Exploration of the Structural, Electronic and Tunable Magnetic Properties of Cu4M (M = Sc-Ni) Clusters

Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
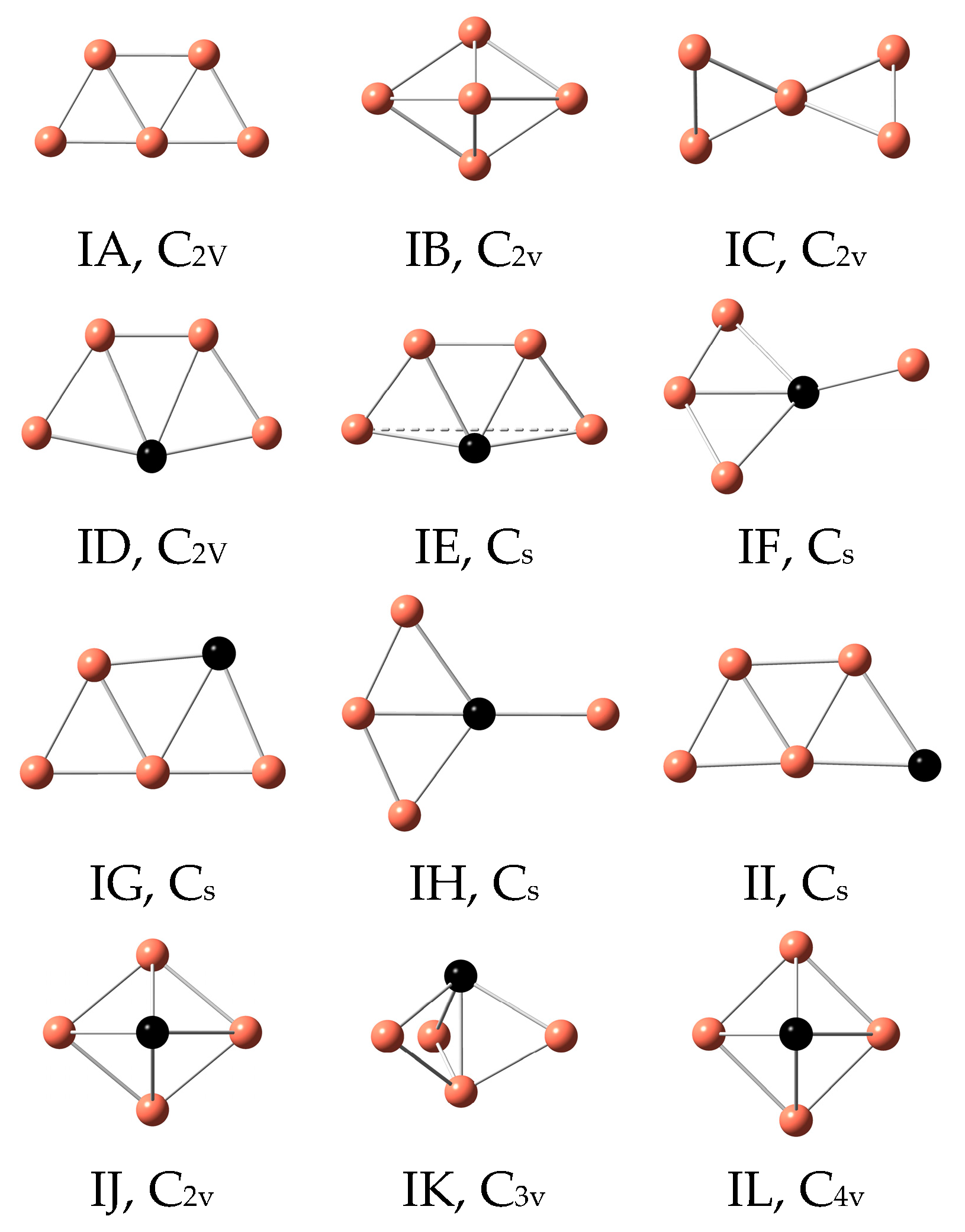
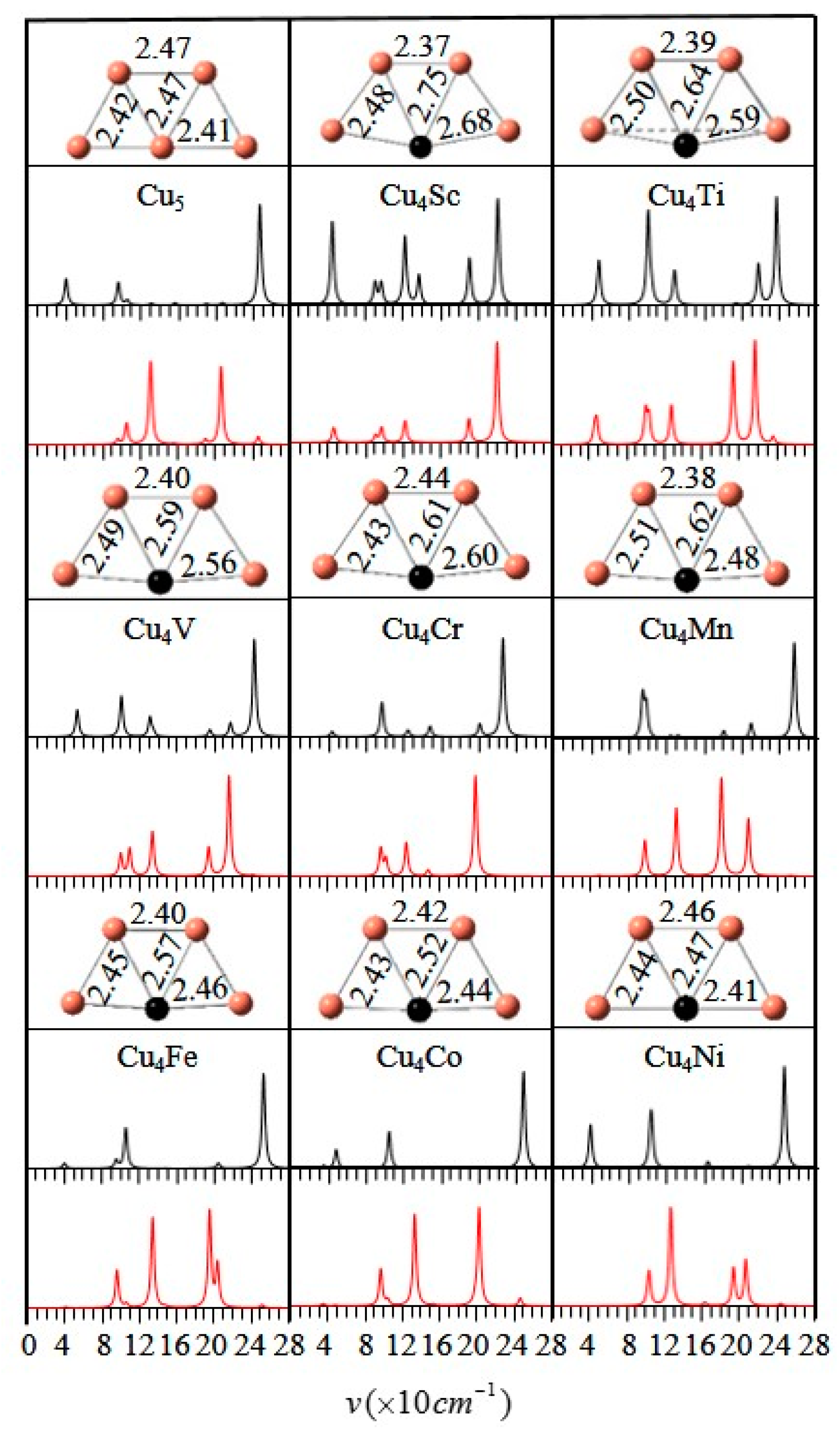
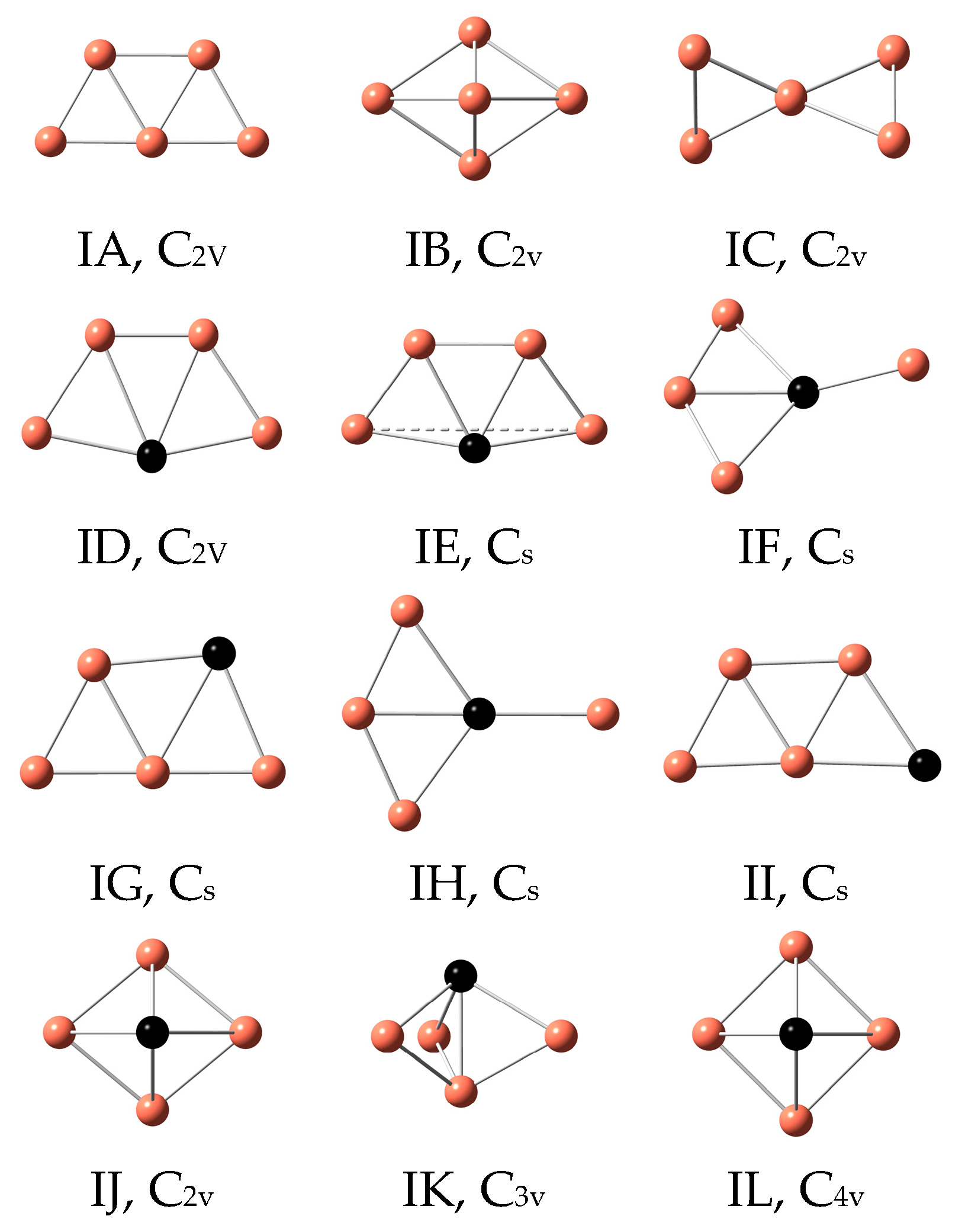
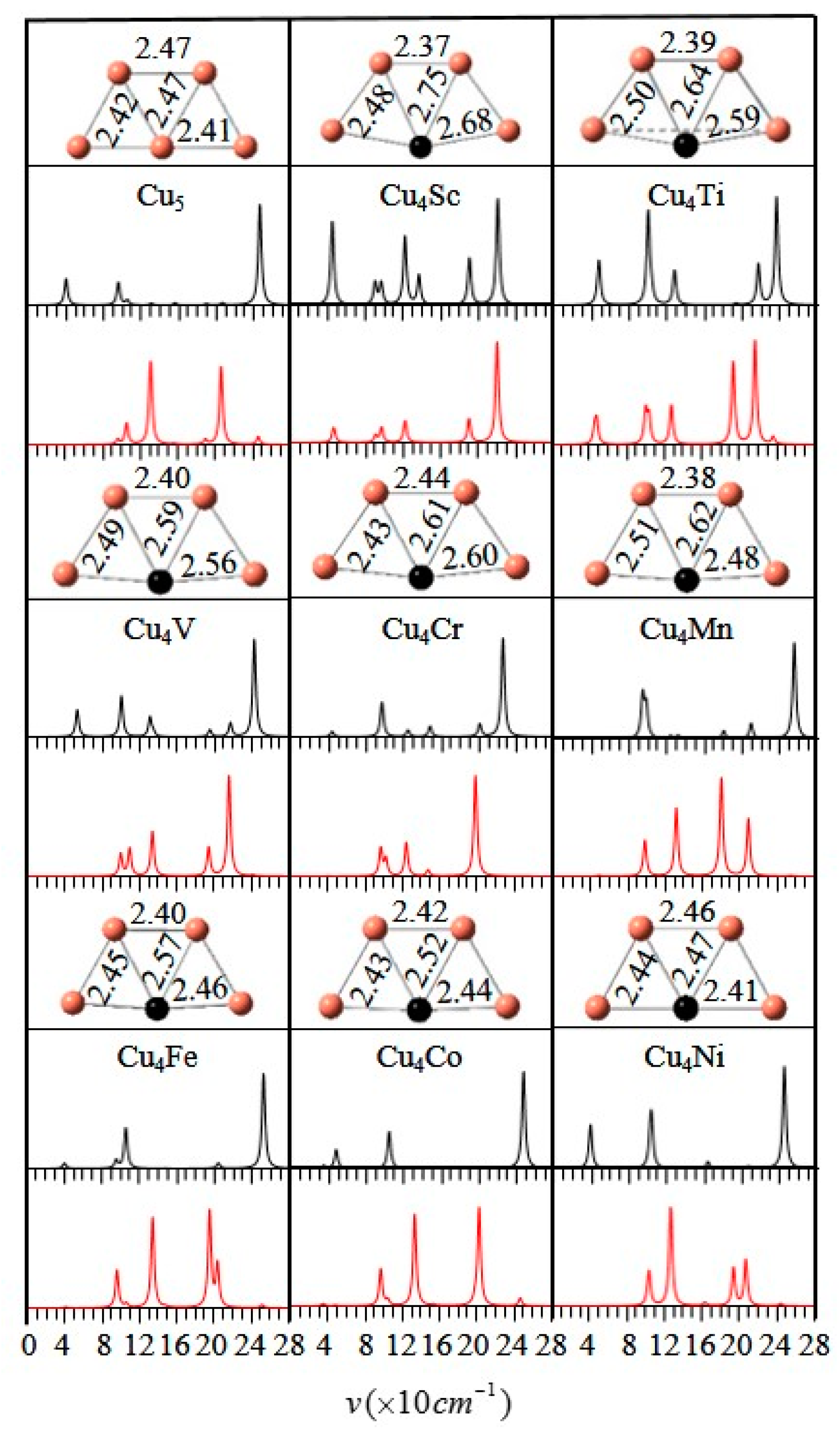
3.1. Geometrical Structures and Vibrational Spectra
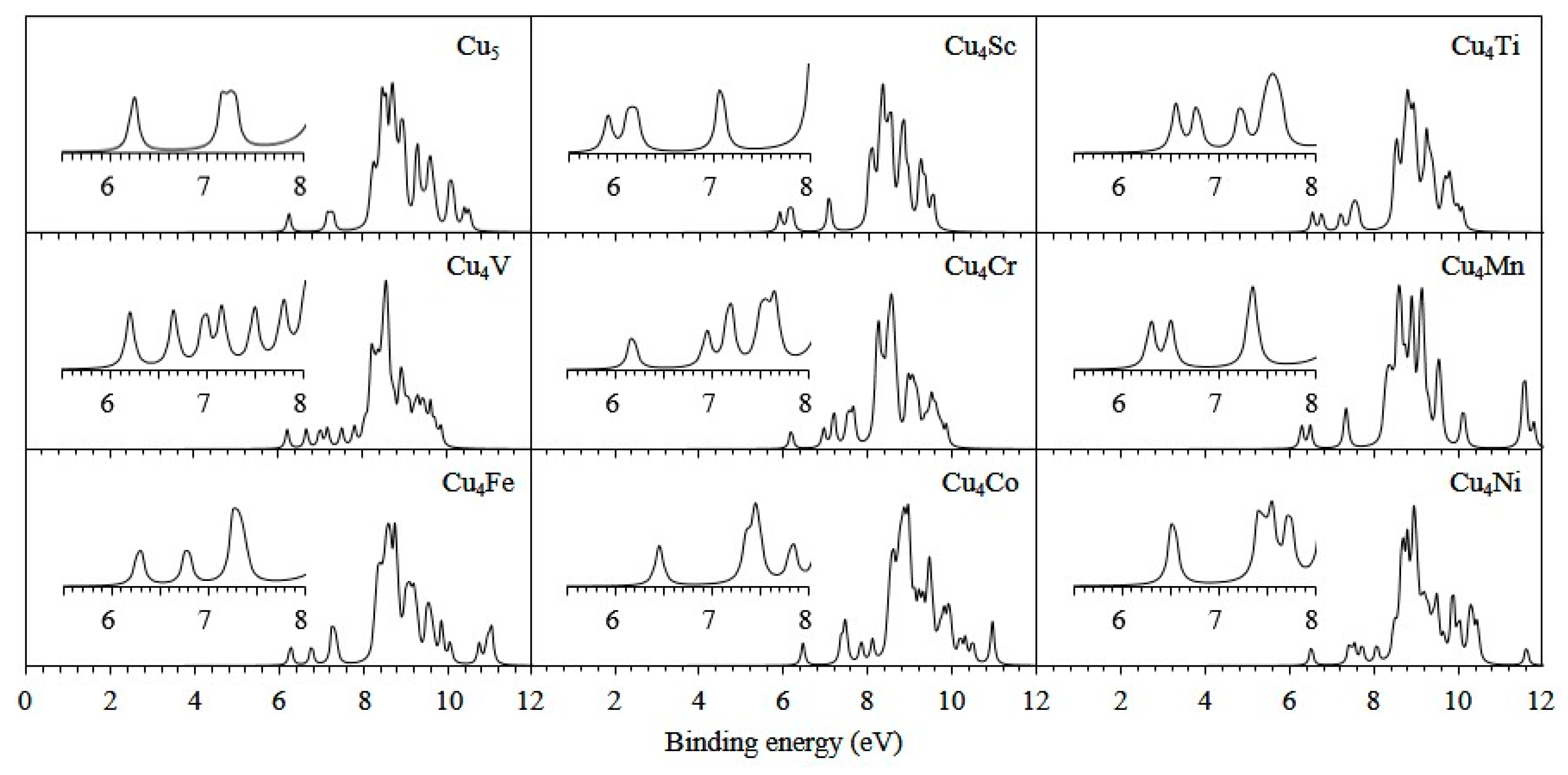
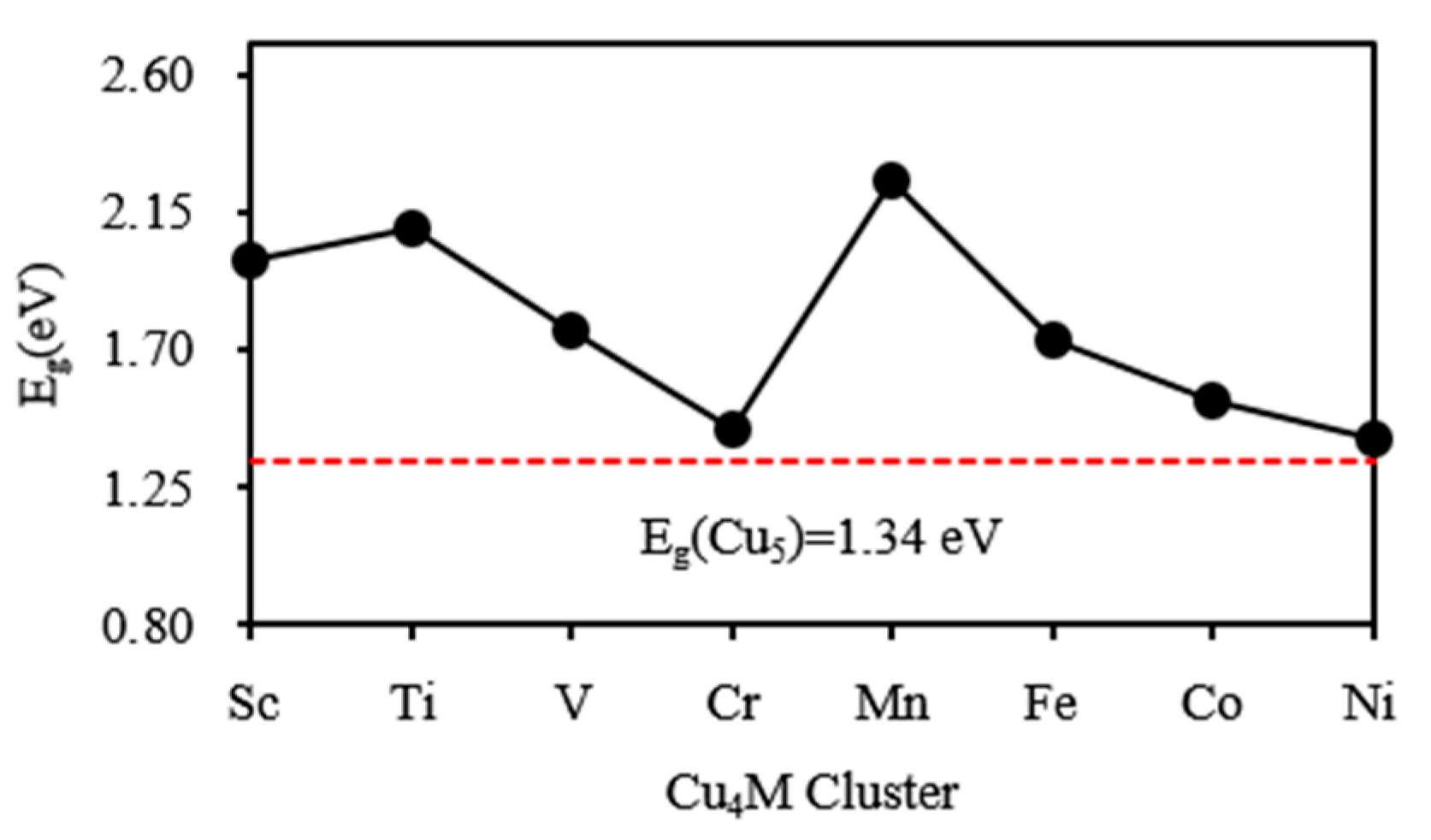
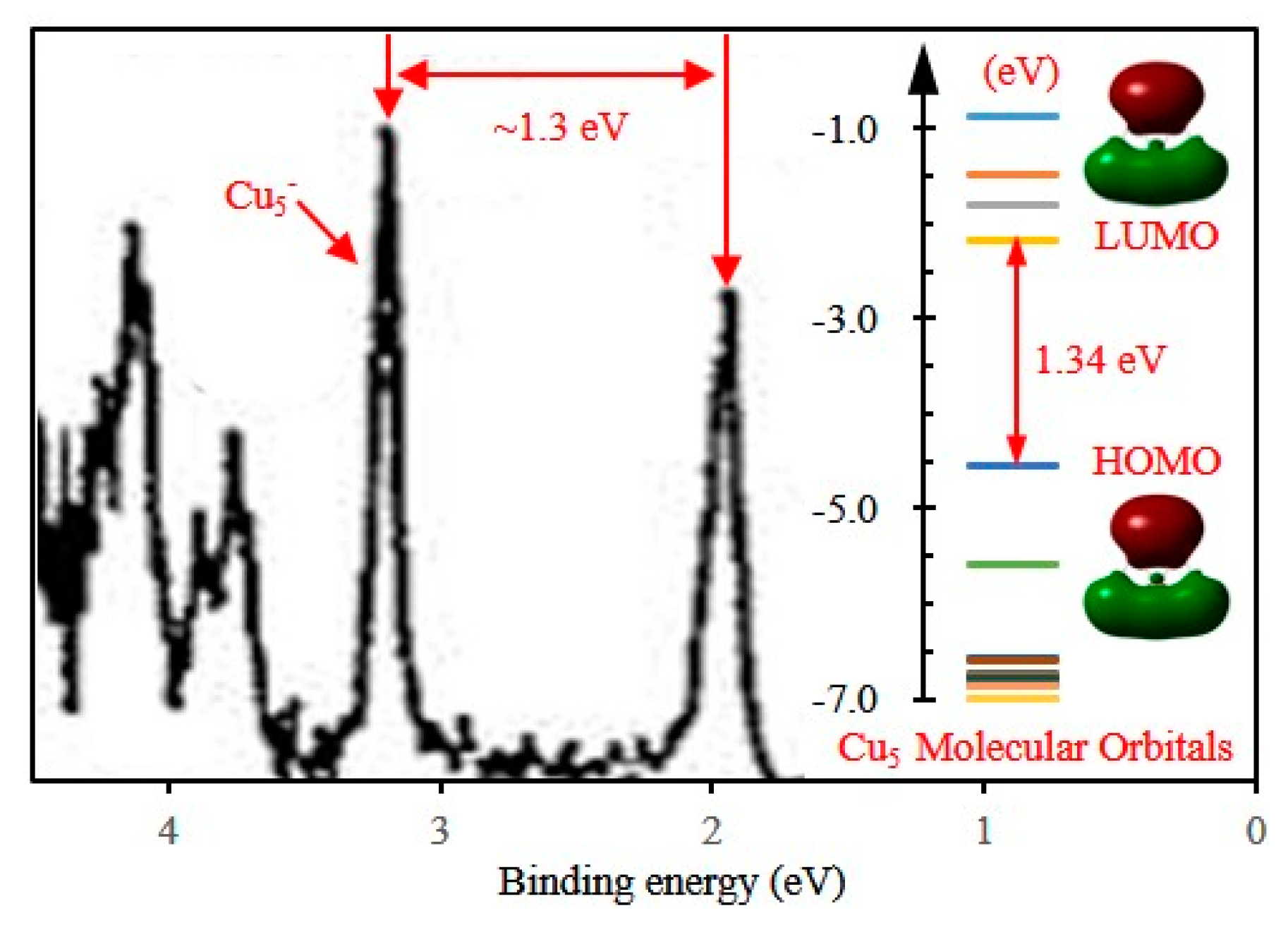
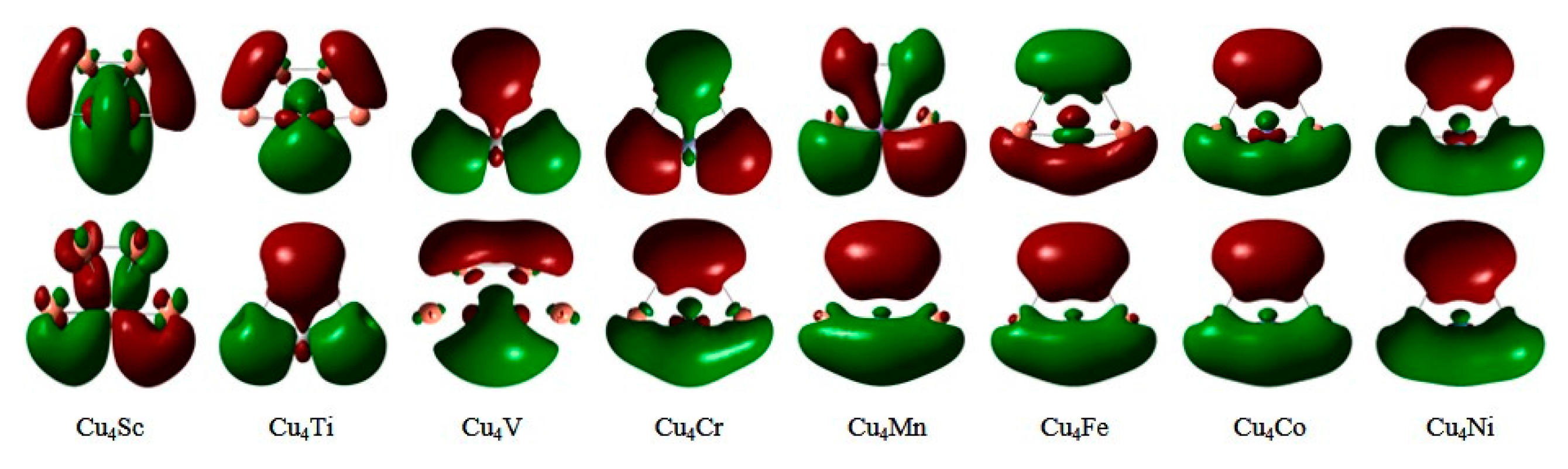
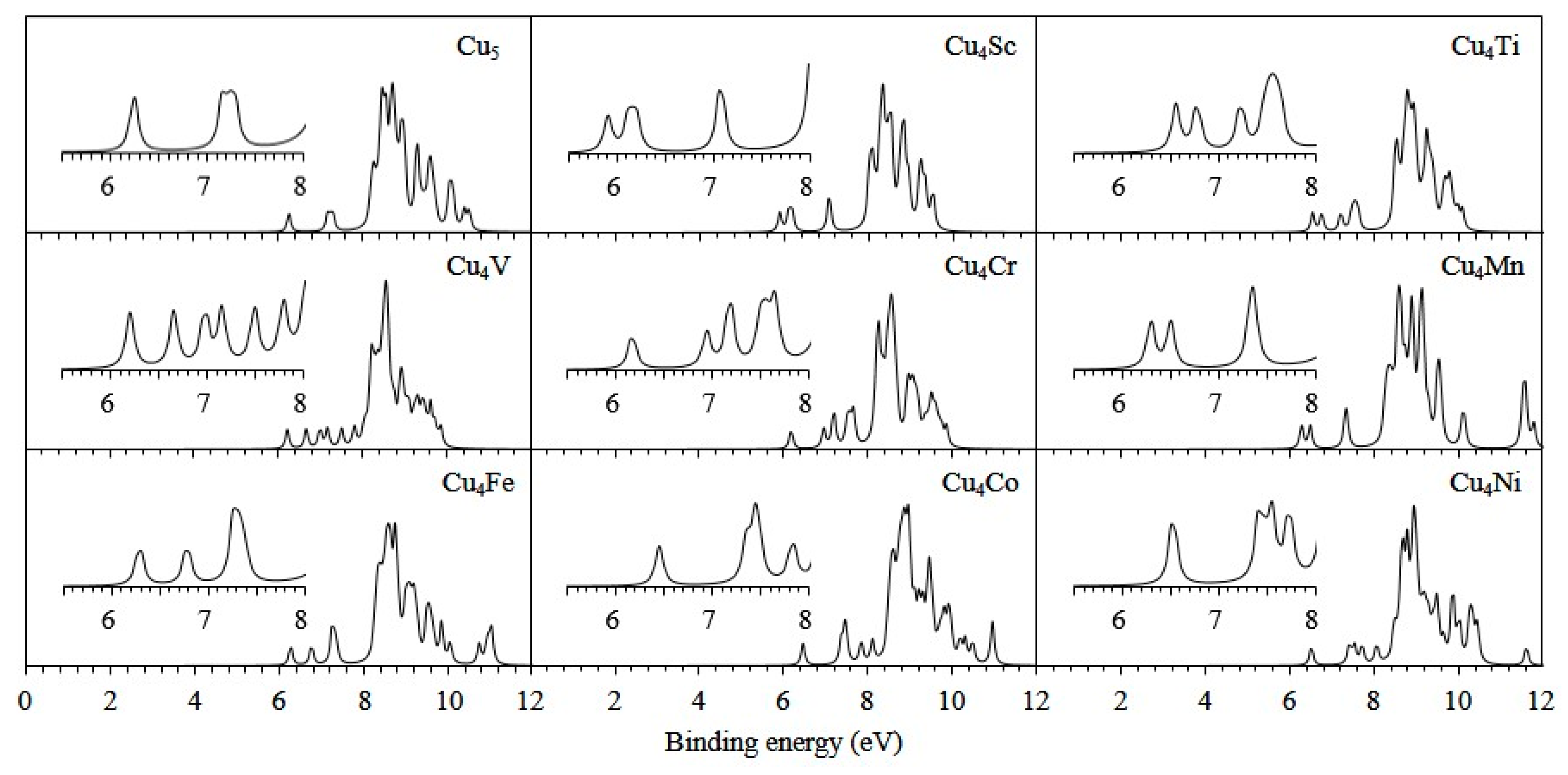
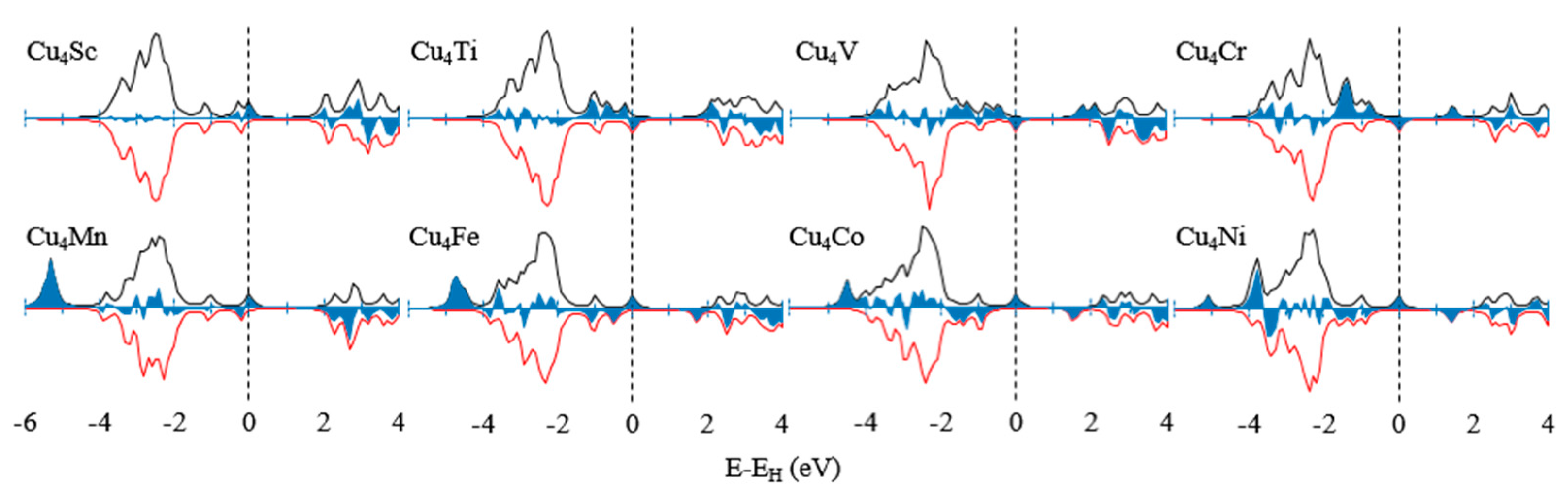
3.2. Electronic Properties
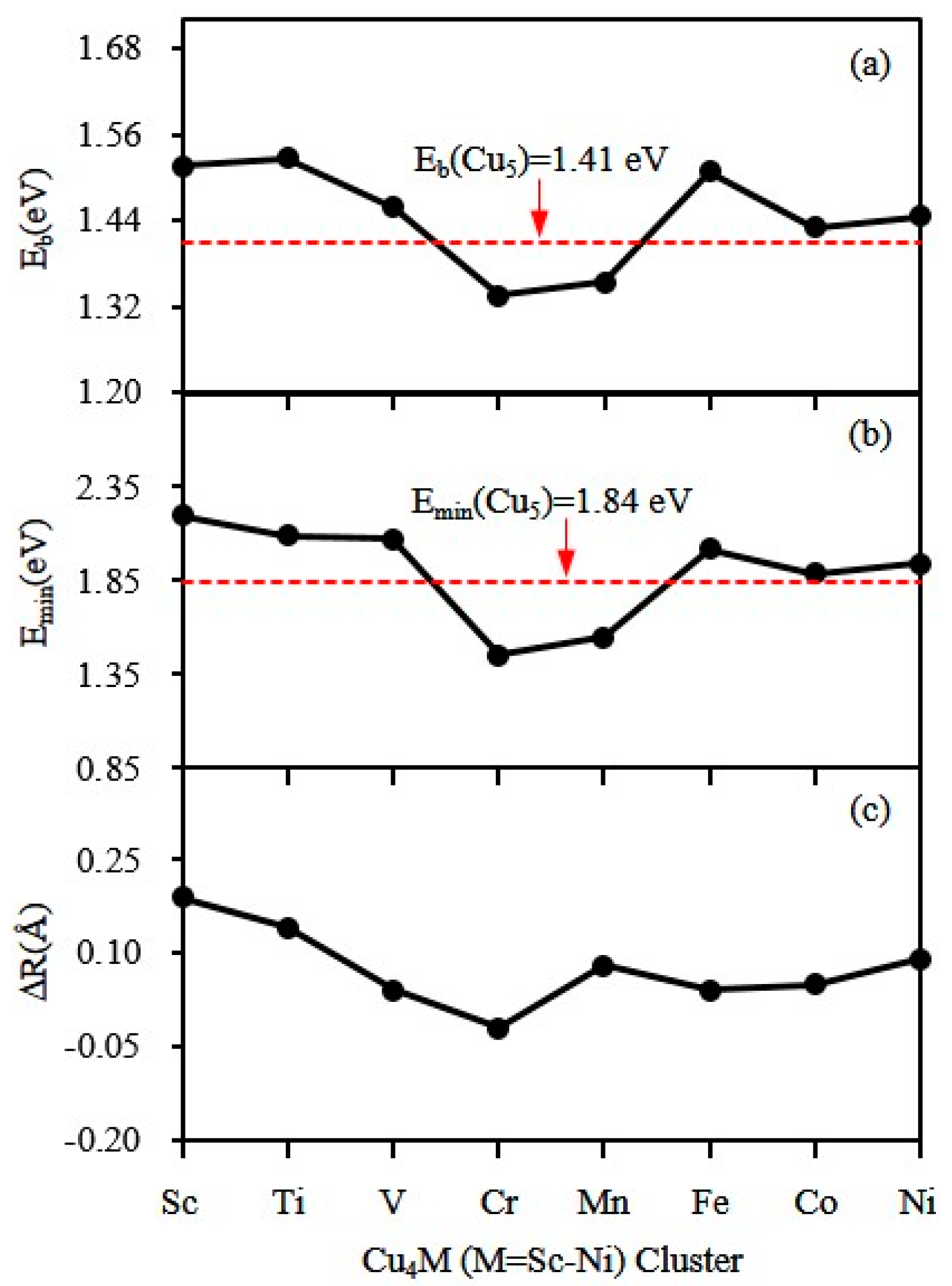
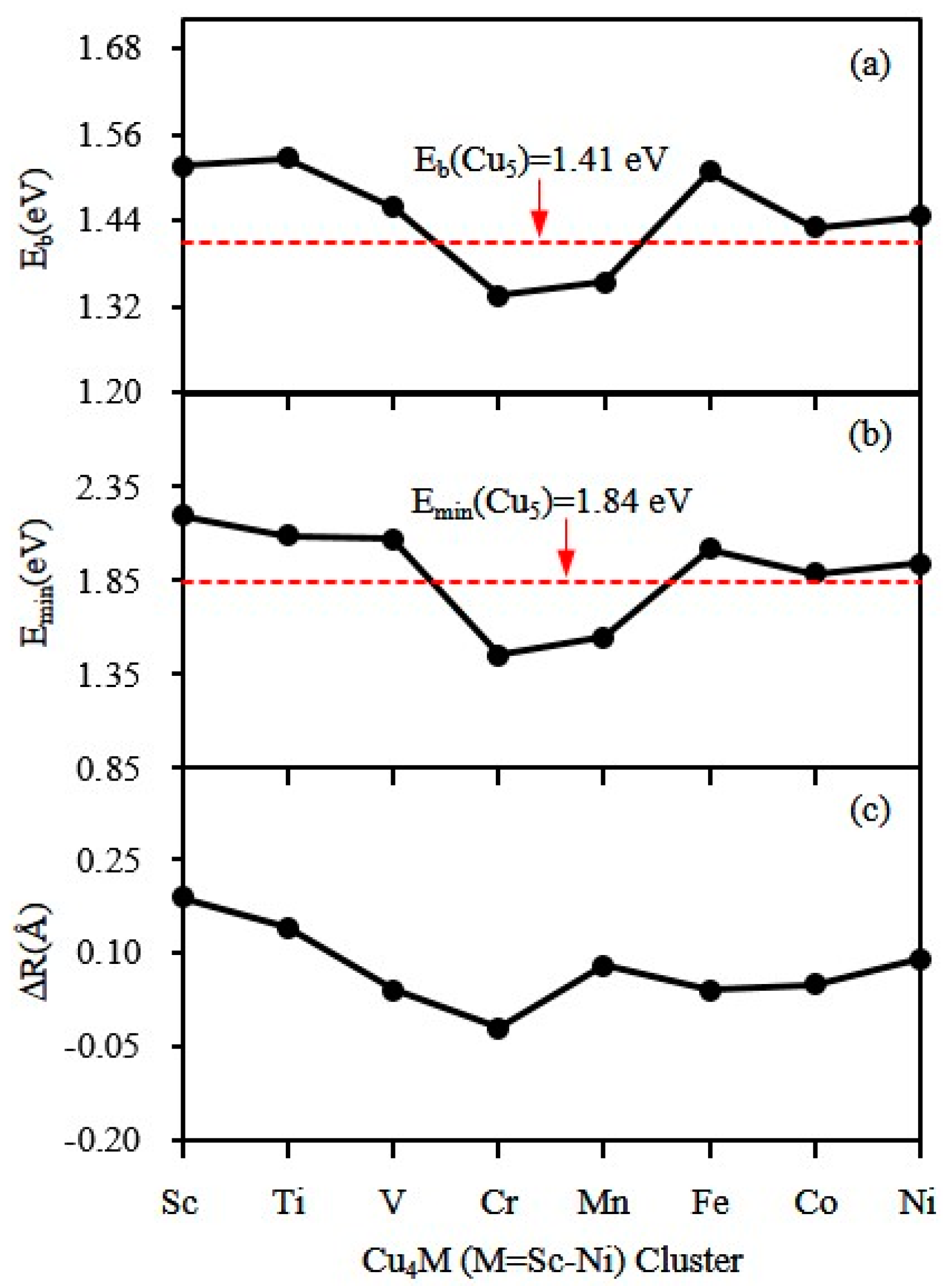
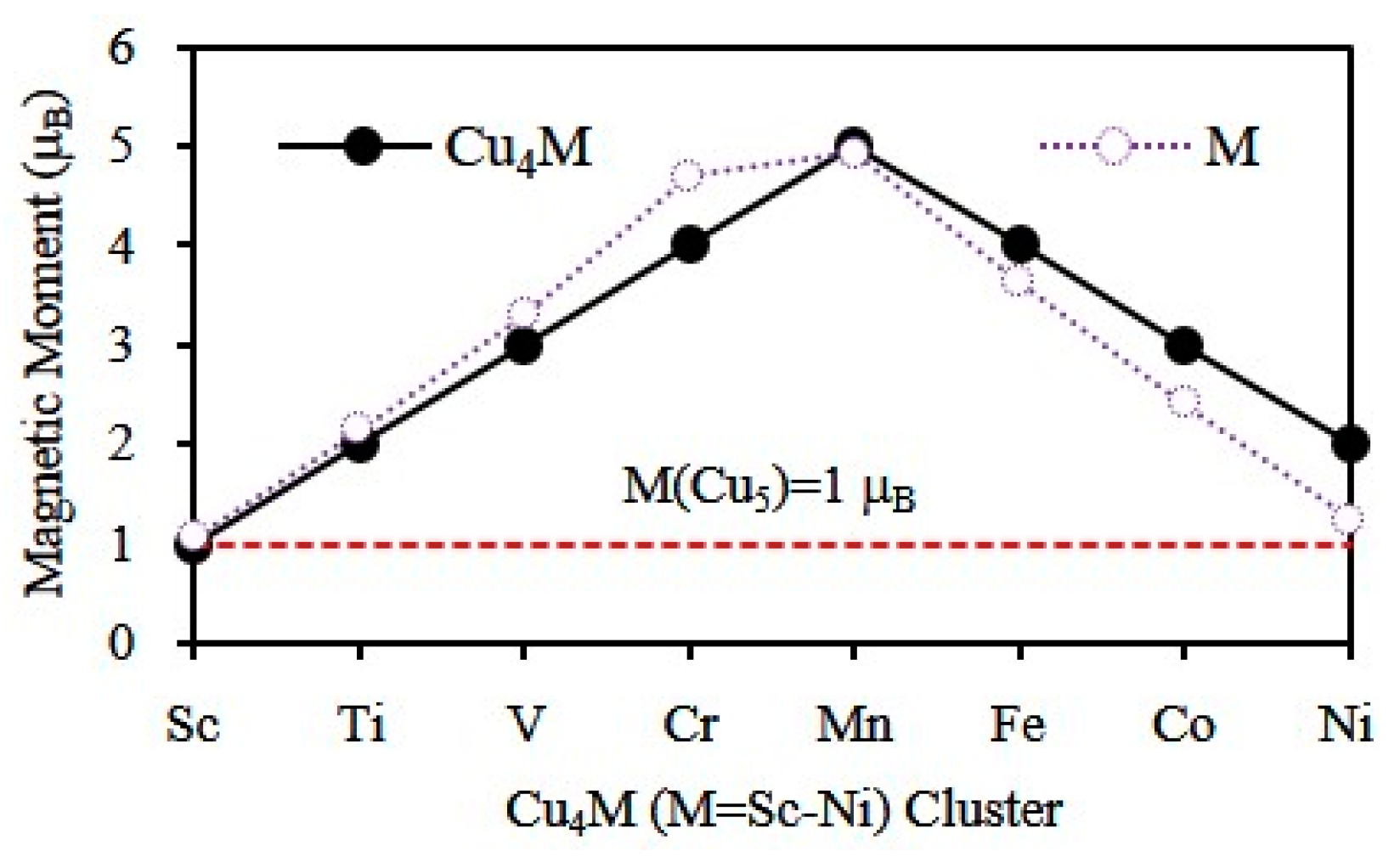
3.3. Magnetic Properties
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xing, X.D.; Wang, J.J.; Kuang, X.Y.; Xia, X.X.; Lu, C.; Maroulis, G. Probing the low-energy structures of aluminum-magnesium alloy clusters: A detailed study. Phys. Chem. Chem. Phys. 2016, 18, 26177–26183. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.J.; Wang, X.Q.; Liu, G.B. A density functional study on the AunAg (n = 1–12) alloy clusters. J. Alloys Compd. 2013, 570, 46–56. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, C.; Dong, H.; Shen, J.R.; Zhao, H.; Dau, J. A synthetic Mn4Ca-cluster mimicking the Oxygen-evolving center of photosynthesis. Science 2015, 348, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Hussain, A.I.; Chatha, S.A.S.; Mansha, A.; Ayub, K. Density functional theory study of geometric and electronic properties of full range of bimetallic AgnYm (n + m = 10) clusters. J. Alloys Compd. 2017, 705, 232–246. [Google Scholar] [CrossRef]
- Eric, C.T.; Stefan, V. Catalysis by clusters with precise numbers of atoms. Nat. Nanotechnol. 2015, 10, 577–588. [Google Scholar]
- Pakiari, A.H.; Jamshidi, Z. Nature and strength of M-S bonds (M = Au, Ag and Cu) in binary alloy gold clusters. J. Phys. Chem. A 2010, 114, 9212–9221. [Google Scholar] [CrossRef] [PubMed]
- Tahzeeb, M.; Ashok, B. Nanoscale alloying: A study in free Cu-Ag bimetallic clusters. J. Alloys Compd. 2013, 559, 24–33. [Google Scholar]
