
Combined X-ray and Raman Studies on the Effect of Cobalt Additives on the Decomposition of Magnesium Borohydride


 ,
,
Abstract
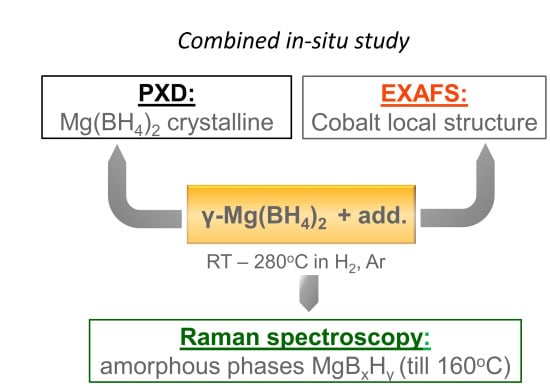
:
1. Introduction
2. Results
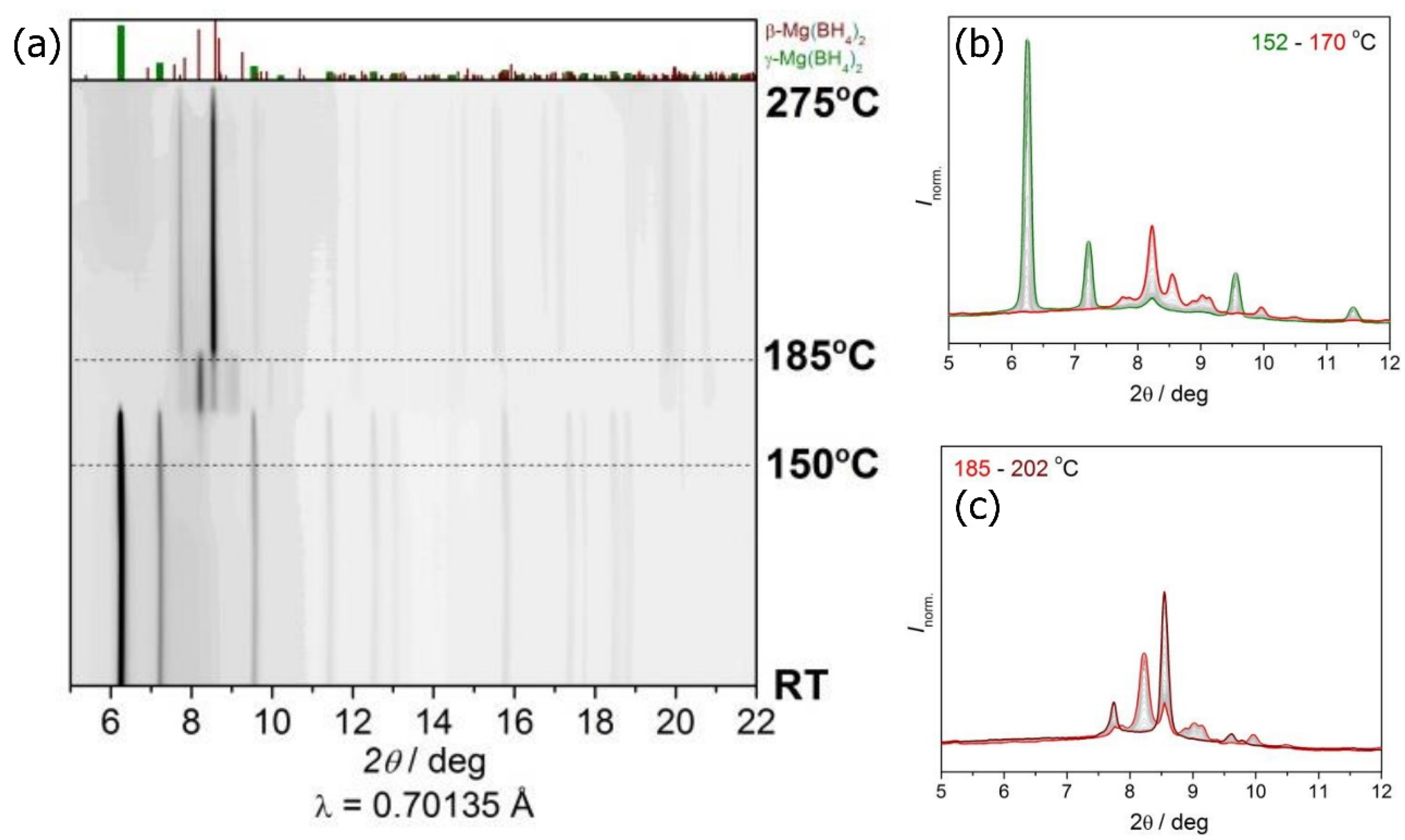
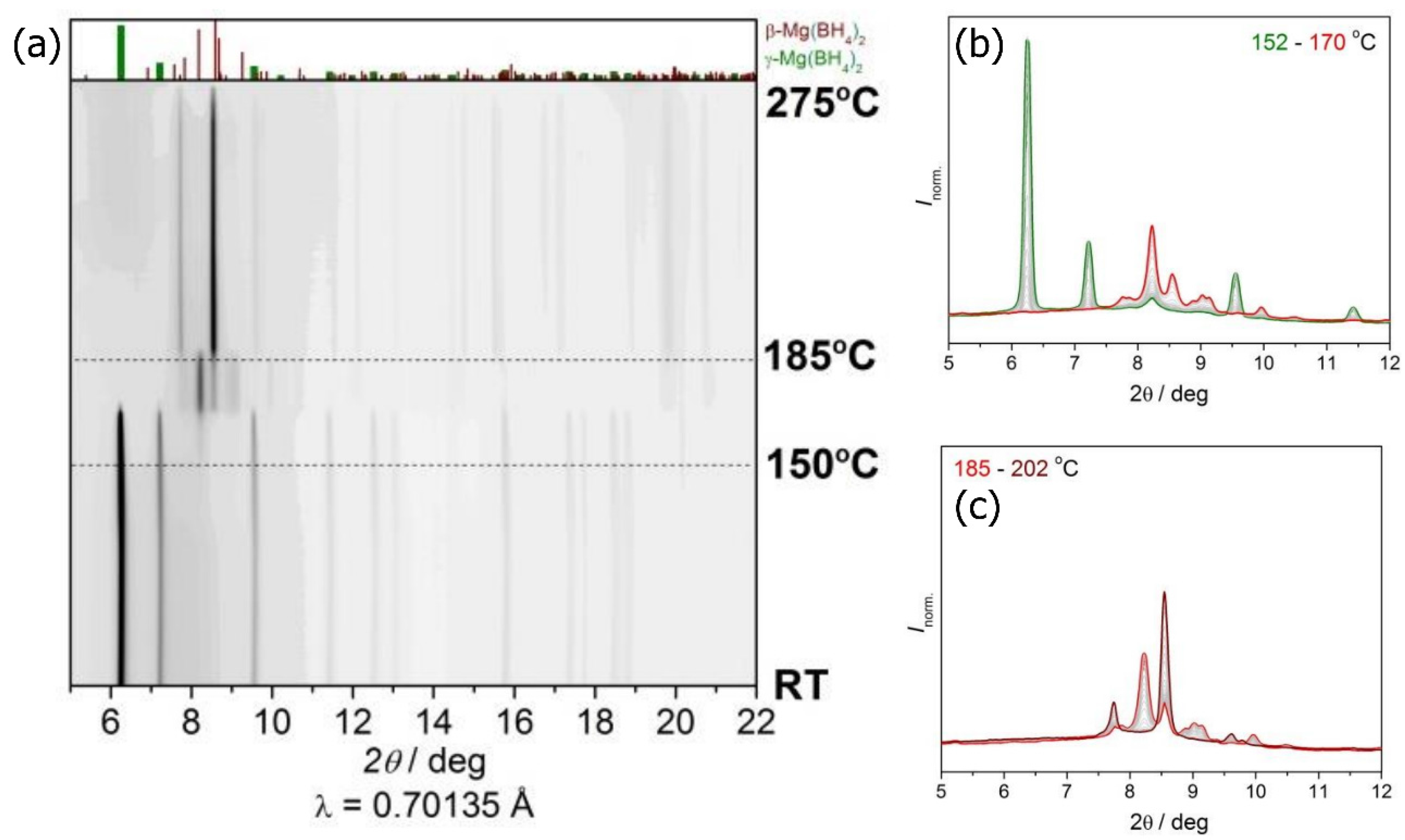
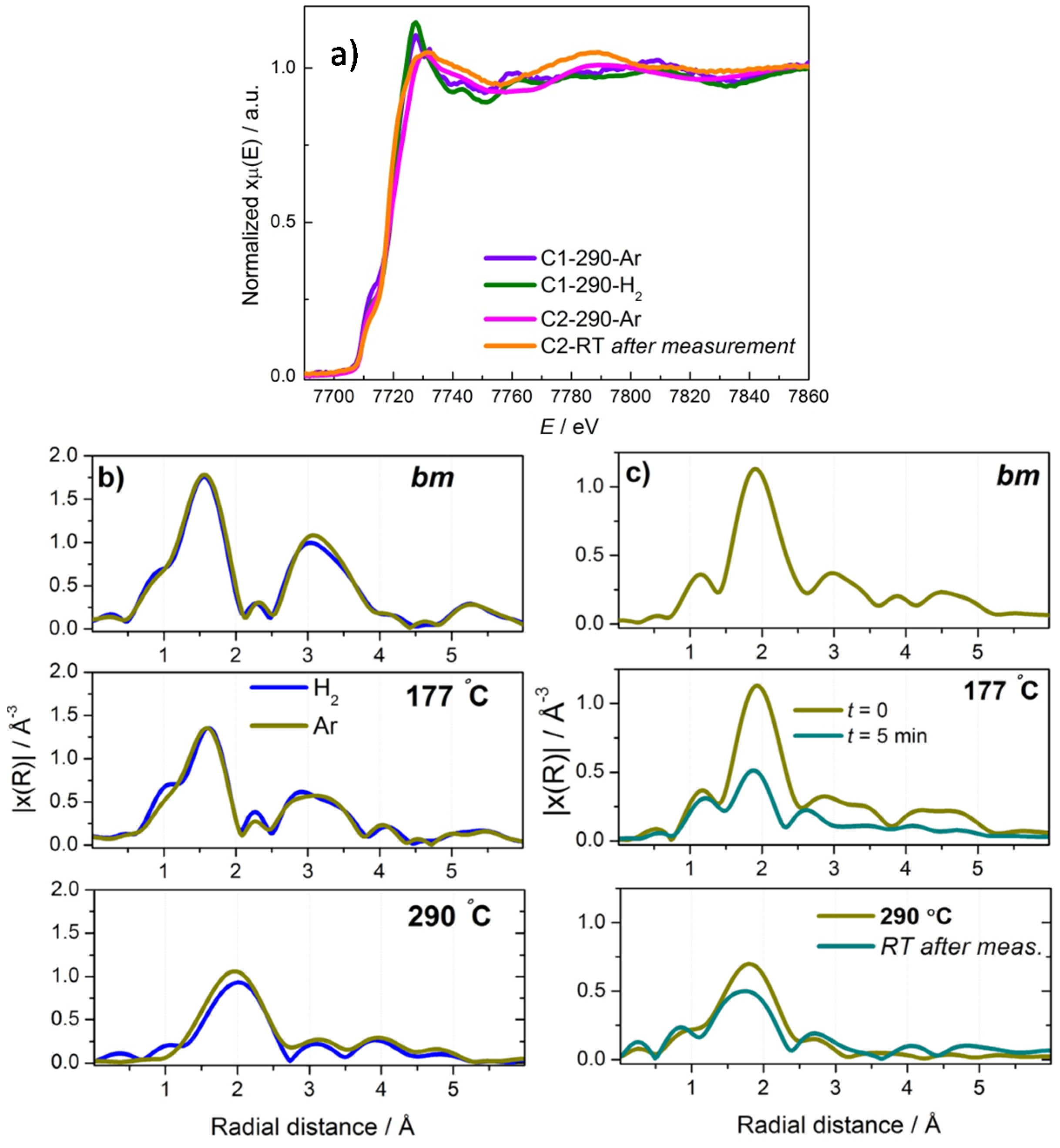
2.1. Synchrotron Radiation Powder X-ray Diffraction (SR-PXD) Study of Mg(BH4)2 Decomposition at 2.5 Bar H2

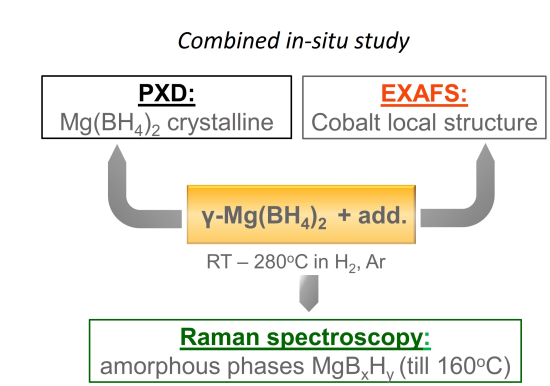
2.2. In-Situ X-ray Diffraction (XRD)/X-ray Absorption Spectroscopy (XAS)/Raman Study of Mg(BH4)2+Coadd Decomposition
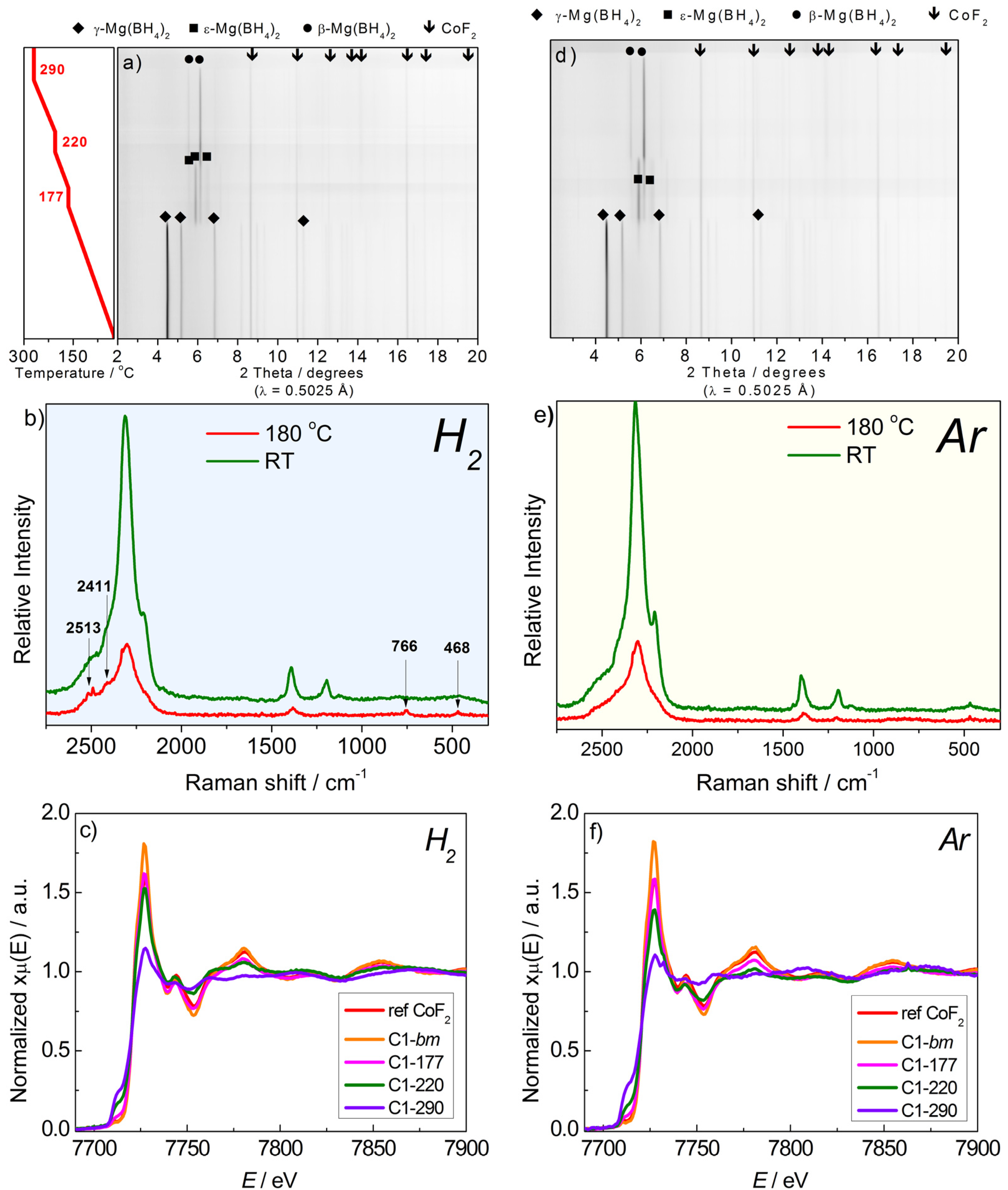
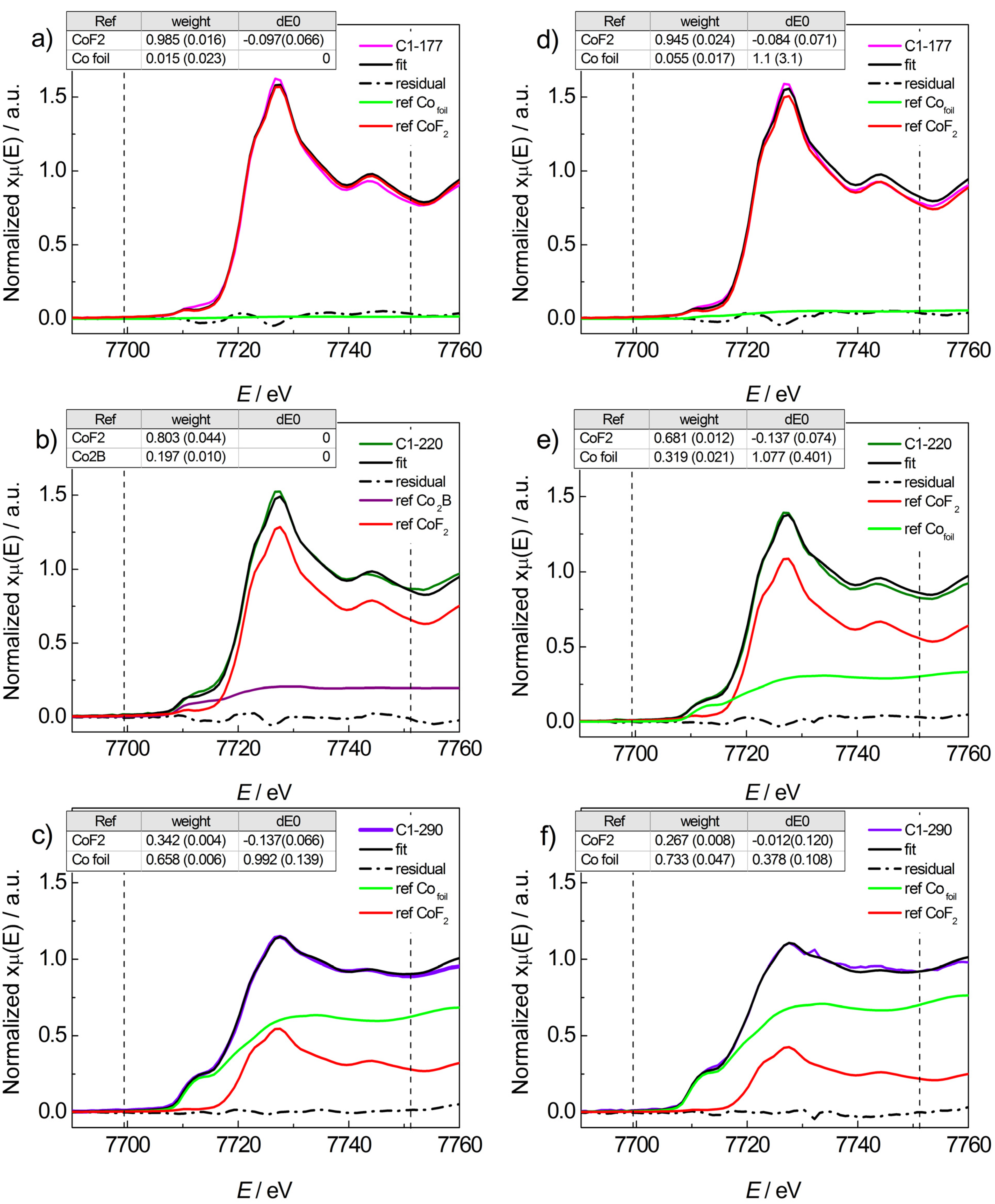
2.2.1. Mg(BH4)2 + CoF2

2.2.2. Mg(BH4)2 + CoCl2
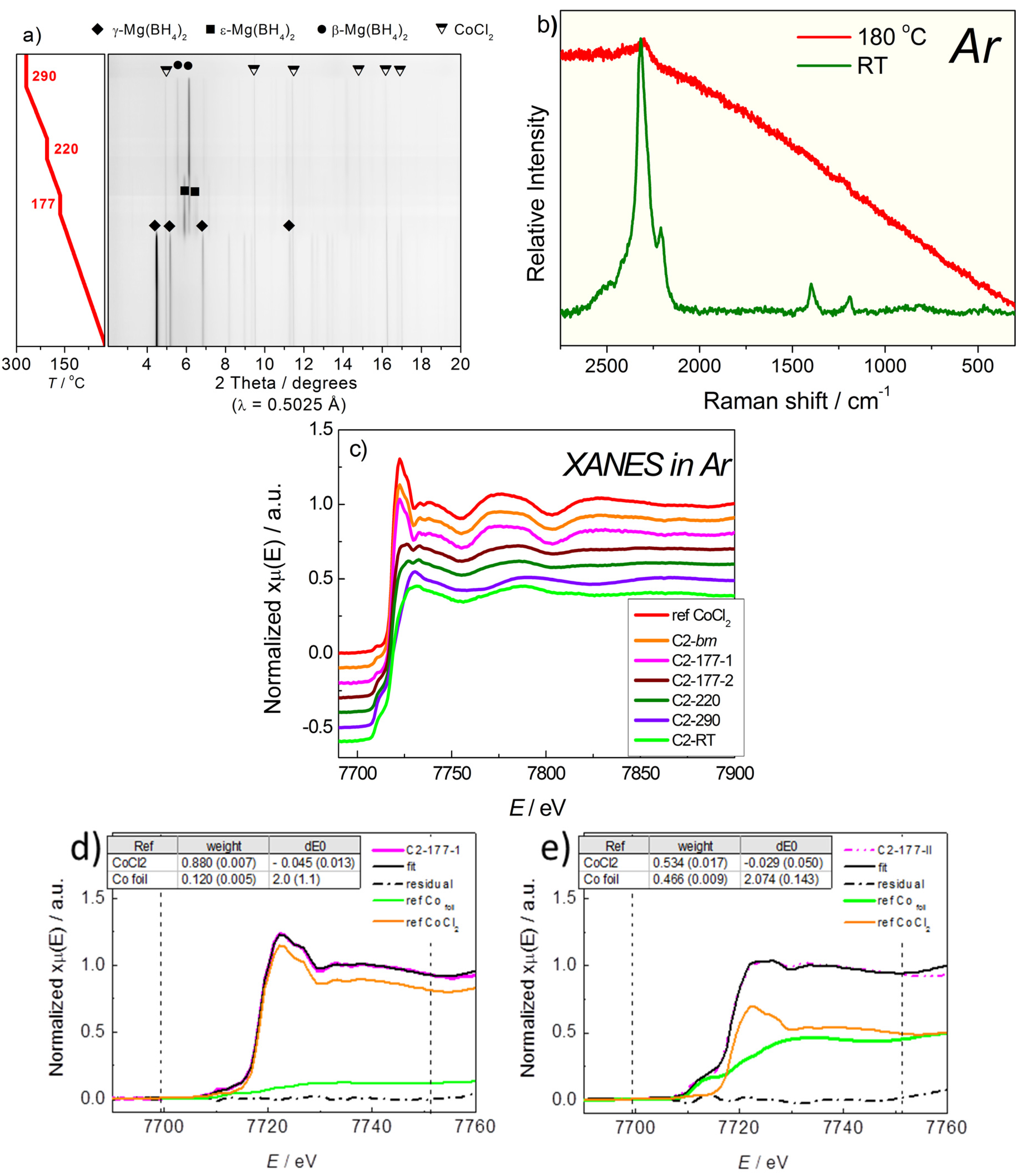

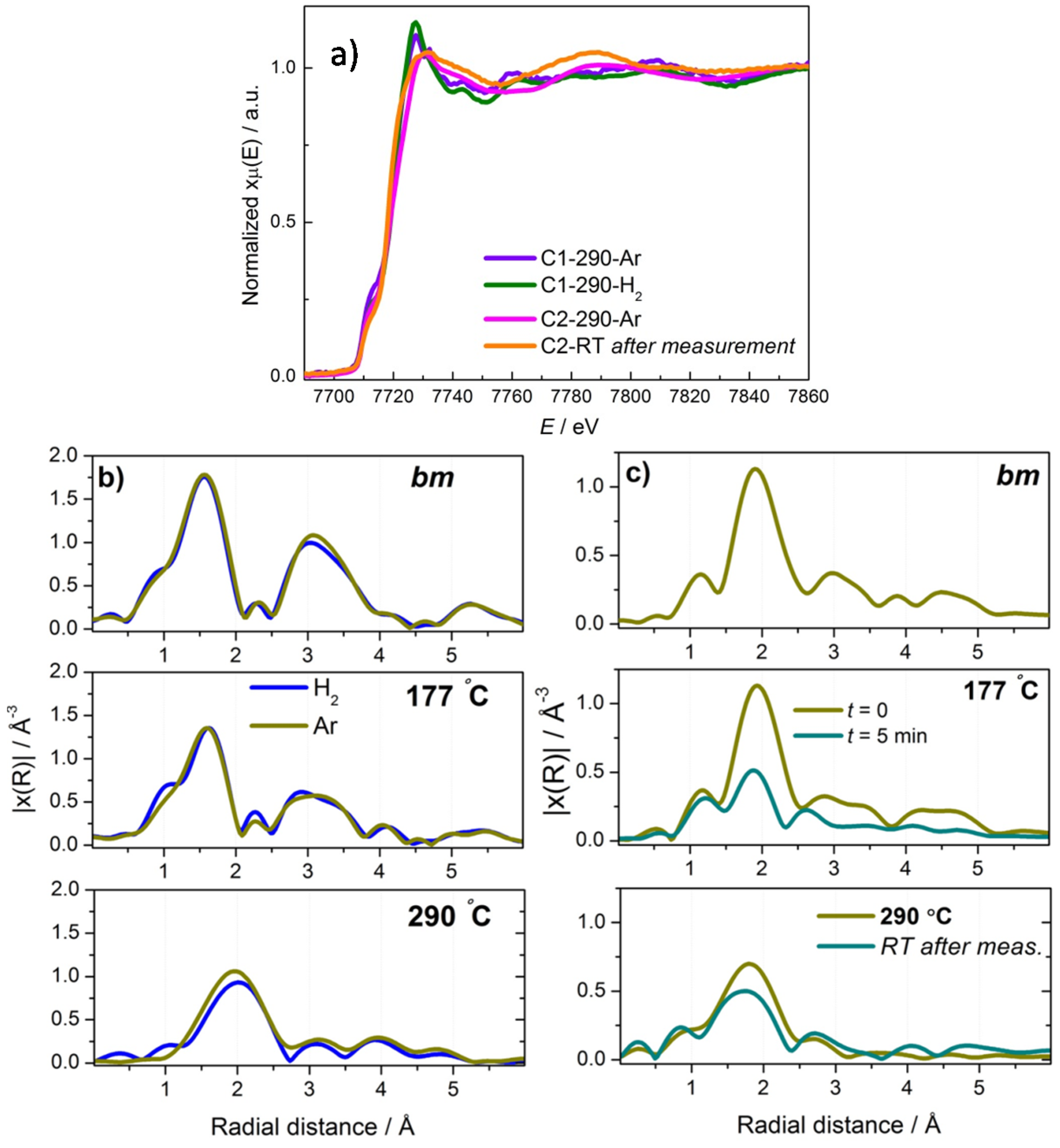
2.3. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Event | Mg(BH4)2 Milled-H2 | C1 (Mg(BH4)2 + 0.2 mol% CoF2)/H2 | C1 (Mg(BH4)2 + 0.2 mol% CoF2)/Ar | C2 (Mg(BH4)2 + 0.2 mol% CoCl2)/Ar |
|---|---|---|---|---|
| γ → ε phase transition | 150–170 °C | 130–175 °C | 105–160 °C | 107–150 °C |
| ε → β′ phase transition | 185–202 °C | 175–190 °C | 175–200 °C | 175–190 °C |
| Decomposition | >200 °C | >220 °C | >200 °C | >200 °C |
3. Experimental Section
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- George, L.; Saxena, S.K. Structural stability of metal hydrides, alanates and borohydrides of alkali and alkali-earth elements: A review. Int. J. Hydrog. Energy 2010, 35, 5454–5470. [Google Scholar] [CrossRef]
- Li, H.-W.; Yan, Y.; Orimo, S.; Zuttel, A.; Jensen, C.M. Recent Progress in Metal Borohydrides for Hydrogen Storage. Energies 2011, 4, 185–214. [Google Scholar] [CrossRef]
