Textural Characterization and Energetics of Porous Solids by Adsorption Calorimetry



Abstract
:1. Introduction
Thermodynamics of Adsorption—Theoretical Considerations [8]
- P = pressure of the adsorptive
- ∆H = enthalpy of the adsorptive, kJ mol−1
- T = temperature, K
- R = the gas constant ¼ 8.314 J mol−1 K−1
- S = entropy, kJ mol−1 K−1
- ns= number of moles adsorbed, mol g−1
- qst = isosteric heat of adsorption (referenced to the adsorptive gas phase), kJ mol−1
- Qi = integral heat of adsorption, kJ
- gi = calorimetric heat measured, Jg−1
- X = moles of an specie of gas
- Es = internal energy of the adsorbent, kJ
- Eg = internal energy of the adsorbate, kJ
- E = internal energy, kJ
- qd =differential heat of adsorption, kJ mol−1
- A = total surface area, m2 g−1
- nd = corresponding number of moles
- Hg = the enthalpy of the adsorptive (in the gas phase), kJ mol−1
- = internal energy parcial molar of solid
- μg = chemical potential of the gas phase
- μs = chemical potential of the solid phase
- Г = moles of gas adsorbed area
- Vg = gas adsorbed in volume
- Ss = entropy of the solid phase
- π = pressure of the adsorbed layer
- = mass
2. Results and Discussion
2.1. Isotherms of Nitrogen at 77 K
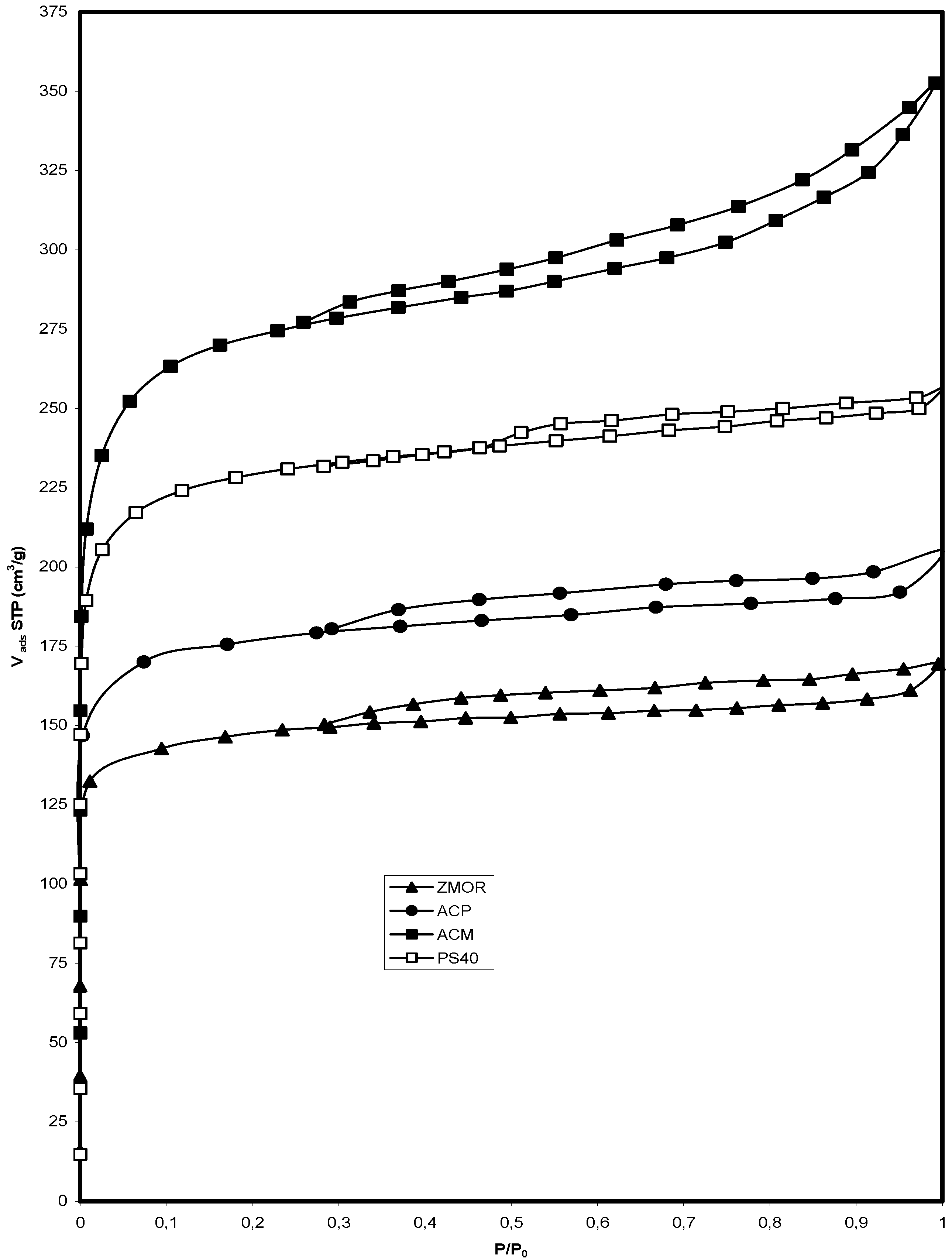

