
Intensification of Processes for the Production of Ethyl Levulinate Using AlCl3·6H2O


Water Research Institute (IRSA), National Research Council (CNR), via F. de Blasio 5, 70132 Bari, Italy
*
Author to whom correspondence should be addressed.
Energies 2021, 14(5), 1273; https://doi.org/10.3390/en14051273
Submission received: 8 January 2021
/
Revised: 17 February 2021
/
Accepted: 20 February 2021
/
Published: 25 February 2021
(This article belongs to the Special Issue Selected Papers from the “20th CIRIAF National Congress—Sustainable Development and Preservation of Environment and Human Health”)
Abstract
:A process for obtaining ethyl levulinate through the direct esterification of levulinic acid and ethanol using AlCl3·6H2O as a catalyst was investigated. AlCl3·6H2O was very active in promoting the reaction and, the correspondent kinetic and thermodynamic data were determined. The reaction followed a homogeneous second-order reversible reaction model: in the temperature range of 318–348 K, Ea was 56.3 kJ·K−1·mol−1, whereas Keq was in the field 2.37–3.31. The activity of AlCl3·6H2O was comparable to that of conventional mineral acids. Besides, AlCl3·6H2O also induced a separation of phases in which ethyl levulinate resulted mainly (>98 wt%) dissolved into the organic upper layer, well separated by most of the co-formed water, which decanted in the bottom. The catalyst resulted wholly dissolved into the aqueous phase (>95 wt%), allowing at the end of a reaction cycle, complete recovery, and possible reuse for several runs. With the increase of the AlCl3·6H2O content (from 1 to 5 mol%), the reaction proceeded fast, and the phases’ separation improved. Such a behavior eventually results in an intensification of processes of reaction and separation of products and catalyst in a single step. The use of AlCl3·6H2O leads to a significant reduction of energy consumed for the final achievement of ethyl levulinate, and a simplification of line-processes can be achieved.
1. Introduction
Fossil fuels have limitations in terms of availability and environmental impact. New routes and technologies based on renewable feedstocks exploitation have been receiving increasing interest. In this perspective, ethyl levulinate (EL) is considered as a critical target product. EL can be applied as fuel or blended as a fuel additive [1] by improving the resulting fuel’s technical properties. For example, when EL is mixed with conventional fossil diesel fuel, a reduction in engine smoke output is determined [2]. Cloud point (277–278 K), pour point (276–277 K), and cold filter plugging point (276 K) resulted in being also improved [3]. Furthermore, EL is a promising alternative/additive to conventional jet fuels [4]. For these reasons, regarding the economic evaluations, EL production alone had a global market size of $10.5 Million in 2019, which is expected to increase to about $11.8 Million by 2022, with a growth rate of 3.6% [5].
EL could be proficiently produced directly from sugars (simple hexoses or their polymeric form, namely starch and cellulose) through ethanolysis. The direct production of alkyl levulinates from glucose could be achieved using catalysts having both base and acid sites [6] or Lewis and Brønsted acidic functionalities [7]. Methyl levulinate can be efficiently obtained from pure sugars and lignocellulosic biomasses by combining aluminum salts and organic sulfonic acids [8]. EL could also be proficiently produced by combining iron and aluminum salts with sulfuric acids under relatively mild conditions (T > 453 K). These integrated systems are robust enough to be applied effectively on very complex feedstocks, namely food waste [9] or sewage sludge [10].
Although the direct obtainment from renewables (carbohydrates) is possible, the current most promising approach for industrial production of EL is still based on the two-step system. Biomasses were first reacted into an acidic aqueous environment to obtain levulinic acid [11], which can be recovered and converted into EL in a second step with ethanol [12] through direct esterification.
The reaction of direct esterification (Equation (1)), known as Fischer esterification, has always attracted attention and investigations.
![Energies 14 01273 i001]()

Direct esterification is a reaction subjected to severe kinetic constraints. The use of a catalyst (usually acid catalysts) results to be mandatory, since autocatalyzed reaction, through autoprotolysis of the organic acid, results too slow and inadequate for industrial purposes [13]. Mineral acids (sulfuric acid, hydrochloric acid and other heteropolyacids) have high effectiveness and meager cost but they are difficult to recover and reuse efficiently [14]. Nevertheless, separation from the final reacted mixture of spent catalysts brings to co-production of waste (salts) to be disposed of at the end of the process, as the relevant sodium salts. The high reactivity makes them too aggressive versus conventional materials so that reactors and pipelines have to be made in Teflon or inert material by negatively influencing the process’s overall economy. Finally, H2SO4 and HCl introduce several constraints in terms of safety and health in the working environment.