- Brito, B.G.A.; Hai, G.Q.; Rabelo, J.N.T.; Candido, L. A quantum Monte Carlo study on electron correlation in all-metal aromatic clusters MAl4− (M = Li, Na, K, Rb, Cu, Ag and Au). Phys. Chem. Chem. Phys. 2014, 16, 8639–8645. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gao, Y.; Xu, H.; Wang, Z.; Li, Z.; Zhang, R.Q. The mechanism of N-Ag bonding determined tunability of surface-enhanced Raman scattering of pyridine on MAg (M = Cu, Ag, Au) diatomic clusters. Phys. Chem. Chem. Phys. 2014, 16, 20665–20671. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, F. Effect of multiple exciton generation on ultraviolet—Visible absorption of Ag-Cu clusters: Ab initio study. J. Alloys Compd. 2014, 607, 238–244. [Google Scholar] [CrossRef]
- Neukermans, S.; Janssens, E.; Tanaka, H.; Silverans, R.E.; Lievens, P. Element- and Size-Dependent Electron Delocalization in AunX+ Clusters (X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni). Phys. Rev. Lett. 2003, 90, 033401. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Tang, Z.; Wang, L.; Wang, Q.; Yang, H.; Chen, S. PdAu alloyed clusters supported by carbon nanosheets as efficient electrocatalysts for Oxygen reduction. Int. J. Hydrogen Energy 2017, 42, 218–227. [Google Scholar] [CrossRef]
- Martins, M. Bimetallic PdM (M = Fe, Ag, Au) alloy nanoparticles assembled on reduced graphene oxide as catalysts for direct borohydride fuel cells. J. Alloys Compd. 2017, 718, 204–214. [Google Scholar] [CrossRef]
- Kaul, I.; Ghosh, P. First principles investigations of small bimetallic PdGa clusters as catalysts for hydrogen dissociation. Chem. Phys. 2017, 487, 87–96. [Google Scholar] [CrossRef]
- Li, G.F.; Zhou, Z.Q.; Chen, X.M.; Wang, J.J. Bimetallic PbnCun (n = 2–14) clusters were investigated by density functional theory. Comput. Theor. Chem. 2017, 1106, 21–27. [Google Scholar] [CrossRef]
- Wu, X.; Sun, Y.; Wei, Z.; Chen, T.J. Influence of noble metal dopants (M = Ag, Au, Pd or Pt) on the stable structures of bimetallic Co-M clusters. J. Alloys Compd. 2017, 701, 447–455. [Google Scholar] [CrossRef]
- Lee, K.E.; Shivhare, A.; Hu, Y.F.; Scott, R.W.J. Supported bimetallic AuPd clusters using activated Au25 clusters. Catal. Today 2017, 280, 259–265. [Google Scholar] [CrossRef]
- Landry, A.M.; Iglesia, E. Displacement-reduction routes to PtPd clusters and mechanistic inferences for the synthesis of other bimetallic compositions. J. Catal. 2016, 344, 389–400. [Google Scholar] [CrossRef]
- Chattaraj, D.; Bhattacharya, S.; Dash, S.; Majumder, C. Atomic, electronic, and magnetic properties of bimetallic ZrCo clusters: A first-principles study. J. Appl. Phys. 2016, 120, 094301. [Google Scholar] [CrossRef]