{kind=link}
{kind=link}
{kind=link}
{kind=link}
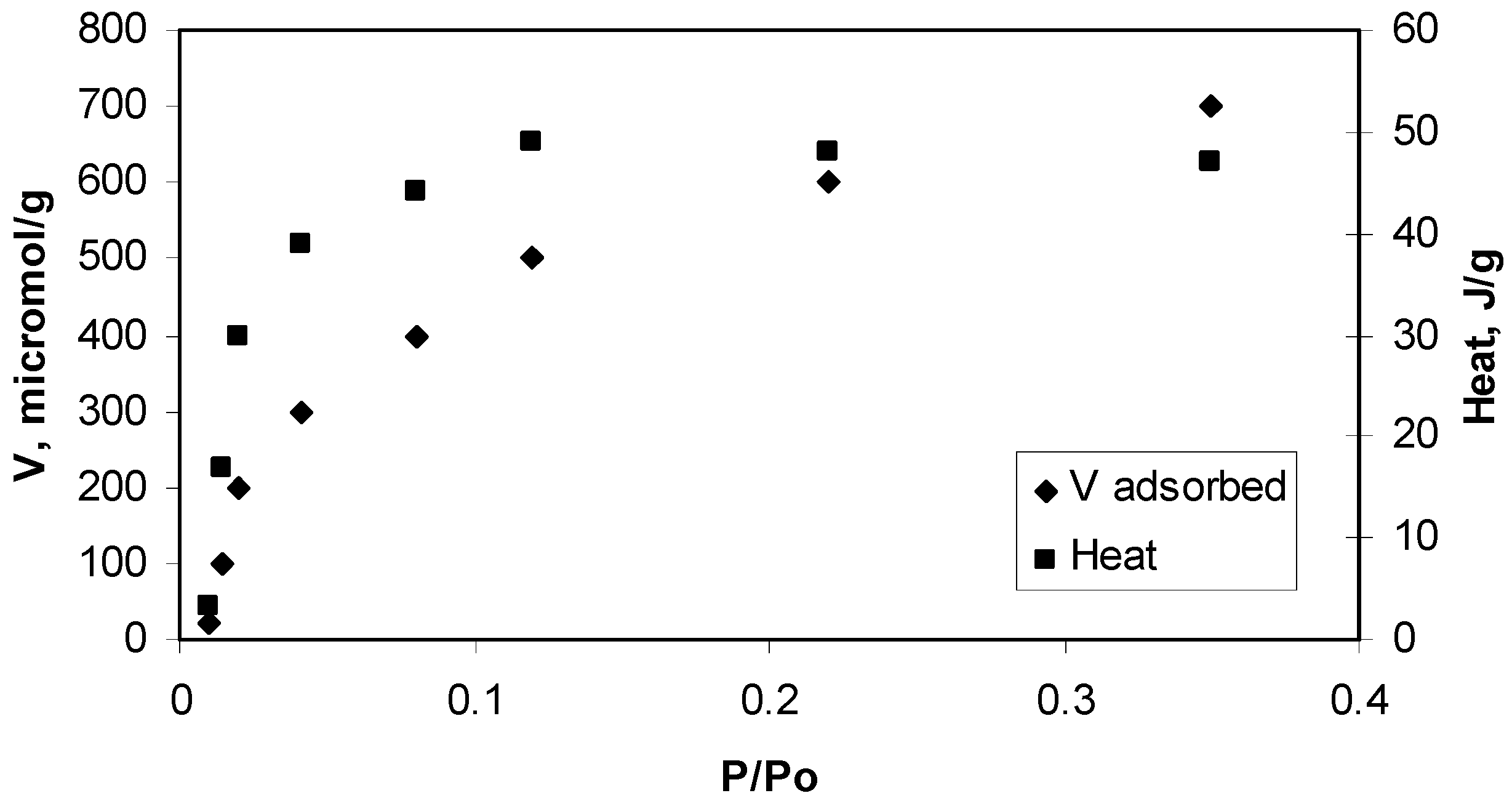{kind=link}
{kind=link}
| Sample | SBET (m2/g) | Smic (m2/g) | Sext (m2/g) | Vmic (cm3/g) | Vmes (cm3/g) | Vtot (cm3/g) | Dp (nm) |
|---|---|---|---|---|---|---|---|
| ZMOR | 510 | 523 | 3 | 0.23 | 0.04 | 0.27 | 2.50 |
| ACP | 810 | 790 | 8 | 0.28 | 0.08 | 0.36 | 1.45 |
| PS40 | 958 | 880 | 11 | 0.36 | 0.06 | 0.42 | 1.02 |
| ACM | 1078 | 890 | 23 | 0.44 | 0.09 | 0.53 | 0.94 |
2.2. Analyze of Chemical Superficial
| Samples | Total Acidic Groups Amount (mmol/g) | pH NH3 |
|---|---|---|
| ZMOR | 0.567 | 9.50 |
| ACP | 1.345 | 3.77 |
| PS40 | 1.998 | 3.66 |
| ACM | 3.124 | 3.34 |
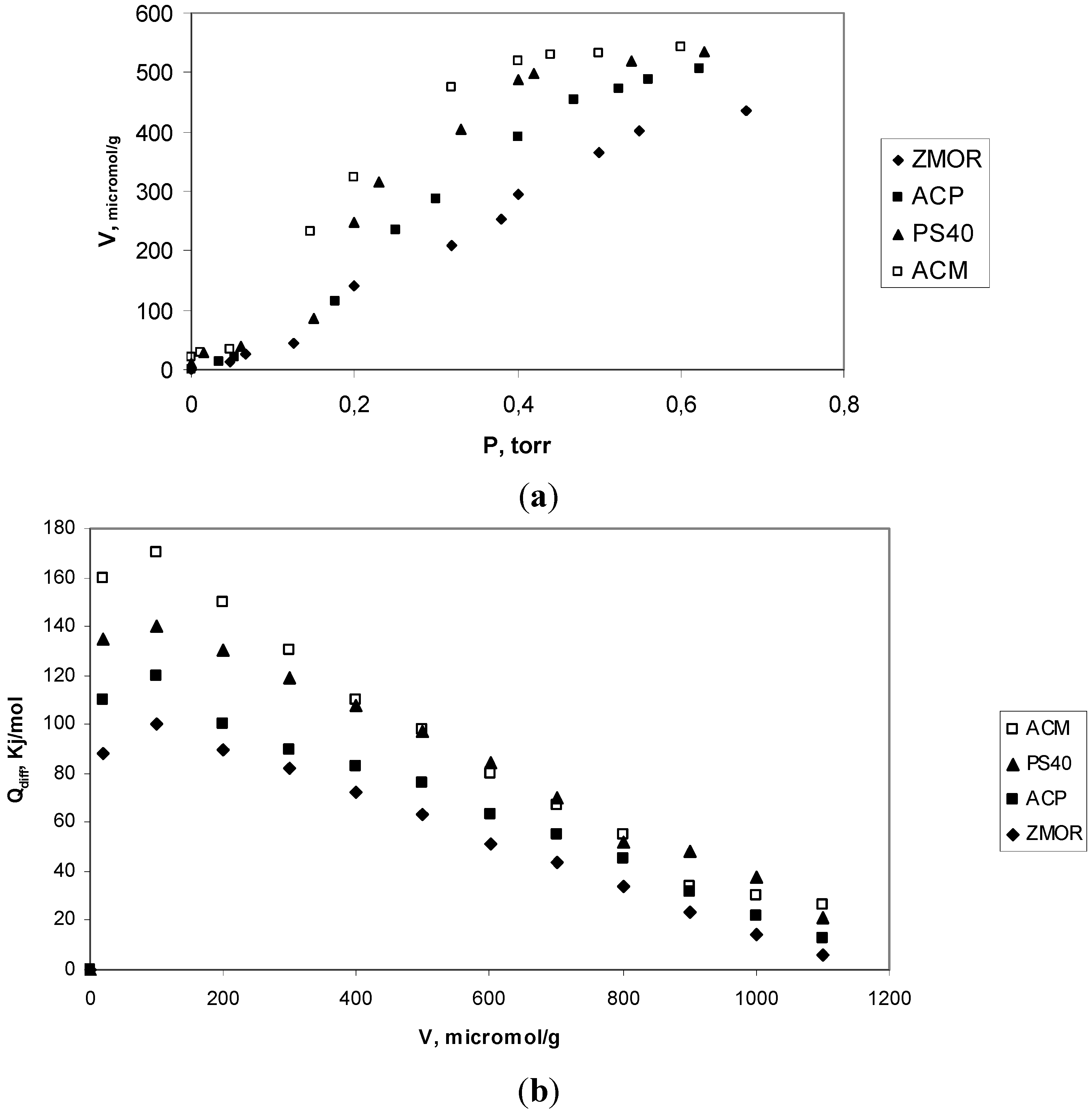
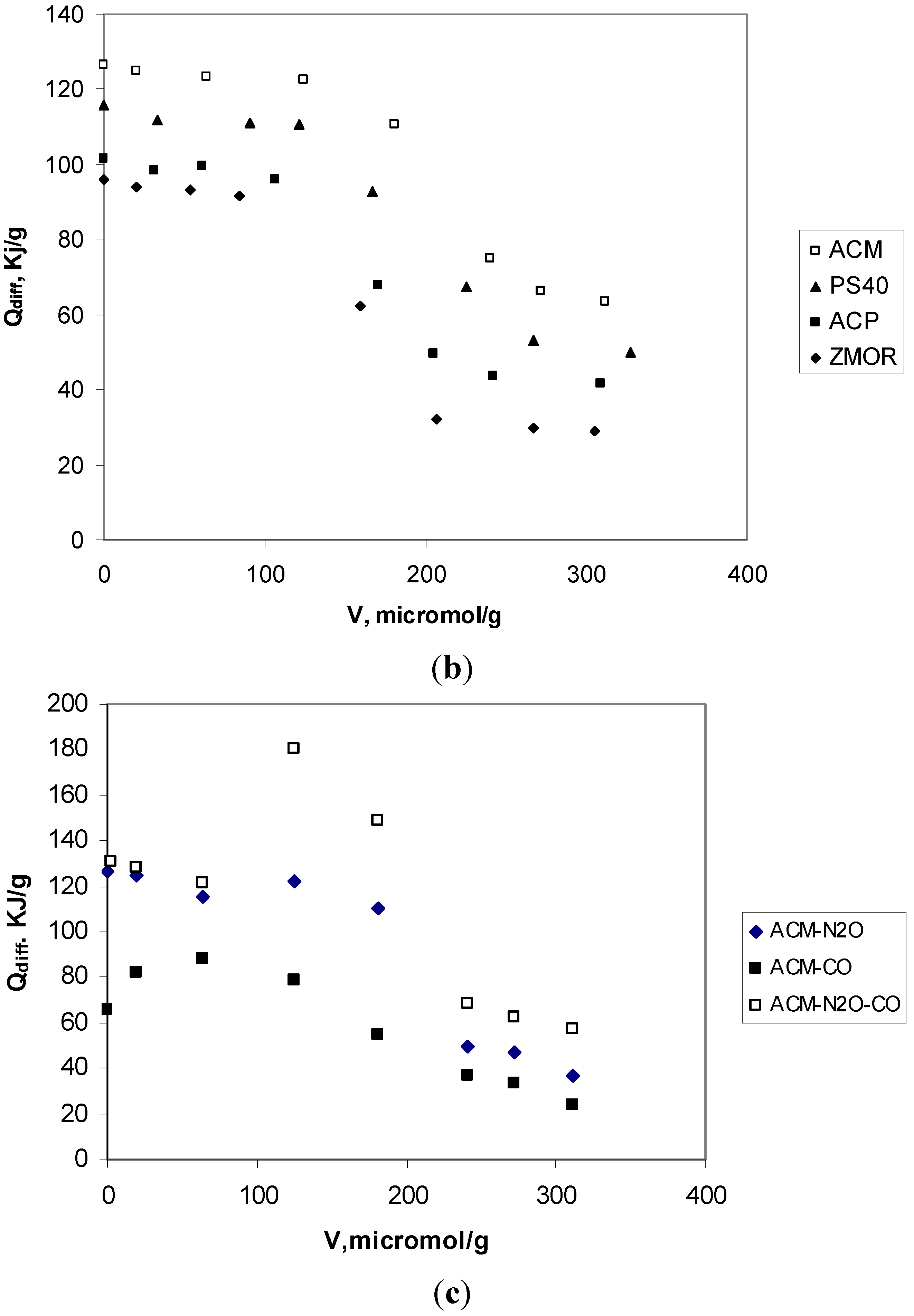
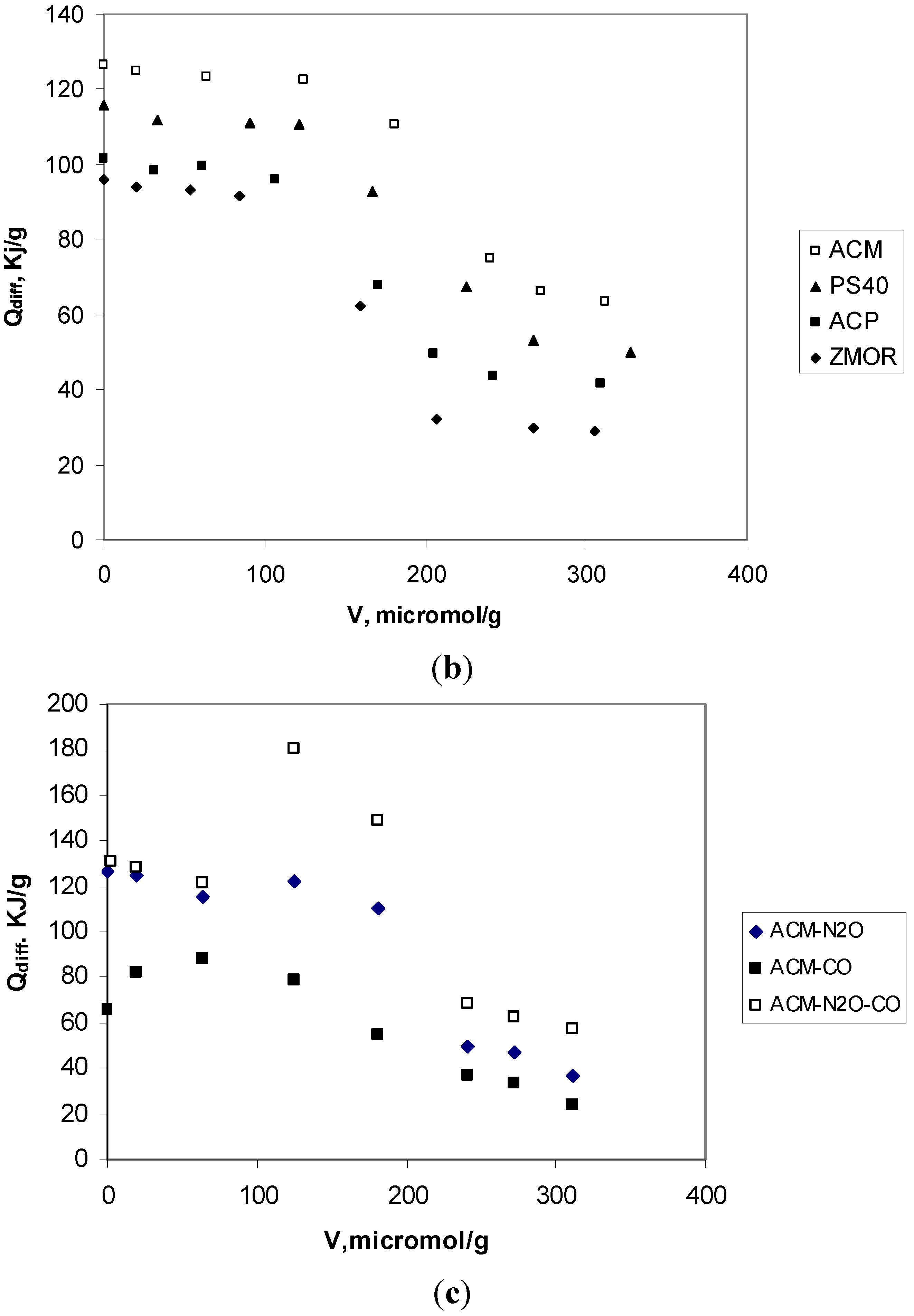
2.3. Analyze of Results Adsorption Calorimetry of Gases
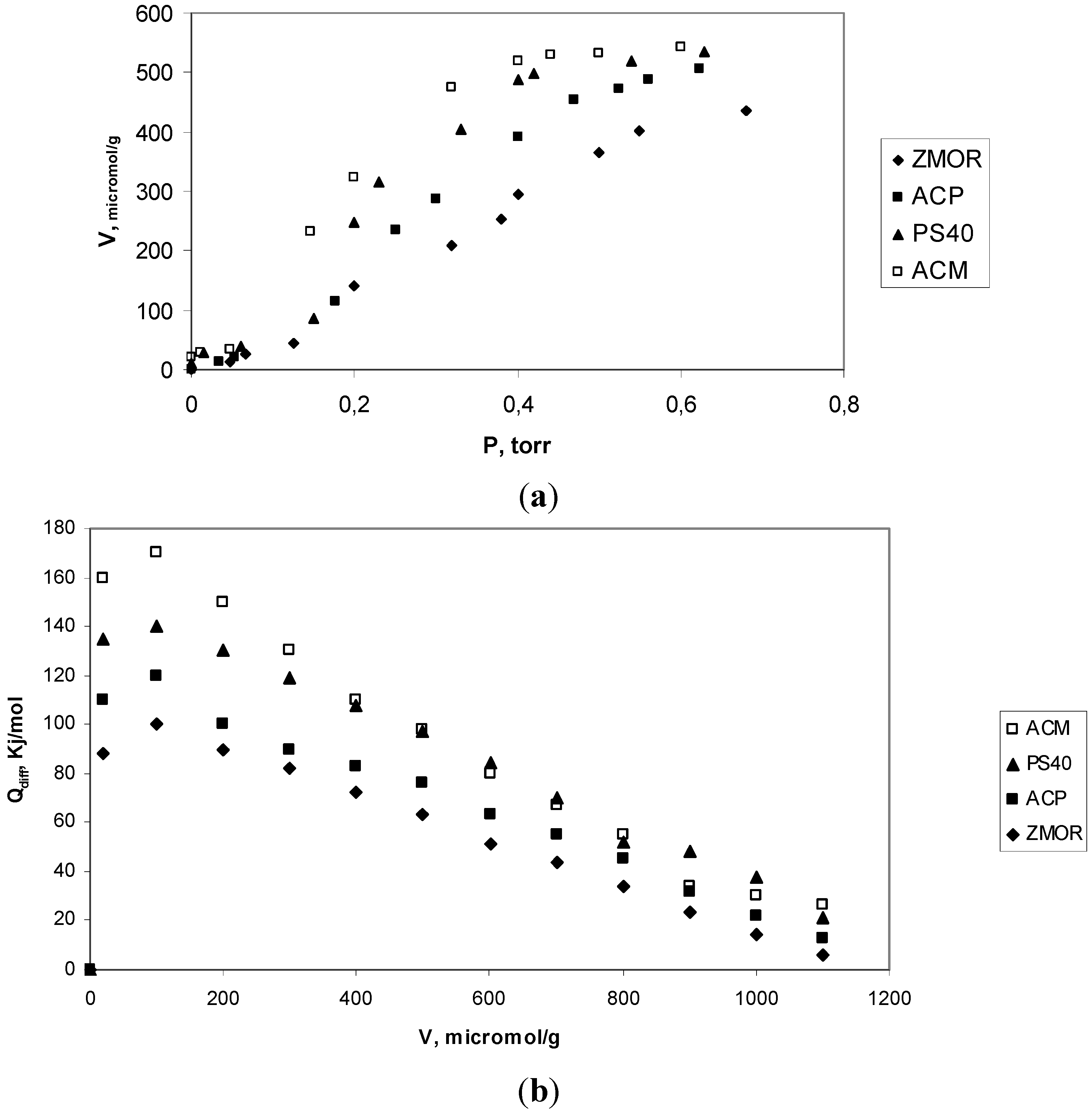
| Sample | Virr (μmol H3/g) | NH3 molecules/group cacarbox | Qint (J/g solid) | Qdiff (kJ/mol) |
|---|---|---|---|---|
| ZMOR | 443 | 0.6 | 54 | 88 |
| ACP | 505.8 | 3.1 ≈ 3 | 94 | 110 |
| PS40 | 510.5 | 2.1 ≈ 2 | 116 | 135 |
| ACM | 534.5 | 1.2 ≈ 1 | 145 | 160 |

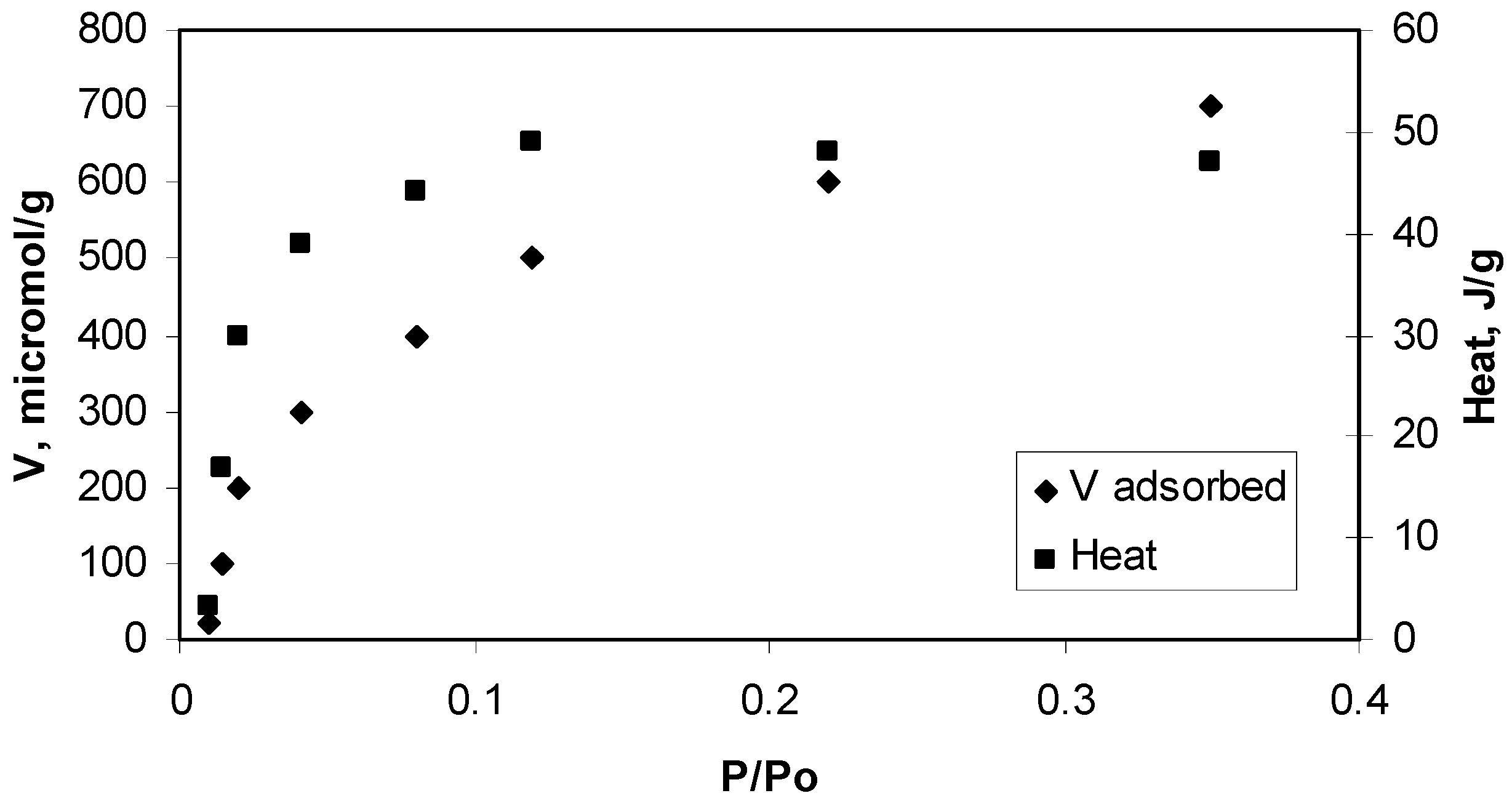
3. Experimental Section