Consequently, most of the efforts have focused on setting up new solutions capable of catalyzing the reaction under heterogeneous conditions (i.e., zeolites, acid resins, molecular sieves and metal oxides [15,16,17,18,19,20]), to favor separation, recoverability and reuse of the catalysts. Anyway, they can be subjected to surface passivation and poisoning. However, even though separation and recovery procedures of exhausted catalysts are easy, their regeneration often consists of high-energy demanding processes or chemical processes that produce new waste.
Very recently, hydrated salts, namely aluminum and iron chlorides and nitrates, have been reported to combine the benefits of homogeneous and heterogeneous catalysis for direct esterification. These catalysts not only resulted very active in promoting the esterification reaction under homogeneous catalysis, but they also induced a favorable separation of the resulting esters by the co-formed water, in two distinguished phases [21,22]. In detail, differently from a conventional acid catalyst, at the end of the process, the hydrated salts were entirely in the aqueous phase, allowing the complete recovery and the direct reuse in a new cycle of reaction [23,24]. They are also less corrosive than HCl and H2SO4, and conventional stainless steel (AISI 316) can be used for reactors and pipelines [25]. Finally, after having been used 4–5 times, in their exhausted form, they can be directly used as coagulants in wastewater treatment plants (WWTPs) for primary sedimentation and/or water clarification [26,27].
In this work, the use of aluminium chloride hexahydrate (AlCl3·6H2O) as a catalyst in the direct esterification of levulinic acid and ethanol was evaluated. The respective kinetic (Ea1, k1, Ea−1 and k−1) and thermodynamic data were determined. Moreover, the effect of reactive conditions (temperature and amount of catalyst) on the conversion of the reactants and on the phases repartition was discussed in detail.
2. Materials and Methods
All reagents used were of analytical grade.
Aluminum chloride hexahydrate (AlCl3·6H2O, 99%) was purchased from Baker (Phillipsburg, NJ, USA). Levulinic acid (CH3C(O)CH2CH2COOH, LA, 99.0%), EL (CH3C(O)CH2CH2COOC2H5, 99.5%), ethanol (C2H5OH, ≥99.9%), sulfuric acid (H2SO4, 98%) and hydrochloric acid (HCl, 37%) were Carlo Erba reagents (Val de Reuil, France). Phenolphthalein (≥99%), potassium dichromate (K2Cr2O7, ≥99%), 0.1 N KOH solution, 0.1 N AgNO3 solution and molecular sieves 3 Å were Sigma-Aldrich (St. Louis, MO, USA) products.
EL, residual ethanol and LA were quantitatively analyzed on a Varian 3800 GC-FID (Varian, Palo Alto, CA, USA), using a DB-FATWAX UI (30 m, 0.25 mm, 0.25 µm) (Agilent Technologies, Santa Clara, CA, USA) and ethylbenzene (C6H5C2H5, ≥99.5% Sigma-Aldrich) as an internal standard. Qualitative analyses were carried out using a Perkin Elmer Clarus 500 gas chromatograph (PerkinElmer, Waltham, MA, USA) equipped with an HP-5MS capillary column (30 m; Ø 0.32 mm; 0.25 µm film) (Agilent Technologies, Santa Clara, CA, USA) coupled with a Clarus 500 spectrometer (GC-MS).
LA content was determined either by gas-chromatography using GC-FID and by titration of the acidity of the samples collected with a 0.1 N KOH solution, using phenolphthalein (≥99%, Sigma-Aldrich) as the indicator.
Aluminum determination was done for the two phases recovered at the end of the reaction using a 7000X ICP-MS instrument (Agilent Technologies, Santa Clara, CA, USA), following the procedure explained in detail in di Bitonto’s work [22].
Chloride analysis was performed by titration with a 0.1 N AgNO3 solution and potassium dichromate as the indicator.
Direct Esterification of LA with Ethanol Using AlCl3·6H2O as a Catalyst
LA (23.2 g, 0.2 mol) and ethanol (8.28 g, 0.18 mol) were initially weighed and introduced into a glass reactor, placed into a thermostatic oil bath (348, 338, 328 and 318 K) and kept under stirring (500 rpm) through a magnetic stirrer. Then, an alcoholic solution of AlCl3·6H2O (0.5 g, 1.5 g or 2.5 g corresponding to 0.002, 0.006 and 0.01 mol in 1 g of ethanol, 0.02 mol) previously prepared, was added to the system, by obtaining the LA:ethanol:catalyst molar ratio required for the test. Two aliquots (250 µL) were up-taken at different reaction times to carry out titration and gas-chromatographic determination of LA and EL content. Once the reaction ended, when 1.5 and 2.5 g of AlCl3·6H2O were used in the reaction, namely 3 and 5% with respect to LA, a bi-phasic system was observed already after the first cycle. The two phases were recovered separately in two different vials by using a glass pipette, weighted and then LA, EL, ethanol, water, aluminum and chloride contents were determined.