- Hummelshoj, J.S.; Landis, D.D.; Voss, J.; Jiang, T.; Tekin, A.; Bork, N.; Dulak, M.; Mortensen, J.J.; Adamska, L.; Andersin, J.; et al. Density functional theory based screening of ternary alkali-transition metal borohydrides: A computational material design project. J. Chem. Phys. 2009, 131. [Google Scholar] [CrossRef] [PubMed]
- Besora, M.; Lledos, A. Coordination modes and hydride exchange dynamics in transition metal tetrahydroborate complexes. In Contemporary Metal Boron Chemistry I: Borylenes, Boryls, Borane; Marder, T.B., Lin, Z., Eds.; Springer: Berlin, Germany, 2008; pp. 149–202. [Google Scholar]
- Marks, T.J.; Kolb, J.R. Covalent transition metal, lanthanide, and actinide tetrahydroborate complexes. Chem. Rev. 1977, 77, 263–293. [Google Scholar] [CrossRef]
- Callini, E.; Borgschulte, A.; Ramirez-Cuesta, A.J.; Zuettela, A. Diborane release and structure distortion in borohydrides. Dalton Trans. 2013, 42, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.; Callini, E.; Borgschulte, A.; Zuettel, A.; Jensen, C.M. Dehydrogenation studies of the bimetallic borohydrides. RSC Adv. 2014, 4, 63933–63940. [Google Scholar] [CrossRef]
- Albanese, E.; Civalleri, B.; Casassa, S.; Baricco, M. Investigation on the Decomposition Enthalpy of Novel Mixed Mg(1−x)Znx(BH4)2 Borohydrides by Means of Periodic DFT Calculations. J. Phys. Chem. C 2014, 118, 23468–23475. [Google Scholar] [CrossRef]
- Schouwink, P.; Ley, M.B.; Tissot, A.; Hagemann, H.; Jensen, T.R.; Smrčok, L.; Černý, R. Structure and properties of complex hydride perovskite materials. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.W.; Chen, P. Amides and borohydrides for high capacity solid-state hydrogen storage—Materials design and kinetics improvement. MRS Bull. 2013, 38, 480–487. [Google Scholar] [CrossRef]