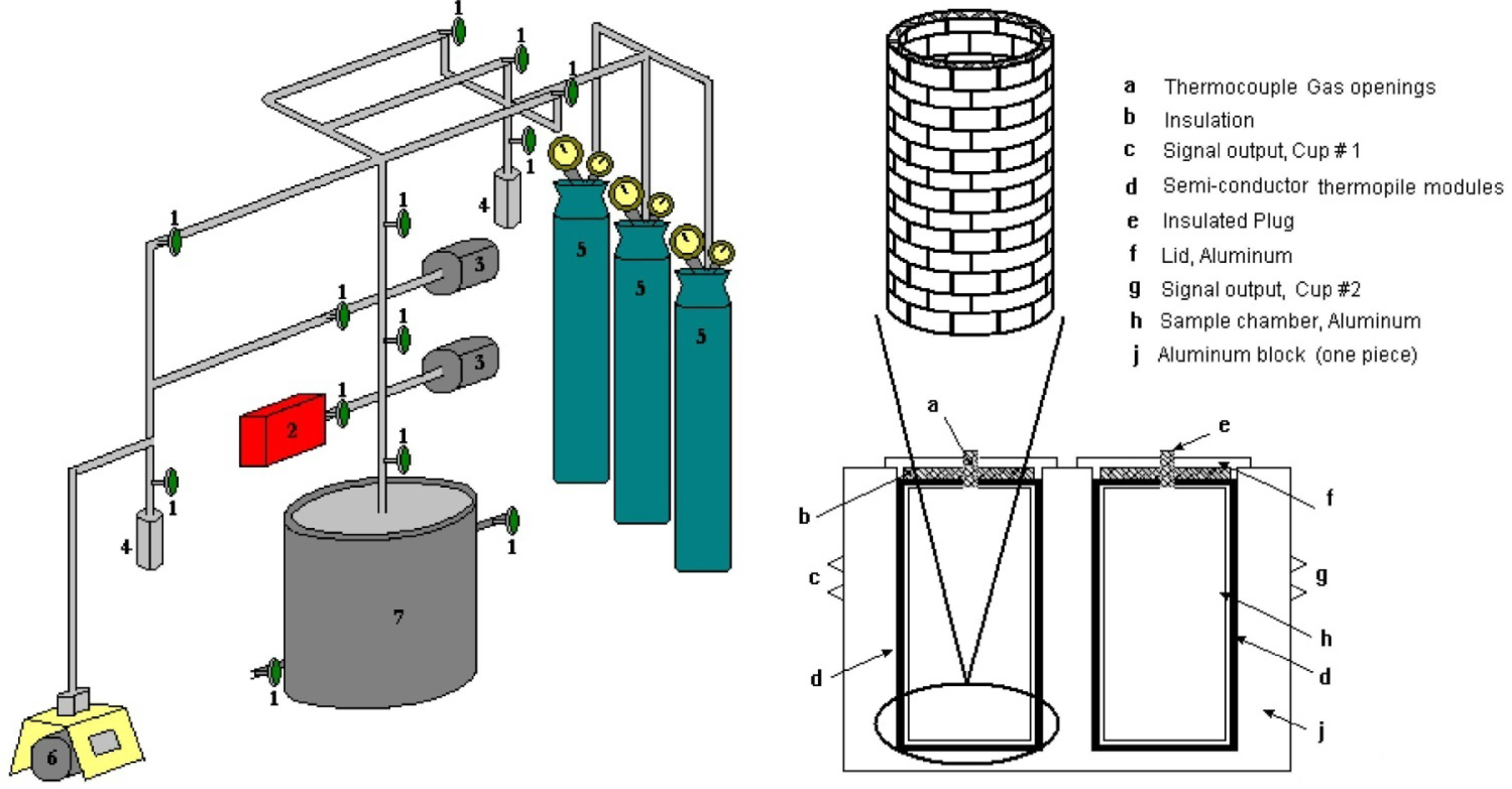
3.1. Description of the Newly Built Microcalorimeter

Design of the Calorimetric Cell
3.2. Samples Used
3.2.1. Preparation of the Carbon Monolith
3.2.2. Granular Activated Carbon
3.2.3. Activated carbon pellets
3.2.4. Zeolite
3.3. Pore Structure Characterization
3.4. Chemical Characterization
3.5. Determination of Adsorption Heats
4. Conclusions
Acknowledgements
References
- Gravelle, P.C. Calorimetry in adsorption and heterogeneous. Catal. Rev. Sci. Eng. 1977, 16, 37–110. [Google Scholar] [CrossRef]
- Sircar, R.; Cao, D.V. Heat of adsorption. Chem. Eng. Technol. 2002, 25, 945–949. [Google Scholar] [CrossRef]
- Auroux, A.; Vedrine, J.C.; Gravelle, P.C. Differential molar heats of adsorption of ammonia on silicious mordenites at high temperature. In Adsorption at the Gas-Solid and Liquid-Solid Interface; Rouquerol, J., Sing, K.S.W., Eds.; Elsevier Science Publishers: Amsterdam, The Netherlands, 1982; pp. 305–322. [Google Scholar]
- Prinsloo, J.J.; Gravelle, P.C. Volumetric and calorimetric study of the adsorption of hydrogen, at 296 K, on silica-supported nickel and nickel-copper catalysts. J. Chem. Soc. Faraday Trans. 1980, 76, 2221–2228. [Google Scholar] [CrossRef]