After the first run of the reaction using 5% AlCl3·6H2O, the catalyst was recycled by removing the organic upper phase and adding fresh 27.3 g AL and 9.8 g EtOH to the bottom aqueous phase. A homogenous solution was finally obtained (in the recycling test with water removal, 5 g molecular sieves were added and left for 12 h at 298 K without agitation). The limpid solution was then recovered and transferred in the reactor at 348 K for 4 h. The reacted mixture initially homogeneous turn back biphasic as the reaction of direct esterification took place. Analogously, the organic phase was also reprocessed by adding new AlCl3·6H2O into fresh ethanol. Initially, the system was homogeneous, but after the reaction, it became biphasic again.
3. Results
3.1. Determination of Kinetics Parameters of the Reaction
LA was reacted with a stoichiometric amount of ethanol in a closed glass reactor at different temperatures (318, 328, 338 and 348 K) in the presence of AlCl3·6H2O (1 mol % concerning the starting acid) ad without any solvents.
The reaction was monitored for about 40 h, determining the LA and EL contents. The reactive trends are reported in Figure 1.
The experiments were done in triplicate and the variability of the experimental data was within 5% of the represented value (mean values were reported in Figure 1).
An evaluation of the reaction order was also done, verifying the fitting of the data in a second-order model for a homogeneous reaction (Equation (2)) [28,29,30]:
where v is the reaction rate, Ccat the molar concentration of the catalyst, k1 the kinetic constant for the forward reaction, Keq the equilibrium constant and the other terms the molar concentration referred to the equilibrium state. Considering that all the experiments were run using an LA:EtOH ratio of 1, the differential Equation (2) can be solved by introducing the Y function [28,29], as defined in Equation (3).
where Xeq represents the acid conversion at the equilibrium (more precisely, the experimental value of X evaluated at the final reaction time), Xt the acid conversion at time t, [RCOOH]t0 the starting molar concentration of the acid k’ the kinetic rate which includes the catalyst concentration [28]. While Xeq, Xt and [RCOOH]t0 were experimentally determined k’ was obtained by plotting Y vs. t, representing the slope of the linear fitting of the experimental data according to Equation (3) (Figure 2).
The values of k1 are listed in Table 1 together with k−1 (the constant rate of the back reaction) and the Keq calculated using Equation (4). The data in Table 1 suggest that Keq was slightly dependent on the temperature.
Arrhenius and van’t Hoff equations were applied (Equations (5) and (6)) for obtaining more detailed information on the kinetics and thermodynamics of the direct esterification of LA mediated by AlCl3·6H2O.
where T is the absolute temperature, A the pre-exponential factor, Ea the activation energy of the reaction, R the universal gas constant, ΔH° the reaction enthalpy and ΔS° the reaction entropy (Figure 3).
3.2. Effect of Catalyst Amount
When the catalyst concentration was increased from 1 to 5 mol%, a clear improvement in the reaction rate was observed (Figure 4).
In Figure 5, the final mixtures related to the different catalyst’s load were depicted.
When 1% of AlCl3·6H2O was used in catalysis, the reacted mixture appeared as a homogenous system, whereas when AlCl3·6H2O was 3 or 5% with respect to LA a biphasic system was eventually obtained (the arrows indicate the separation of phases). The upper yellowish part is the organic fraction, whereas the uncolored part on the bottom was the aqueous phase.
Reaction mixtures obtained by the use of 3 and 5% AlCl3·6H2O were separated into upper organic and bottom aqueous phases and thoroughly analyzed. Details on their composition were reported in Table 3.
4. Discussion
Thermodynamics and kinetics of direct esterification strongly depend on the reacting acid’s nature, the temperature and the EtOH:acid molar ratio.
The reaction rate and the final equilibrium composition are negatively correlated with the size of the alkyl tail of the carboxylic acid [22,31]. The reaction profile (Figure 1) of the synthesis of EL appears to be coherent to this point, since slower than acetic, propionic and butyric acid conversion carried out under the same conditions [22].
The kinetic profiles in Figure 1 suggest a positive effect of temperature: an increase in operating temperature improved the reaction rate.
The absolute value of Ea1 of this reaction is lower than that one calculated for heterogeneous catalysts (over 70 kJ K−1 mol−1) [32]. At the same time, it is comparable to the value determined by using a conventional homogenous catalyst, namely H2SO4 (54.3 kJ K−1 mol−1) [33]. Regarding the thermodynamics, direct esterification is an endothermic reaction favored by heat and high temperature and the ΔH0 and ΔS0 estimated matched previous determinations for the same reaction [32,33]. FeCl3 was also proved to catalyze the direct esterification, but it is less active than AlCl3·6H2O [21,26]. This reactivity contrasts with pKa values of FeCl3 and AlCl3·6H2O determined in aqueous solutions, but it was perfectly coherent with the strength of acidity of these species in an alcoholic media [21]. This could be explained by the partial substitution of water molecules in the starting hexa-aquo-aluminum complex with alcohols, which produce mixed aqua-alcohol aluminum complexes, more acid than the starting species [25].