- Li, H.-W.; Kikuchi, K.; Sato, T.; Nakamori, Y.; Ohba, N.; Aoki, M.; Miwa, K.; Towata, S.; Orimo, S. Synthesis and Hydrogen Storage Properties of a Single-Phase Magnesium Borohydride Mg(BH4)2. Mater. Trans. 2008, 49, 2224–2228. [Google Scholar] [CrossRef]
- Matsurtaga, T.; Buchter, F.; Miwa, K.; Towata, S.; Orimo, S.; Zuttel, A. Magnesium borohydride: A new hydrogen storage material. Renew. Energy 2008, 33, 193–196. [Google Scholar] [CrossRef]
- Filinchuk, Y.; Richter, B.; Jensen, T.R.; Dmitriev, V.; Chernyshov, D.; Hagemann, H. Porous and Dense Magnesium Borohydride Frameworks: Synthesis, Stability, and Reversible Absorption of Guest Species. Angew. Chem. Int. Ed. 2011, 50, 11162–11166. [Google Scholar] [CrossRef] [PubMed]
- Roennebro, E. Development of group II borohydrides as hydrogen storage materials. Curr. Opin. Solid State Mater. Sci. 2011, 15, 44–51. [Google Scholar] [CrossRef]
- Ozolins, V.; Akbarzadeh, A.R.; Gunaydin, H.; Michel, K.; Wolverton, C.; Majzoub, E.H. First-principles computational discovery of materials for hydrogen storage. J. Phys. Conf. Ser. 2009, 180. [Google Scholar] [CrossRef]
- Ozolins, V.; Majzoub, E.H.; Wolverton, C. First-principles prediction of a ground state crystal structure of magnesium borohydride. Phys. Rev. Lett. 2008, 100. [Google Scholar] [CrossRef]
- Van Setten, M.J.; de Wijs, G.A.; Fichtner, M.; Brocks, G. A density functional study of α-Mg(BH4)2. Chem. Mater. 2008, 20, 4952–4956. [Google Scholar] [CrossRef]
- Pinatel, E.R.; Albanese, E.; Civalleri, B.; Baricco, M. Thermodynamic modelling of Mg(BH4)2. J. Alloys Compd. 2015, 645, S64–S68. [Google Scholar] [CrossRef]
