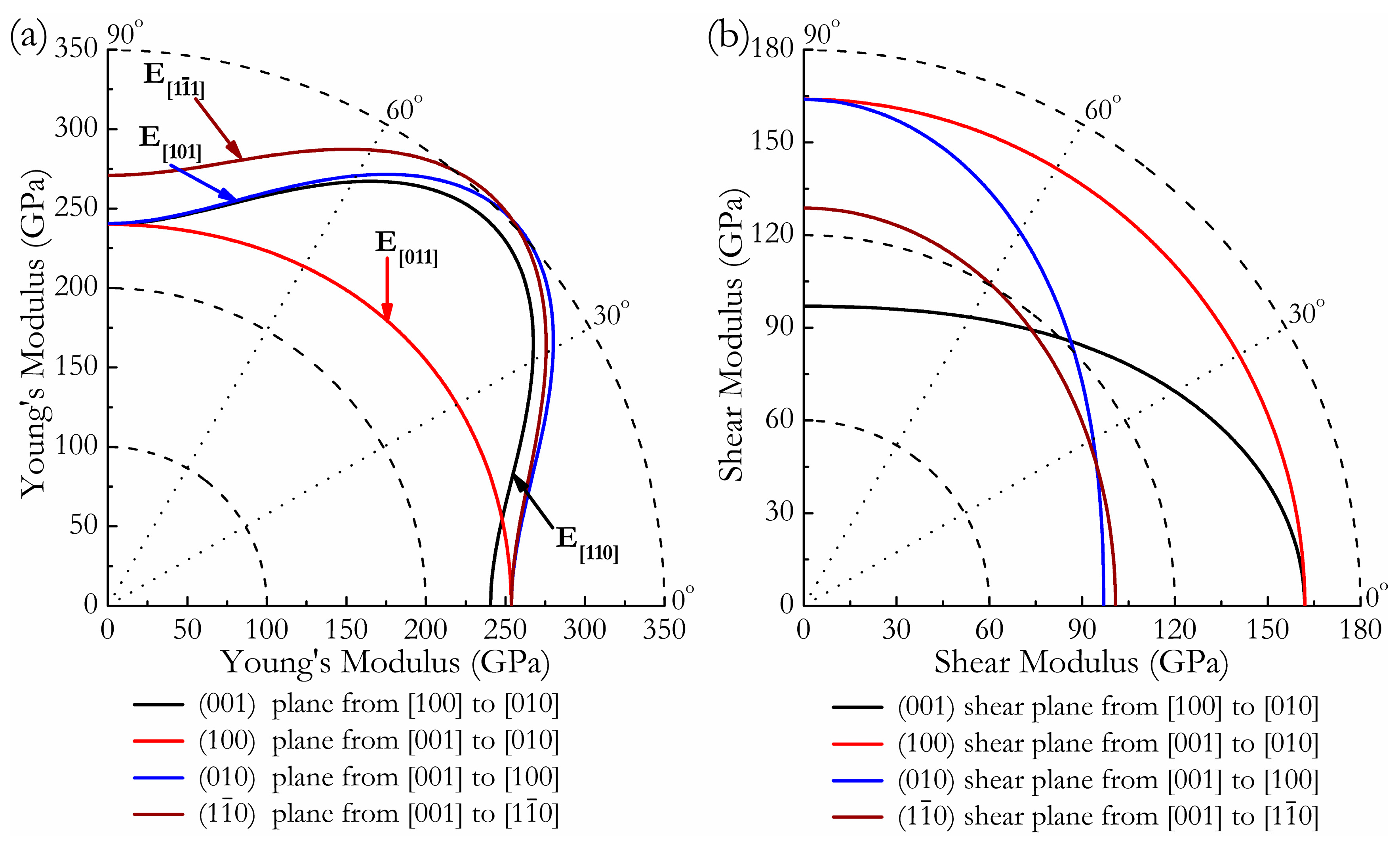
Figure 5.
Calculated total and partial DOS of Cu2Mg-Ir2Zr (a) and Cmmm-Ir2Zr (b). The vertical dash line is the EF.
Figure 5.
Calculated total and partial DOS of Cu2Mg-Ir2Zr (a) and Cmmm-Ir2Zr (b). The vertical dash line is the EF.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated single elastic constants Cij (in GPa) of Cu2Mg-type and Cmmm phases for Ir2Zr intermetallic compound.
Table 1.
Calculated single elastic constants Cij (in GPa) of Cu2Mg-type and Cmmm phases for Ir2Zr intermetallic compound.
| Ir2Zr | Source | C11 | C22 | C33 | C44 | C55 | C66 | C12 | C13 | C23 |
|---|---|---|---|---|---|---|---|---|---|---|
| Cu2Mg-type | This work | 379 | - | - | 152 | - | - | 181 | - | - |
| - | Theory [18] | 379 | - | - | 151 | - | - | 180 | - | - |
| Cmmm | This work | 362 | 348 | 378 | 97 | 162 | 164 | 173 | 188 | 174 |
Table 2.
Calculated polycrystalline bulk modulus B, shear modulus G, Young’s modulus E, and hardness H (in units of GPa) for Ir2Zr. Also shown are Poisson’s ratio v and B/G ratio.
Table 2.
Calculated polycrystalline bulk modulus B, shear modulus G, Young’s modulus E, and hardness H (in units of GPa) for Ir2Zr. Also shown are Poisson’s ratio v and B/G ratio.
| Ir2Zr | Source | B | G | E | v | B/G | H |
|---|---|---|---|---|---|---|---|
| Cu2Mg-type | This work | 248 | 129 | 323 | 0.283 | 1.972 | 18.9 |
| - | Theory [18] | 246 | 128 | 327 | 0.278 | 1.920 | 20.3 |
| Cmmm | This work | 239 | 117 | 302 | 0.289 | 2.038 | 18.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, M.; Cao, R.; Zhao, M.; Du, J.; Cheng, K. Unexpected Ground-State Structure and Mechanical Properties of Ir2Zr Intermetallic Compound. Materials 2018, 11, 103. https://doi.org/10.3390/ma11010103
AMA Style
Zhang M, Cao R, Zhao M, Du J, Cheng K. Unexpected Ground-State Structure and Mechanical Properties of Ir2Zr Intermetallic Compound. Materials. 2018; 11(1):103. https://doi.org/10.3390/ma11010103
Chicago/Turabian StyleZhang, Meiguang, Rui Cao, Meijie Zhao, Juan Du, and Ke Cheng. 2018. "Unexpected Ground-State Structure and Mechanical Properties of Ir2Zr Intermetallic Compound" Materials 11, no. 1: 103. https://doi.org/10.3390/ma11010103
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.