The effect of the amount of AlCl3·6H2O on the direct esterification was also evaluated. The importance of the presence of the catalyst is already evident by looking for the data reported in Figure 1, where 1 mol% AlCl3·6H2O was used. Without the use of a catalyst, the reaction progressed very slowly. After 20 h at 348 K, the final molar conversion was 9.8%, while after 200 h, the yield was about 20%. When the catalyst concentration was increased from 1 to 5 mol%, a clear improvement in the reaction rate was evaluated (Figure 4).
At 348 K, using a catalyst concentration of 5 mol%, the reactive system reached the equilibrium composition after only 120 min. When the reaction was carried out with 1 mol% AlCl3·6H2O, it took more than 12 h to get the same yield of EL. In any case, the reaction rate did not follow a proportional increase with respect to the amount of the catalyst. When 3% and 5% AlCl3·6H2O were used instead of 1%, the reaction rate calculated in the first twenty minutes, proceeded faster only by a factor of 2.2 and 3, respectively. This aspect is connected with the concomitant separation of phases. In fact, after 20 min a yield of around 50% was already obtained for a reaction run with 3 and 5% of catalyst, with the solution that already appeared as an emulsion (separation of phases). This separation could justify the slight slowdown of reaction rate with respect to the reaction catalyzed by 1% AlCl3·6H2O, due to a less efficient contact among reactants (mostly dissolved into the organic phase) and the catalyst (dissolved into the aqueous phase) [21].
Figure 5 shows the biphasic systems obtained at the end of the reaction using different catalysts’ content.
When 1 mol% AlCl3·6H2O was used for the catalysis, the final system remained homogeneous, while biphasic system was obtained if further AlCl3·6H2O was added on the reacted mixtures, confirming that the catalyst induces the separation of EL from the water produced by the reaction (Equation (1)). In addition, increasing amounts of AlCl3·6H2O, from 3 to 5 mol% with respect to LA, led to an increase of the bottom phase level. The phases were then isolated in separated vials, weighed and analyzed in terms of LA, EL, water, ethanol, aluminum, and chloride contents. The trend of distribution among the two phases already reported for acetic, propionic and butyric acids [22] was confirmed for the case of LA (Table 3). In detail, when 5 mol% AlCl3·6H2O was used, the organic phase was 83.6 wt% with respect to the overall system. More than 98% of the produced EL was dissolved into the upper organic part, together with most of the unreacted LA (>94%). Unreacted ethanol was also mostly dissolved into the organic upper layer, whereas AlCl3·6H2O (>99%) and water (>90%) were mainly into the uncolored bottom phase. Such a separation resulted in a convenient intensification of processes. Besides the reaction of direct esterification, the separation of products (EL, AL and ethanol from water and catalyst) took place.
The recovery of AlCl3·6H2O from the aqueous phase generated by the direct esterification is a challenging aspect related to the use of this catalytic system. On the contrary, the direct reuse of such a layer in a new cycle of reaction with fresh alcohol and acids was already demonstrated to be effective for the direct esterification of FFAs to produce biodiesel [23]. In this specific case, after a cycle of reaction with 5% AlCl3·6H2O. (EL molar yield of 69.7%), the bottom phase was directly reacted again with fresh LA and ethanol to replenish the initial reactive conditions (AL:EtOH:AlCl3·6H2O = 1:1:0.05). After 4 h at 348 K a yield of 61.9% of EL was obtained, while in the third cycle the final yield was 57.5%. The most crucial reason of the deactivation of the catalyst was the increasing amount of water in the reactive system. Consequently, to maximize the (re)use of the catalyst, the removal of water generated by the reaction plays a crucial role. Different strategies have been already proposed to address this issue. The first option consisted of using a series of reactors fed in countercurrent [24] to allow the water content to be maximized in the reactor, where also the acid content has the highest value. This condition allowed the reaction to take place efficiently in all the three-step. In the alternative, the hydro-alcoholic phase enriched by AlCl3·6H2O was proved to be benefited by preliminary dehydration before being recycled: zeolites, silica and molecular sieves were found effective in this sense [21]. In this specific case, after a cycle of reaction with 5% AlCl3·6H2O. (EL molar yield of 69.7%), the bottom phase was directly reacted again with fresh LA and ethanol to replenish the initial reactive conditions (AL:EtOH:AlCl3·6H2O = 1:1:0.05) again. After 4 h at 348 K, a yield of 61.9% of EL was obtained. In contrast, in the third cycle, the final yield was 57.5%. According to these data, a new scheme of production of EL from LA and ethanol was designed and evaluated, based on three subsequent batch reactors. The organic phase obtained from the first cycle was recovered and again reacted with fresh 5 mol% of AlCl3·6H2O in ethanol at 348 K. This operation was repeated for two times. At the end of the third cycle, a technical grade product could be directly achieved without using any high-energetic post-processing operations, namely distillation, typically adopted to purify ethyl esters from direct esterification [22]. The final organic phase should already contain an EL amount corresponding to a conversion of 90 wt% of the initial LA. This organic phase would be already free of catalyst and water and have only residual unreacted ethanol and LA.