- Riktor, M.D.; Sorby, M.H.; Chlopek, K.; Fichtner, M.; Buchter, F.; Zuttel, A.; Hauback, B.C. In situ synchrotron diffraction studies of phase transitions and thermal decomposition of Mg(BH4)2 and Ca(BH4)2. J. Mater. Chem. 2007, 17, 4939–4942. [Google Scholar] [CrossRef]
- Hanada, N.; Chlopek, K.; Frommen, C.; Lohstroh, W.; Fichtner, M. Thermal decomposition of Mg(BH4)2 under He flow and H2 pressure. J. Mater. Chem. 2008, 18, 2611–2614. [Google Scholar] [CrossRef]
- Soloveichik, G.L.; Gao, Y.; Rijssenbeek, J.; Andrus, M.; Kniajanski, S.; Bowman, R.C., Jr.; Hwan, S.-J.; Zhao, J.-C. Magnesium borohydride as a hydrogen storage material: Properties and dehydrogenation pathway of unsolvated Mg(BH4)2. Int. J. Hydrog. Energy 2009, 34, 916–928. [Google Scholar] [CrossRef]
- Yan, Y.; Li, H.-W.; Maekawa, H.; Aoki, M.; Noritake, T.; Matsumoto, M.; Miwa, K.; Towata, S.; Orimo, S. Formation Process of [B12H12]2− from [BH4]− during the Dehydrogenation Reaction of Mg(BH4)2. Mater. Trans. 2011, 52, 1443–1446. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Zheng, J.; Song, P.; Li, X. Decomposition pathway of Mg(BH4)2 under pressure: Metastable phases and thermodynamic parameters. Scr. Mater. 2011, 64, 225–228. [Google Scholar] [CrossRef]
- Paskevicius, M.; Pitt, M.P.; Webb, C.J.; Sheppard, D.A.; Filso, U.; Gray, E.M.; Buckley, C.E. In-Situ X-ray Diffraction Study of γ-Mg(BH4)2 Decomposition. J Phys. Chem. C 2012, 116, 15231–15240. [Google Scholar] [CrossRef]
- Guo, S.; Chan, H.Y.L.; Reed, D.; Book, D. Investigation of dehydrogenation processes in disordered γ-Mg(BH4)2. J. Alloys Compd. 2013, 580, S296–S300. [Google Scholar] [CrossRef]
- Zavorotynska, O.; Saldan, I.; Hino, S.; Humphries, T.D.; Deledda, S.; Hauback, B.C. Hydrogen cycling in γ-Mg(BH4)2 with cobalt-based additives. J. Mater. Chem. A 2015, 3, 6592–6602. [Google Scholar] [CrossRef]