- Landry, A.M.; Iglesia, E. Synthesis of bimetallic AuPt clusters with clean surfaces via sequential displacement-reduction processes. Chem. Mater. 2016, 28, 5872–5886. [Google Scholar] [CrossRef]
- Oh, E.; Delehanty, J.B.; Field, L.D.; Makinen, A.J.; Goswami, R.; Huston, A.L.; Medintz, I.L. Synthesis and characterization of pegylated luminescent gold nanoclusters doped with silver and other metals. Chem. Mater. 2016, 28, 8676–8688. [Google Scholar] [CrossRef]
- Li, W.L.; Liu, C.; Abroshan, H.; Ge, Q.J.; Yang, X.J.; Xu, H.Y.; Li, G. Catalytic CO Oxidation using bimetallic MxAu25–x clusters: A combined experimental and computational study on doping effects. J. Phys. Chem. C 2016, 120, 10261–10267. [Google Scholar] [CrossRef]
- Sels, A.; Salassa, G.; Pollitt, S.; Guglieri, C.; Rupprechter, G.; Barrabes, N.; Burgi, T. Structural investigation of the ligand exchange reaction with rigid dithiol on doped (Pt, Pd) Au25 Clusters. J. Phys. Chem. C 2017, 121, 10919–10926. [Google Scholar] [CrossRef]
- Elisa, M.S.H.; Juan, M.M.C.; Pedro, G.A.L. Global minimum structures, stability and electronic properties of small NixSny (x + y ≤ 5) bimetallic clusters; A DFT study. Eur. Phys. J. D 2016, 70, 208. [Google Scholar]
- Carretta, S.; Amoretti, G.; Santini, P.; Mougel, V.; Mazzanti, M.; Gambarelli, S.; Colineau, E.; Caciuffo, R. Magnetic properties and chiral states of a trimetallic uranium complex. J. Phys. Condens. Matter 2013, 25, 486001. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, A.; Carretta, S.; Santini, P.; Amoretti, G.; Pavarini, E. Many-body ab initio study of antiferromagnetic {Cr7M} molecular rings. Phys. Rev. B 2016, 94, 224422. [Google Scholar] [CrossRef]
- Adams, R.D.; Zhang, Q.; Yang, X.Z. Two-dimensional bimetallic carbonyl cluster complexes with new properties and reactivities. J. Am. Chem. Soc. 2011, 133, 15950–15953. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, R.; Chantrell, R.W. Electronic and magnetic properties of bimetallic L1(0) cuboctahedral clusters by means of fully relativistic density-functional-based calculations. Phys. Rev. B 2012, 86, 224415. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, D.J. Role of Composition and geometric relaxation in CO2 binding to Cu-Ni bimetallic clusters. J. Phys. Chem. C 2014, 118, 250–258. [Google Scholar] [CrossRef]