The direct esterification between ethanol and LA promoted by AlCl3·6H2O can efficiently complete the conversion of carbohydrates (glucose, cellulose, lignocellulose, etc.) into EL. The two-step process consisting of (i) the consolidate method through sulfuric acids [34], which allows LA to be obtained from biomasses, followed by (ii) the direct esterification of LA with ethanol in the presence of AlCl3·6H2O could unlock the potential of renewable feedstocks for the industrial production of EL. Not only because the first step is already conducted on an industrial scale [34], but also because the proposed strategy has several advantages if compared with the direct alcoholysis of biomasses [35]. Direct ethanolysis is an alternative route to obtain EL from carbohydrates. It is carried out in the presence of Lewis-Brønsted acid catalysts and occurs at high temperature (160–200 °C) and pressure, using alcohol as a reagent and solvent media. Besides relatively high EL yields (>50%), a considerable amount of alcohol (up to 60% [36]) can be lost as di-alkyl ether. Finally, the direct use of biomasses is possible, only after the total preliminary removal of water and humidity due to the relevant dampening effect [9,10]. Under these constraints, the two-step process looks to have the only drawback of being operated in two separated phases, but on the other hand, this aspect can even represent a point of strength. Considering that, the first step is a thermal treatment performed in an aqueous environment, very simple pretreatments of starting biomass are demanded, and effective desiccation is not essential. Even the authorization practices for the realization of industrial plants can be simplified with respect to alcohol. Capital costs for the equipment are reduced and do not suffer from safety requirements (ATEX rules) typical of processes that use organic solvents at high temperatures and pressure. Finally, the two-step process has a very high atoms’ economy referred to the alcohol, since it is used in stoichiometric amounts to LA and specifically consumed to obtain EL.
All these aspects make the two-step strategy prompt for sustainable industrial production of EL from biomasses.
5. Conclusions
In this work, a novel approach was tested using AlCl3·6H2O as a catalyst in the direct esterification of LA and ethanol. The esterification reaction was studied, collecting the kinetic and thermodynamic data. There is a positive effect of the temperature on kinetic and thermodynamic parameters. Ea1 value was lower than those calculated using heterogeneous catalysts (56.3 kJ K−1 mol−1), confirming a better efficiency of the process, comparable to H2SO4. Differently from conventional mineral acids (HCl, H2SO4, p-toluene-sulfonic acid), AlCl3·6H2O also induced a favorable final separation of EL (recovery > 99%wt) by the co-formed water, in two distinguished phases. With the increase of AlCl3·6H2O (up to 5 mol%), besides improving the kinetics, a more advantageous separation was observed, since most of the water was contained in the lower phase and most of starting catalyst. This repartition opens to possible direct production of EL without needing high-energy demanding purification.
Author Contributions
Conceptualization, C.P.; methodology, C.P.; software, C.P. and V.D.; validation, C.P.; writing—original draft preparation, C.P.; writing—review and editing, C.P. and V.D.; supervision, C.P.; project administration, C.P.; funding acquisition, C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MIUR, grant number Code: PE8-11775-2017FWC3WC_004 PRIN-VISION Project.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dutta, S.; De, S.; Saha, B.; Alam, M.I. Advances in conversion of hemicellulosic biomass to furfural and upgrading to biofuels. Catal. Sci. Technol. 2012, 2, 2025–2036. [Google Scholar] [CrossRef]
- Christensen, E.; Williams, A.; Paul, S.; Burton, S.; McCormick, R.L. Properties and performance of levulinate esters as diesel blend components. Energy Fuels 2011, 25, 5422–5428. [Google Scholar] [CrossRef]
- Joshi, H.; Moser, B.R.; Toler, J.; Smith, W.F.; Walker, T. Ethyl levulinate: A potential bio-based diluent for biodiesel which improves cold flow properties. Biomass Bioenergy 2011, 35, 3262–3266. [Google Scholar] [CrossRef]
- Chuck, C.J.; Donnelly, J. The compatibility of potential bioderived fuels with Jet A-1 aviation kerosene. Appl. Energy 2014, 118, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Ethyl Levulinate Market Size, Share & Trends Analysis Report by Application (Flavors, Fragrance), by Region (North America, Europe, Asia Pacific, Middle East & Africa, Central & South America) and Segment Forecast, 2015–2022. Available online: https://www.grandviewresearch.com/industry-analysis/ethyl-levulinate-market (accessed on 24 February 2021).