- Chong, M.; Karkamkar, A.; Autrey, T.; Orimo, S.-I.; Jalisatgi, S.; Jensen, C.M. Reversible dehydrogenation of magnesium borohydride to magnesium triborane in the solid state under moderate conditions. Chem. Commun. 2011, 47, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- Zavorotynska, O.; Deledda, S.; Li, G.; Matsuo, M.; Orimo, S.-I.; Hauback, B.C. Isotopic Exchange in Porous and Dense Magnesium Borohydride. Angew. Chem. Int. Ed. 2015. [Google Scholar] [CrossRef]
- Brown, W.G.; Kaplan, L.; Wilzbach, K.E. The exchange of hydrogen gas with lithium and sodium borohydrides. J. Am. Chem. Soc. 1952, 74, 1343–1344. [Google Scholar] [CrossRef]
- Gremaud, R.; Lodziana, Z.; Hug, P.; Willenberg, B.; Racu, A.-M.; Schoenes, J.; Ramirez-Cuesta, A.J.; Clark, S.J.; Refson, K.; Züttel, A.; et al. Evidence for hydrogen transport in deuterated LiBH4 from low-temperature Raman-scattering measurements and first-principle calculations. Phys. Rev. B 2009, 80, 1–4. [Google Scholar] [CrossRef]
- Mesmer, R.E.; Jolly, W.L. The Exchange of Deuterium with Solid Potassium Hydroborate. J. Am. Chem. Soc. 1962, 84, 2039–2042. [Google Scholar] [CrossRef]
- Van Setten, M.J.; Lohstroh, W.; Fichtner, M. A new phase in the decomposition of Mg(BH4)2: First-principles simulated annealing. J. Mater. Chem. 2009, 19, 7081–7087. [Google Scholar] [CrossRef]
- Bogdanovic, B.; Schwickardi, M. Ti-doped alkali metal aluminium hydrides as potential novel reversible hydrogen storage materials. J. Alloys Compd. 1997, 253–254, 1–9. [Google Scholar] [CrossRef]
- Mao, J.F.; Yu, X.B.; Guo, Z.P.; Liu, H.K.; Wu, Z.; Ni, J. Enhanced hydrogen storage performances of NaBH4-MgH2 system. J. Alloys Compd. 2009, 479, 619–623. [Google Scholar] [CrossRef]