- Auroux, A.; Gravelle, P.C. Comparative study of the bond energy of oxygen at the surface of supported silver catalysts and of the activity of these catalysis for ethylene epoxidation. Thermochim. Acta 1981, 47, 333–342. [Google Scholar]
- Gravelle, P.C.; Teichner, S.J. Carbon monoxide oxidation and related reactions on a highly divided nickel oxide. Advan. Catal. 1969, 20, 167–266. [Google Scholar]
- Gravelle, P.C. Netsu, Ondo Sokutei to Netsu Bunseki; Kagaku Gijitsu-Sha Pub: Tokyo, Japan, 1980; pp. 21–42. [Google Scholar]
- Adamson, A.W. Physical Chemistry of Surface; Interscience Publishers: London, UK, 1960. [Google Scholar]
- Brunauer, S. The Adsorption of Gases and Vapors, Vol I; Princeton University Press: Princeton, NJ, USA, 1945. [Google Scholar]
- Hill, T.L. statistical mechanics of adsorption V. thermodynamics and heat of adsorption. J. Chem. Phys. 1949, 17, 520–536. [Google Scholar] [CrossRef]
- Hill, T.L. Statistical mechanics of adsorption IX. Adsorption thermodynamics and solution thermodynamics. J. Chem. Phys. 1950, 18, 246–257. [Google Scholar] [CrossRef]
- Everett, D.H. The thermodynamics of adsorption. Part III—Analysis and discussion of experimental data. Trans. Faraday Soc. 1957, 46, 957–973. [Google Scholar] [CrossRef]