- Mondal, K.; Banerjee, A.; Ghanty, T.K. Structural and chemical properties of subnanometer-sized bimetallic Au19Pt cluster. J. Phys. Chem. C 2014, 118, 11935–11945. [Google Scholar] [CrossRef]
- Chen, L.; Gao, Y.; Cheng, Y.K.; Su, Y.B.; Wang, Z.G.; Li, Z.Q.; Zhang, R.Q. Strong core@shell dependence in surface-enhanced raman scattering of pyridine on sTable 13-atom silver-caged bimetallic clusters. J. Phys. Chem. C 2015, 119, 17429–17437. [Google Scholar] [CrossRef]
- Pellarin, M.; Issa, I.; Langlois, C.; Lebeault, M.A.; Ramade, J.; Lerme, J.; Broyer, M.; Cottancin, E. Plasmon spectroscopy and chemical structure of small bimetallic Cu(1−x)Agx clusters. J. Phys. Chem. C 2015, 119, 5002–5012. [Google Scholar] [CrossRef]
- Li, H.F.; Wang, H.Q. Probing the stability of neutral and anionic transition-metal-doped golden cage nanoclusters: M@Au16 (M = Sc, Ti, V). Phys. Chem. Chem. Phys. 2014, 16, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Kuang, X.Y.; Lu, Z.W.; Mao, A.J.; Ma, Y.M. Determination of structures, stabilities, and electronic properties for bimetallic cesium-doped gold clusters: A density functional theory study. J. Phys. Chem. A 2011, 115, 9273–9281. [Google Scholar]
- Irena, E.; Moshe, S. DFT study of small bimetallic palladium-copper clusters. Chem. Phys. Lett. 2005, 401, 232–240. [Google Scholar]
- Pham, H.T.; Cuong, N.T.; Tam, N.M.; Tung, N.T. A systematic investigation on CrCun clusters with n = 9–16: Noble gas and tunable magnetic property. J. Phys. Chem. A 2016, 120, 7335–7343. [Google Scholar] [CrossRef] [PubMed]
- Holtzl, T.; Janssens, E.; Veldeman, N.; Veszpremi, T.; Lievens, P.; Nguyen, M.T. The Cu7Sc cluster is a stable s-aromatic seven-membered ring. ChemPhysChem 2008, 9, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Holtzl, T.; Veldeman, N.; De, H.J.; Veszpremi, T.; Lievens, P.; Nguyen, M.T. Growth mechanism and chemical bonding in scandium-doped copper clusters: Experimental and theoretical study in concert. Chem. Eur. J. 2009, 15, 3970–3982. [Google Scholar] [CrossRef] [PubMed]