- Zhang, B.J.; Liu, B.H. Hydrogen desorption from LiBH4 destabilized by chlorides of transition metal Fe, Co, and Ni. Int. J. Hydrog. Energy 2010, 35, 7288–7294. [Google Scholar] [CrossRef]
- Wahab, M.A.; Jia, Y.; Yang, D.; Zhao, H.; Yao, X. Enhanced hydrogen desorption from Mg(BH4)2 by combining nanoconfinement and a Ni catalyst. J. Mater. Chem. A 2013, 1, 3471–3478. [Google Scholar] [CrossRef]
- Xu, J.; Qi, Z.; Cao, J.; Meng, R.; Gu, X.; Wang, W.; Chen, Z. Reversible hydrogen desorption from LiBH4 catalyzed by graphene supported Pt nanoparticles. Dalton Trans. 2013, 42, 12926–12933. [Google Scholar] [CrossRef] [PubMed]
- Humphries, T.D.; Kalantzopoulos, G.N.; Llamas-Jansa, I.; Olsen, J.E.; Hauback, B.C. Reversible Hydrogenation Studies of NaBH4 Milled with Ni-Containing Additives. J. Phys. Chem. C 2013, 117, 6060–6065. [Google Scholar] [CrossRef] [Green Version]
- Au, Y.S.; Yan, Y.; de Jong, K.P.; Remhof, A.; de Jongh, P.E. Pore Confined Synthesis of Magnesium Boron Hydride Nanoparticles. J. Phys. Chem. C 2014, 118, 20832–20839. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Wang, H.; Liu, J.W.; Zhu, M. Thermal decomposition behaviors of magnesium borohydride doped with metal fluoride additives. Thermochim. Acta 2013, 560, 82–88. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Pang, Y.; Gao, M.; Pan, H. Role of Co3O4 in improving the hydrogen storage properties of a LiBH4-2LiNH2 composite. J. Mater. Chem. A 2014, 2, 11155–11161. [Google Scholar] [CrossRef]
- Saldan, I.; Hino, S.; Humphries, T.D.; Zavorotynska, O.; Chong, M.; Jensen, C.M.; Deledda, S.; Hauback, B.C. Structural changes observed during the reversible hydrogenation of Mg(BH4)2 with Ni-based additives. J. Phys. Chem. C 2014, 118, 23376–23384. [Google Scholar] [CrossRef]