- Li, H.; Fang, Z.; Luo, J.; Yang, S. Direct conversion of biomass components to the biofuel methyl levulinate scatalysed by acid-base bifunctional zirconia-zeolites. Appl. Cat. B Environ. 2017, 200, 182–191. [Google Scholar] [CrossRef]
- Peng, L.; Lin, L.; Zhang, J.; Zhuang, J.; Zhang, B.; Gong, Y. Catalytic conversion of cellulose to levulinic acid by metal chlorides. Molecules 2010, 15, 5258–5272. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, K.; Nemoto, K.; Kamimura, Y.; Yamada, A.; Yamamoto, Y.; Sato, K. A practical and efficient synthesis of methyl levulinate from cellulosic biomass catalyzed by an aluminum-based mixed acid catalyst system. RSC Adv. 2016, 6, 65119–65124. [Google Scholar] [CrossRef] [Green Version]
- di Bitonto, L.; Antonopoulou, G.; Braguglia, M.C.; Campanale, C.; Gallipoli, A.; Lyberatos, G.; Ntaikou, I.; Pastore, C. Lewis-Brønsted acid scatalysed ethanolysis of the organic fraction of municipal solid waste for efficient production of biofuels. Bioresour. Technol. 2018, 266, 297–305. [Google Scholar] [CrossRef] [PubMed]
- di Bitonto, L.; Locaputo, V.; D’Ambrosio, V.; Pastore, C. Direct Lewis-Brønsted acid ethanolysis of sewage sludge for production of liquid fuels. Appl. Energy 2020, 259, 114163. [Google Scholar] [CrossRef]
- Hayes, D.J.; Fitzpatrick, S.; Hayes, M.H.B.; Ross, J.R.H. The Biofine process- production of levulinic acid, furfural and formic acid from lignocellulosic feedstocks. Bioref. Ind. Proc. Prod. 2006, 1, 139–164. [Google Scholar]
- Raspolli Galletti, A.M.; Antonetti, C.; De Luise, V.; Licursi, D.; Nassi, N. Levulinic acid from waste. BioResources 2012, 7, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Bankole, K.S. Uncatalysed Esterification of Biomass-Derived Carboxylic Acids. Ph.D. Thesis, University of Iowa, Iowa City, IA, USA, 2011. [Google Scholar]
- de la Iglesia, Ó.; Mallada, R.; Menéndez, M.; Coronas, J. Continuous zeolite membrane reactor for esterification of ethanol and acetic acid. Chem. Eng. J. 2007, 131, 35–39. [Google Scholar] [CrossRef]
- Zainol, M.M.; Amin, N.A.S.; Asmadi, M. Kinetics and thermodynamic analysis of levulinic acid esterification using lignin-furfural carbon cryogel catalyst. Renew. Energy 2019, 130, 547–557. [Google Scholar] [CrossRef]
- Kuwaharaa, Y.; Fujitania, T.; Yamashita, H. Esterification of levulinic acid with ethanol over sulfated mesoporous zirconosilicates: Influences of the preparation conditions on the structural properties and catalytic performances. Catalysis Today 2014, 237, 18–28. [Google Scholar] [CrossRef]
- Pasquale, G.; Vázquez, P.; Romanelli, G.; Baronetti, G. Catalytic upgrading of levulinic acid to ethyl levulinate using reusable silica-included Wells-Dawson heteropolyacid as catalyst. Catal. Commun. 2012, 18, 115–120. [Google Scholar] [CrossRef]
- Yan, K.; Wu, G.; Wen, J.; Chen, A. One-step synthesis of mesoporous H4SiW12O40-SiO2 catalysts for the production of methyl and ethyl levulinate biodiesel. Catal. Commun. 2013, 34, 58–63. [Google Scholar] [CrossRef]
- Unlu, D.; Ilgen, O.; Hilmioglu, N.D. Biodiesel additive ethyl levulinate synthesis by catalytic membrane: SO4−2/ZrO2 loaded hydroxyethyl cellulose. Chem. Eng. J. 2016, 302, 260–268. [Google Scholar] [CrossRef]
- Russo, V.; Hrobar, V.; Mäki-Arvela, P.; Eränen, K.; Sandelin, F.; Di Serio, M.; Salmi, T. Kinetics and Modelling of Levulinic Acid Esterification in Batch and Continuous Reactors. Topics Catal. 2018, 61, 1856–1865. [Google Scholar] [CrossRef]
- di Bitonto, L.; Pastore, C. Metal hydrated-salts as efficient and reusable catalysts for pre-treating waste cooking oils and animal fats for an effective production of biodiesel. Renew. Energy 2019, 143, 1193–1200. [Google Scholar] [CrossRef]