- Veldeman, N.; Holtzl, T.; Neukermans, S.; Veszpremi, T.; Nguyen, M.T.; Lievens, P. Experimental observation and computational identification of Sc@Cu16+ a stable dopant-encapsulated copper cage. Phys. Rev. A 2007, 76, 011201. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Ichihashi, M. Reactions of Ti- and V-doped Cu cluster cations with nitric oxide and oxygen: Size dependence and preferential NO adsorption. J. Phys. Chem. A 2016, 120, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Cutrano, C.S.; Lekka, C.E. Structural, magnetic and electronic properties of Cu-Fe nanoclusters by density functional theory calculations. J. Alloys Compd. 2017, 707, 114–119. [Google Scholar] [CrossRef]
- Li, C.G.; Zhang, J.; Yuan, Y.Q.; Tang, Y.N.; Ren, B.Z.; Chen, W.G. Geometries, stabilities and electronic properties of copper and selenium doped copper clusters: Density functional theory study. Phys. E 2017, 86, 303–310. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lee, K.H.; Li, S.T.; Chu, S.Y. Structures and charge distributions of cationic and neutral Cun−1Ag clusters (n = 2–8). Phys. Rev. B 2006, 73, 235423. [Google Scholar] [CrossRef]
- Han, S.L.; Xue, X.L.; Nie, X.C.; Zhai, H.; Wang, F.; Sun, Q.; Jia, Y.; Li, S.F.; Guo, Z.X. First-principles calculations on the role of Ni-doping in Cun clusters: From geometric and electronic structures to chemical activities towards CO2. Phys. Lett. A 2010, 374, 4324–4330. [Google Scholar] [CrossRef]
- Kahnouji, H.; Najafvandzadeh, H.; Hashemifar, S.J.; Alaei, M.; Akbarzadeh, H. Density-functional study of the pure and palladium doped small copper and silver clusters. Chem. Phys. Lett. 2015, 630, 101–105. [Google Scholar] [CrossRef]
- Ricardo-Chavez, J.L.; Pastor, G.M. First principles calculations on Ni impurities in Cu clusters. J. Magn. Magn. Mater. 2005, 294, 122–126. [Google Scholar] [CrossRef]
- Li, C.G.; Shen, Z.G.; Hu, Y.F.; Tang, Y.N.; Chen, W.G.; Ren, B.Z. Insights into the structures and electronic properties of Cun+1μ and CunSμ (n = 1–12; μ = 0 ± 1) clusters. Sci. Rep. 2017, 7, 1345. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, Y.Q.; Zhang, J.J.; Liang, X.G.; Duan, H.M. Density functional theory study of the adsorption of hydrogen atoms on Cu2X (X = 3d) clusters. Chem. Phys. Lett. 2016, 651, 137–143. [Google Scholar] [CrossRef]
- Cao, Z.; Guo, L.; Liu, N. A theoretical study of the water—Gas-shift reaction on Cu6TM (TM = Co, Ni, Cu, Rh, Pd, Ag, Ir, Pt, Au) Clusters. J. Clust. Sci. 2016, 27, 523–535. [Google Scholar] [CrossRef]
- Sargolzaei, M.; Lotfizadeh, N. Spin and orbital magnetism of a single 3d transition-metal atom doped into icosahedral coinage-metal clusters X12 (X = Cu, Ag, Au). Phys. Rev. B 2011, 83, 155404. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 09 Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–311. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 2010, 82, 094116. [Google Scholar] [CrossRef]