- Ngene, P.; Verkuijlen, M.H.W.; Zheng, Q.; Kragten, J.; van Bentum, P.J.M.; Bitter, J.H.; de Jongh, P.E. The role of Ni in increasing the reversibility of the hydrogen release from nanoconfined LiBH4. Faraday Discuss. 2011, 151, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ban, V.; Soloninin, A.V.; Skripov, A.V.; Hadermann, J.; Abakumov, A.; Filinchuk, Y. Pressure-Collapsed Amorphous Mg(BH4)2: An Ultradense Complex Hydride Showing a Reversible Transition to the Porous Framework. J. Phys. Chem. C 2014, 118, 23402–23408. [Google Scholar] [CrossRef]
- David, W.I.F.; Callear, S.K.; Jones, M.O.; Aeberhard, P.C.; Culligan, S.D.; Pohl, A.H.; Johnson, S.R.; Ryan, K.R.; Parker, J.E.; Edwards, P.P.; et al. The structure, thermal properties and phase transformations of the cubic polymorph of magnesium tetrahydroborate. Phys. Chem. Chem. Phys. 2012, 14, 11800–11807. [Google Scholar] [CrossRef] [PubMed]
- Giannasi, A.; Colognesi, D.; Ulivi, L.; Zoppi, M.; Ramirez-Cuesta, A.J.; Bardaji, E.G.; Roehm, E.; Fichtner, M. High Resolution Raman and Neutron Investigation of Mg(BH4)2 in an Extensive Temperature Range. J. Phys. Chem. A 2010, 114, 2788–2793. [Google Scholar] [CrossRef] [PubMed]
- Shimanouchi, T.; Nakagawa, I. Infrared spectroscopic study on the co-ordination bond-I: Infrared spectra of cobalt hexammine, pentammine and trans-tetraammine complexes. Spectrochim. Acta 1962, 18, 89–100. [Google Scholar] [CrossRef]
- Tomkinson, J.; Ludman, C.J.; Waddington, T.C. IR Raman and Inelastic Neutron-Scattering Spectra of Cesium Octahydroborate, CSB3H8. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1979, 35, 117–122. [Google Scholar] [CrossRef]