- di Bitonto, L.; Menegatti, S.; Pastore, C. Process intensification for the production of the ethyl esters of volatile fatty acids using aluminium chloride hexahydrate as a catalyst. J. Clean. Product. 2019, 239, 118122. [Google Scholar] [CrossRef] [Green Version]
- Pastore, C.; Barca, E.; Del Moro, G.; Lopez, A.; Mininni, G.; Mascolo, G. Recoverable and reusable aluminium solvated species used as a homogeneous catalyst for biodiesel production from brown grease. Appl. Catal. A Gen. 2015, 501, 48–55. [Google Scholar] [CrossRef]
- di Bitonto, L.; Lopez, A.; Mascolo, G.; Mininni, G.; Pastore, C. Efficient solvent-less separation of lipids from municipal wet sewage scum and their sustainable conversion into biodiesel. Renew. Energy 2016, 90, 55–61. [Google Scholar] [CrossRef]
- Nickel Institute. Available online: https://nickelinstitute.org/media/1662/corrosionresistanceoftheausteniticchromium_nickelstainlesssteelsinchemicalenvironments_2828_.pdf (accessed on 24 February 2021).
- Pastore, C.; Lopez, A.; Mascolo, G. Efficient conversion of brown grease produced by municipal wastewater treatment plant into biofuel using aluminium chloride hexahydrate under very mild conditions. Bioresour. Technol. 2014, 155, 91–97. [Google Scholar] [CrossRef]
- Pastore, C.; Pagano, M.; Lopez, A.; Mininni, G.; Mascolo, G. Fat, oil and grease waste from municipal wastewater: Characterisation, activation and sustainable conversion into biofuel. Water Sci. Technol. 2015, 71, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Banchero, M.; Gozzelino, G. A Simple Pseudo-Homogeneous Reversible Kinetic Model for the Esterification of Different Fatty Acids with Methanol in the Presence of Amberlyst-15. Energies 2018, 11, 1843. [Google Scholar] [CrossRef] [Green Version]
- Mekala, M.; Goli, V.R. Kinetics of esterification of methanol and acetic acid with mineral homogeneous acid catalyst. Chin. J. Chem. Eng. 2015, 23, 100–105. [Google Scholar] [CrossRef]
- Akyalçin, S.; Altıokka, M.R. Kinetics of esterification of acetic acid with 1-octanol in the presence of Amberlyst 36. Appl. Catal. A. Gen. 2012, 429−430, 79–84. [Google Scholar] [CrossRef]
- Liu, Y.; Lotero, E.; Goodwin, J.G., Jr. Effect of carbon chain length on esterification of carboxylic acids with methanol using acid catalysis. J. Catal. 2006, 243, 221–228. [Google Scholar] [CrossRef]
- Bart, H.J.; Reidetschlagerj, J.; Schatkaj, K.; Lehmannt, A. Kinetics of Esterification of Levulinic Acid with n-Butanol by Homogeneous Catalysis. Ind. Eng. Chem. Res. 1994, 33, 21–25. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Niphadkar, P.S.; Deshpande, S.S.; Bokade, V.V. Esterification of renewable levulinic acid to ethyl levulinate biodiesel scatalysed by highly active and reusable desilicated H-ZSM-5. J. Chem. Technol. Biotechnol. 2014, 89, 1507–1515. [Google Scholar] [CrossRef]
- Hayes, D.J.; Fitzpatrick, S.; Hayes, M.H.B.; Ross, J.R.H. The Biofine Process—Production of Levulinic Acid, Furfural, and Formic Acid from Lignocellulosic Feedstocks. In Biorefineries-Industrial Processes and Products; Kamm, B., Gruber, P.R., Kamm, M., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005. [Google Scholar] [CrossRef]
- Raspolli Galletti, A.M.; Antonetti, C.; Fulignati, S.; Licursi, D. Direct Alcoholysis of Carbohydrate Precursors and Real Cellulosic Biomasses to Alkyl Levulinates: A Critical Review. Catalysts 2020, 10, 1221. [Google Scholar] [CrossRef]
- Peng, L.; Lin, L.; Li, H.; Yang, Q. Conversion of carbohydrates biomass into levulinate esters using heterogeneous catalysts. Appl. Energy 2011, 88, 4590–4596. [Google Scholar] [CrossRef]
Figure 1.