- Guzmán-Ramírez, G.; Aguilera-Granja, F.; Robles, J. DFT and GEGA genetic algorithm optimized structures of clusters. Eur. Phys. J. D 2010, 57, 49–60. [Google Scholar] [CrossRef]
- Wang, S.Y.; Yu, J.Z.; Mizuseki, H.; Sun, Q.; Wang, C.Y.; Kawazoe, Y. Energetics and local spin magnetic moment of single 3, 4d impurities encapsulated in an icosahedral Au12 cage. Phys. Rev. B 2004, 70, 165413. [Google Scholar] [CrossRef]
- Wu, X.; Ray, A.K. A density functional study of small neutral and cationic vanadium clusters Vn and Vn+ (n = 2–9). J. Chem. Phys. 1999, 110, 2437. [Google Scholar] [CrossRef]
- Simard, B.; Lebeaultdorget, M.A.; Marijnissen, A.; Meulen, J.J.T. Photoionization spectroscopy of dichromium and dimolybdenum: Ionization potentials and bond energies. J. Chem. Phys. 1998, 108, 9668–9674. [Google Scholar] [CrossRef]
- Ma, Q.M.; Xie, Z.; Wang, J.; Liu, Y.; Li, Y.C. Structures, binding energies and magnetic moments of small iron clusters: A study based on all-electron DFT. Solid State Commun. 2007, 142, 114. [Google Scholar] [CrossRef]
- Morse, M.D. Clusters of transition-metal atoms. Chem. Rev. 1988, 86, 1049–1109. [Google Scholar] [CrossRef]
- Gagliardi, L. When does gold behave as a halogen? Predicted uranium tetraauride and other MAu4 tetrahedral species, (M = Ti, Zr, Hf, Th). J. Am. Chem. Soc. 2003, 125, 7504. [Google Scholar] [CrossRef] [PubMed]
- Cha, C.; Ganteför, G.; Eberhardt, W. Photoelectron spectroscopy of Cun− clusters: Comparison with jellium model predictions. J. Chem. Phys. 1993, 99, 6308–6312. [Google Scholar] [CrossRef]
- Ingolfsson, O.; Busolt, U.; Sugawara, K. Energy-resolved collision-induced dissociation of Cun+ (n = 2–9): Stability and fragmentation pathways. J. Chem. Phys. 2000, 112, 4613–4620. [Google Scholar] [CrossRef]
- Rienstra-Kiracofe, J.C.; Tschumper, G.S.; Schaefer, H.F.; Nandi, S.; Ellison, G.B. Atomic and molecular electron affinities: Photoelectron experiments and theoretical computations. Chem. Rev. 2002, 102, 231–282. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dimer | Functional/Basis Set | R (Å) | De (eV) | (cm−1) | VIP (eV) | EA (eV) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Calc. | Expt. | Calc. | Expt. | Calc. | Expt. | Calc. | Expt. | Calc. | Expt. | ||