- Newton, M.A.; van Beek, W. Combining synchrotron-based X-ray techniques with vibrational spectroscopies for the in situ study of heterogeneous catalysts: A view from a bridge. Chem. Soc. Rev. 2010, 39, 4845–4863. [Google Scholar] [CrossRef] [PubMed]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Rehr, J.J.; Albers, R.C. Theoretical approaches to x-ray absorption fine structure. Rev. Mod. Phys. 2000, 72, 621–654. [Google Scholar] [CrossRef]
- Vitillo, J.G.; Bordiga, S.; Baricco, M. Spectroscopic and structural characterization of thermal decomposition of γ-Mg(BH4)2: Dynamic vacuum vs. H2 atmosphere. J. Phys. Chem. C 2015, in press. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavorotynska, O.; Deledda, S.; Vitillo, J.G.; Saldan, I.; Guzik, M.N.; Baricco, M.; Walmsley, J.C.; Muller, J.; Hauback, B.C. Combined X-ray and Raman Studies on the Effect of Cobalt Additives on the Decomposition of Magnesium Borohydride. Energies 2015, 8, 9173-9190. https://doi.org/10.3390/en8099173
Zavorotynska O, Deledda S, Vitillo JG, Saldan I, Guzik MN, Baricco M, Walmsley JC, Muller J, Hauback BC. Combined X-ray and Raman Studies on the Effect of Cobalt Additives on the Decomposition of Magnesium Borohydride. Energies. 2015; 8(9):9173-9190. https://doi.org/10.3390/en8099173
Chicago/Turabian StyleZavorotynska, Olena, Stefano Deledda, Jenny G. Vitillo, Ivan Saldan, Matylda N. Guzik, Marcello Baricco, John C. Walmsley, Jiri Muller, and Bjørn C. Hauback. 2015. "Combined X-ray and Raman Studies on the Effect of Cobalt Additives on the Decomposition of Magnesium Borohydride" Energies 8, no. 9: 9173-9190. https://doi.org/10.3390/en8099173