- Hill, T.L.; Emmett, P.H.; Joyner, L.G. calculation of thermodynamic functions of adsorbed molecules from adsorption isotherm measurements: Nitrogen on graphon. J. Am. Chem. Soc. 1951, 73, 5102–5110. [Google Scholar] [CrossRef]
- Rouquerol, F.; Rouquerol, J.; Sing, K.S.W. Adsorption by Powders and Porous Solids; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Rouquerol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.M.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the characterization of porous solids (Technical Report). Pure Appl. Chem. 1994, 66, 1739–1758. [Google Scholar]
- Do, D.D. Adsorption Analysis: Equilibria and Kinetics; Imperial College Press: Singapore, 1998. [Google Scholar]
- Moreno, J.C.; Giraldo, L. Setups for simultaneous measurement of isotherms and adsorption heats. Rev. Sci. Instr. 2005, 76, 1–8. [Google Scholar] [CrossRef]
- Huertemendia, M.; Giraldo, L.; Parra, D.; Moreno, J.C. Adsorption microcalorimeter and its software: Design for the establishment of parameters corresponding to different models of adsorption isotherms. Instrum. Sci. Technol. 2005, 33, 645–660. [Google Scholar] [CrossRef]
- Steckler, D.K.; Goldberg, R.N.; Ferrari, Y.B.; Buckley, T.J. High precision microcalorimetry: Apparatus, procedures, and biochemical applications. J. Res. Nat. Bur. Stand. 1986, 91, 113–121. [Google Scholar] [CrossRef]
- Gunn, S.R. comparison standards for solution calorimetry. J. Am. Chem. Soc. 1965, 69, 2902–2913. [Google Scholar]
- Russell, D.J.; Thomas, D.; Hansen, L.D. Batch calorimetry with solids, liquids and gases in less than 1 mL total volume. Thermochim. Acta 2006, 446, 161–167. [Google Scholar] [CrossRef]
- Lerchner, J.; Wolf, G.; Auguet, C.; Torra, V. Accuracy in integrated circuit (IC) calorimeters. Thermochim. Acta 2002, 382, 65–76. [Google Scholar]
- Lerchner, J.; Wolf, A.; Wolf, G.; Fernandez, I. Chip calorimeters for the investigation of liquid phase reactions: Design rules. Thermochim. Acta 2006, 446, 168–175. [Google Scholar] [CrossRef]
- Moreno, J.C.; Giraldo, L. influence of thermal insulation of the surroundings on the response of the output electric signal in a heat conduction calorimetric unit. Instrum. Sci. Technol. 2005, 33, 415–425. [Google Scholar] [CrossRef]
- Garrone, E.; Ghiotti, G.; Giamello, E.; Fubini, B. Entropy of adsorption by microcalorimetry. Part 1. J. Chem. Soc. Faraday Trans. 1 1981, 77, 2613–2620. [Google Scholar] [CrossRef]