| Cu2 | B3LYP/LanL2DZ | 2.26 | 2.22 a | 2.02 | 2.01 a | 256 | 264 a | 7.99 | 7.90 a | 0.63 | 0.83 a |
| Blyp/6-311+G(d) | 2.27 | 2.01 | 244 | 8.20 | 0.83 | ||||||
| Ti2 | B3LYP/LanL2DZ | 1.91 | 1.94 b | 1.42 | 1.54 ± 0.19 b | ||||||
| V2 | B3LYP/LanL2DZ | 1.75 | 1.77 c | 1.94 | 2.47 ± 0.22 c | 6.39 | 6.35 c | ||||
| Cr2 | B3LYP/LanL2DZ | 6.22 | 6.4 ± 0.2 d | ||||||||
| Fe2 | B3LYP/LanL2DZ | 2.15 | 2.02 e | 1.28 | 1.30 e | 6.24 | 6.30 e | ||||
| Ni2 | B3LYP/LanL2DZ | 2.38 | 2.20 f | 1.59 | 2.06 f | ||||||
| Clusters | B3LYP/LanL2DZ | Blyp/6-311+G(d) | ||||
|---|---|---|---|---|---|---|
| LI | ΔE (eV) | SM | LI | ΔE (eV) | SM | |
| Cu5 | IA | 0 | 2 | IA | 0 | 2 |
| IB | 0.54 | 2 | IB | 0.44 | 2 | |
| IC | 0.88 | 2 | IC | 0.91 | 2 | |
| Cu4Sc | ID | 0 | 2 | IE | 0 | 2 |
| IE | 0.07 | 2 | IF | 0.07 | 2 | |
| IF | 0.21 | 2 | IJ | 0.09 | 2 | |
| Cu4Ti | IE | 0 | 3 | IE | 0 | 3 |
| ID | 0.15 | 3 | ID | 0.15 | 3 | |
| IH | 0.22 | 3 | IL | 0.28 | 3 | |
| Cu4V | ID | 0 | 4 | ID | 0 | 4 |
| IG | 0.36 | 4 | IE | 0.28 | 4 | |
| IL | 0.43 | 4 | IK | 0.49 | 4 | |
| Cu4Cr | ID | 0 | 5 | ID | 0 | 5 |
| IG | 0.13 | 7 | IG | 0.36 | 5 | |
| II | 0.24 | 7 | IJ | 0.43 | 5 | |
| Cu4Mn | ID | 0 | 6 | ID | 0 | 6 |
| IG | 0.16 | 6 | IG | 0.23 | 6 | |
| IH | 0.24 | 6 | IH | 0.26 | 6 | |
| Cu4Fe | ID | 0 | 5 | ID | 0 | 5 |
| IG | 0.11 | 5 | IG | 0.18 | 5 | |
| II | 0.25 | 5 | IJ | 0.33 | 5 | |
| Cu4Co | IE | 0 | 4 | IE | 0 | 4 |
| ID | 0.01 | 4 | IG | 0.16 | 4 | |
| IG | 0.18 | 2 | II | 0.31 | 4 | |
| Cu4Ni | ID | 0 | 3 | ID | 0 | 3 |
| IG | 0.09 | 3 | IG | 0.10 | 3 | |
| II | 0.31 | 3 | IL | 0.12 | 3 | |
| Clusters | µ (D) | (a.u.) | (a.u.) | (a.u.) | (a.u.) | ZPE (J/mol) | (Å) |
|---|---|---|---|---|---|---|---|
| Cu5 | 0.01 | 282.6 | 199.3 | 105.0 | 195.6 | 7265.7 | |
| Cu4Sc | 2.22 | 314.1 | 223.6 | 182.7 | 240.1 | 7020.5 | 2.72 |
| Cu4Ti | 1.54 | 128.1 | 206.6 | 304.1 | 212.9 | 7204.7 | 2.61 |
| Cu4V | 1.07 | 299.2 | 200.6 | 118.6 | 206.1 | 7383.0 | 2.58 |
| Cu4Cr | 0.83 | 299.3 | 196.2 | 115.6 | 203.7 | 7077.0 | 2.60 |
| Cu4Mn | 0.56 | 116.0 | 286.7 | 217.2 | 206.6 | 7148.5 | 2.55 |
| Cu4Fe | 0.37 | 110.7 | 283.8 | 206.7 | 200.4 | 7319.2 | 2.51 |
| Cu4Co | 0.27 | 108.1 | 202.5 | 283.1 | 197.9 | 7315.1 | 2.48 |
| Cu4Ni | 0.08 | 283.2 | 201.9 | 106.4 | 197.2 | 7532.0 | 2.44 |
| Dissociation Channels | DE (eV) | ||||