- Giraldo, L.; Huertas, J.I.; Valencia, A.; Moreno, J.C. A Heat conduction microcalorimeter for the determination of the immersion heats of activated carbon into phenol aqueous solutions. Instrum. Sci. Technol. 2003, 31, 385–397. [Google Scholar] [CrossRef]
- Yff, B.T.S.; Royall, P.G.; Brown, M.B.; Martin, G.P. An investigation of calibration methods for solution calorimetry. Int. J. Pharm. 2004, 269, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Barata-Rodríguez, P.M.; Mays, T.J.; Moggridge, G.D. Structured carbon adsorbents from clay, zeolite and mesoporous aluminosilicate templates. Carbon 2003, 41, 2231–2246. [Google Scholar] [CrossRef]
- Garcia-Cuello, V.S.; Moreno-Piraján, J.C.; Giraldo-Gutierréz, L.; Sapag, K.; Zgrablich, G. A new microcalorimeter of adsorption for the determination of differential enthalpies. Microporous Mesoporous Mater. 2009, 120, 239–245. [Google Scholar] [CrossRef]
- Garcia-Cuello, V.S.; Moreno-Piraján, J.C.; Giraldo-Gutierréz, L.; Sapag, K.; Zgrablich, G. Adsorption microcalorimeter design and electric calibration. J. Therm. Anal. Calorim. 2009, 97, 711–715. [Google Scholar] [CrossRef]