|---|---|---|---|---|---|
| m = 1 | m = 2 | m = 3 | m = 4 | ||
| Cu5 = Cum + Cu5−m | 1.84 | 2.03 | |||
| Cu4Sc= Cum + Cu4−mSc | 2.23 | 2.20 | 3.10 | 2.36 | |
| Cu4Ti = Cum + Cu4−mTi | 2.20 | 2.09 | 2.92 | 2.41 | |
| Cu4V = Cum + Cu4−mV | 2.12 | 2.10 | 2.81 | 2.07 | |
| Cu4Cr = Cum + Cu4−mCr | 1.86 | 2.03 | 2.17 | 1.45 | |
| Cu4Mn = Cum + Cu4−mMn | 2.18 | 1.73 | 2.78 | 1.55 | |
| Cu4Fe = Cum + Cu4−mFe | 2.02 | 2.11 | 2.47 | 2.32 | |
| Cu4Co = Cum + Cu4−mCo | 1.89 | 2.08 | 2.28 | 1.93 | |
| Cu4Ni = Cum + Cu4−mNi | 1.94 | 2.11 | 2.34 | 2.00 | |
| Clusters | VIP (eV) | EA (eV) | M-3d | M-4s | M-4p | M-5p | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Q (e) | M (μB) | Q (e) | M (μB) | Q (e) | M (μB) | Q (e) | M (μB) | |||
| Cu5 | 6.24 (6.30) | 1.70 (1.82) | ||||||||
| Cu4Sc | 5.89 | 1.08 | 1.65 | 0.96 | 0.63 | 0.09 | 0.04 | 0 | 0.21 | 0.02 |
| Cu4Ti | 6.55 | 1.23 | 2.70 | 2.08 | 0.60 | 0.04 | 0.56 | 0.02 | 0.30 | 0 |
| Cu4V | 6.20 | 1.41 | 3.73 | 3.27 | 0.59 | 0.01 | 0.55 | 0.01 | 0 | 0 |
| Cu4Cr | 6.17 | 1.61 | 4.92 | 4.66 | 0.55 | 0.03 | 0.43 | 0 | 0 | 0 |
| Cu4Mn | 6.29 | 0.95 | 5.35 | 4.61 | 0.89 | 0.21 | 0.77 | 0.11 | 0 | 0 |
| Cu4Fe | 6.28 | 1.10 | 6.66 | 3.30 | 0.86 | 0.20 | 0.64 | 0.12 | 0 | 0 |
| Cu4Co | 6.45 | 1.63 | 7.8 | 2.13 | 0.81 | 0.17 | 0.57 | 0.11 | 0 | 0 |
| Cu4Ni | 6.52 | 1.57 | 8.95 | 0.99 | 0.78 | 0.14 | 0.22 | 0.08 | 0.31 | 0.03 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Die, D.; Zheng, B.-X.; Kuang, X.-Y.; Zhao, Z.-Q.; Guo, J.-J.; Du, Q. Exploration of the Structural, Electronic and Tunable Magnetic Properties of Cu4M (M = Sc-Ni) Clusters. Materials 2017, 10, 946. https://doi.org/10.3390/ma10080946
Die D, Zheng B-X, Kuang X-Y, Zhao Z-Q, Guo J-J, Du Q. Exploration of the Structural, Electronic and Tunable Magnetic Properties of Cu4M (M = Sc-Ni) Clusters. Materials. 2017; 10(8):946. https://doi.org/10.3390/ma10080946
Chicago/Turabian StyleDie, Dong, Ben-Xia Zheng, Xiao-Yu Kuang, Zheng-Quan Zhao, Jian-Jun Guo, and Quan Du. 2017. "Exploration of the Structural, Electronic and Tunable Magnetic Properties of Cu4M (M = Sc-Ni) Clusters" Materials 10, no. 8: 946. https://doi.org/10.3390/ma10080946
APA StyleDie, D., Zheng, B.-X., Kuang, X.-Y., Zhao, Z.-Q., Guo, J.-J., & Du, Q. (2017). Exploration of the Structural, Electronic and Tunable Magnetic Properties of Cu4M (M = Sc-Ni) Clusters. Materials, 10(8), 946. https://doi.org/10.3390/ma10080946