- Baquero, M.C.; Giraldo, L.; Moreno, J.C.; Suarez-Garcia, F.; Martinez-Alonso, A.; Tascon, J.M.D. Activated carbons by pyrolysis of coffee bean husk in presence of phosphoric acid. J. Anal. Appl. Pyrol. 2003, 70, 779–784. [Google Scholar] [CrossRef]
- Pudipeddi, M.; Sokoloski, T.D.; Duddu, S.P.; Carstensen, J.T. quantitative characterization of adsorption isotherms using isothermal microcalorimetry. J. Pharm. Sci. 1996, 85, 381–386. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Garcia-Cuello, V.S.; Giraldo, L.; Moreno-Pirajan, J.C. Textural Characterization and Energetics of Porous Solids by Adsorption Calorimetry. Energies 2011, 4, 928-947. https://doi.org/10.3390/en4060928
Garcia-Cuello VS, Giraldo L, Moreno-Pirajan JC. Textural Characterization and Energetics of Porous Solids by Adsorption Calorimetry. Energies. 2011; 4(6):928-947. https://doi.org/10.3390/en4060928
Chicago/Turabian StyleGarcia-Cuello, Vanessa Silenia, Liliana Giraldo, and Juan C. Moreno-Pirajan. 2011. "Textural Characterization and Energetics of Porous Solids by Adsorption Calorimetry" Energies 4, no. 6: 928-947. https://doi.org/10.3390/en4060928
APA StyleGarcia-Cuello, V. S., Giraldo, L., & Moreno-Pirajan, J. C. (2011). Textural Characterization and Energetics of Porous Solids by Adsorption Calorimetry. Energies, 4(6), 928-947. https://doi.org/10.3390/en4060928