Direct esterification of Levulinic Acid (LA) with ethanol evaluated at different times and temperatures. Yields of ethyl levulinate (EL) were plotted vs. time. Reaction conditions: molar ratio ethanol:LA:AlCl3·6H2O = 1:1:0.01, temperatures from 318 to 348 K, time = 40 h.
Figure 1.
Direct esterification of Levulinic Acid (LA) with ethanol evaluated at different times and temperatures. Yields of ethyl levulinate (EL) were plotted vs. time. Reaction conditions: molar ratio ethanol:LA:AlCl3·6H2O = 1:1:0.01, temperatures from 318 to 348 K, time = 40 h.
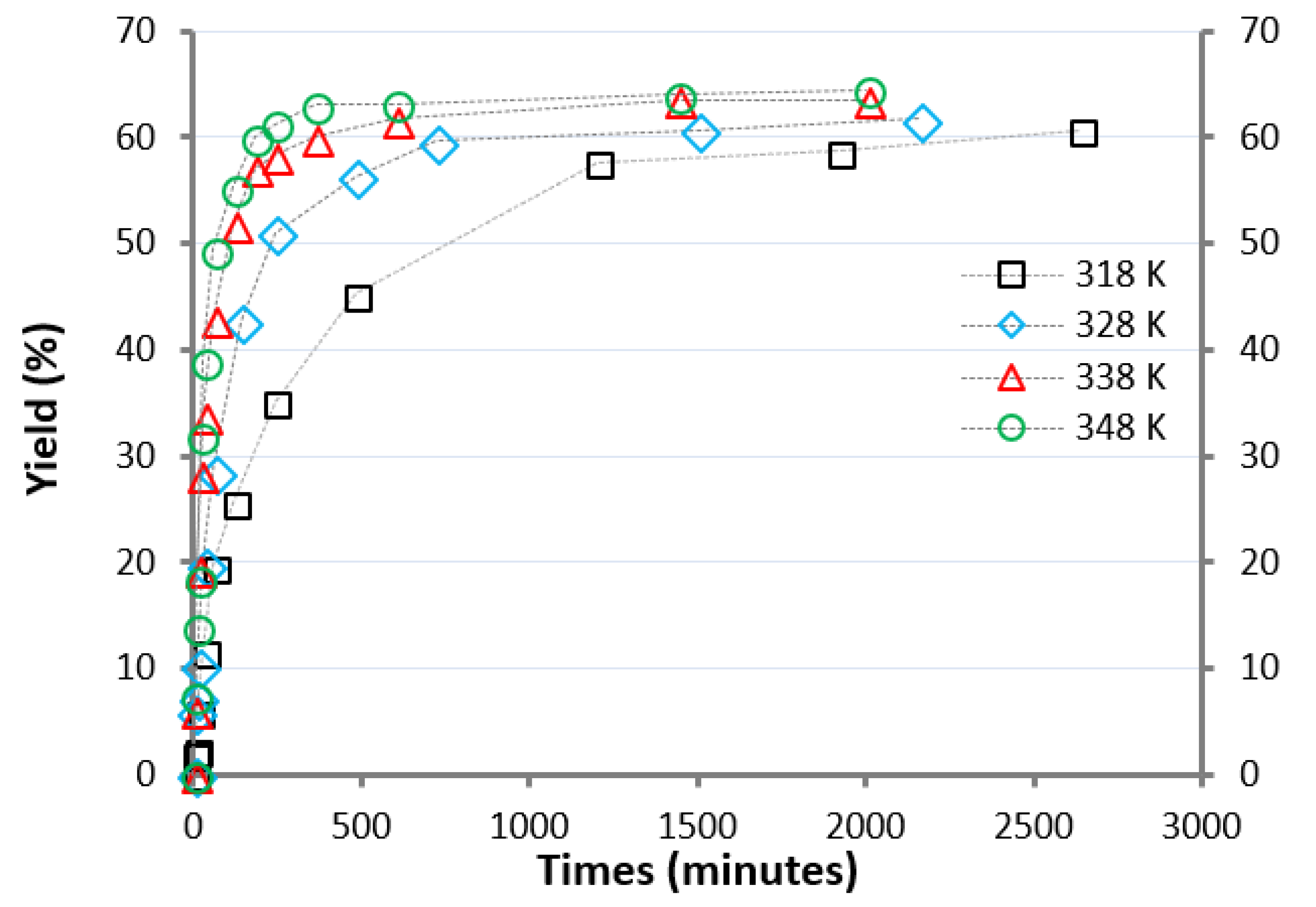
Figure 2.
Evaluation of the kinetic constant at different temperatures (from 318 to 348 K) for the forward reaction (k’) for LA and ethanol.
Figure 2.
Evaluation of the kinetic constant at different temperatures (from 318 to 348 K) for the forward reaction (k’) for LA and ethanol.
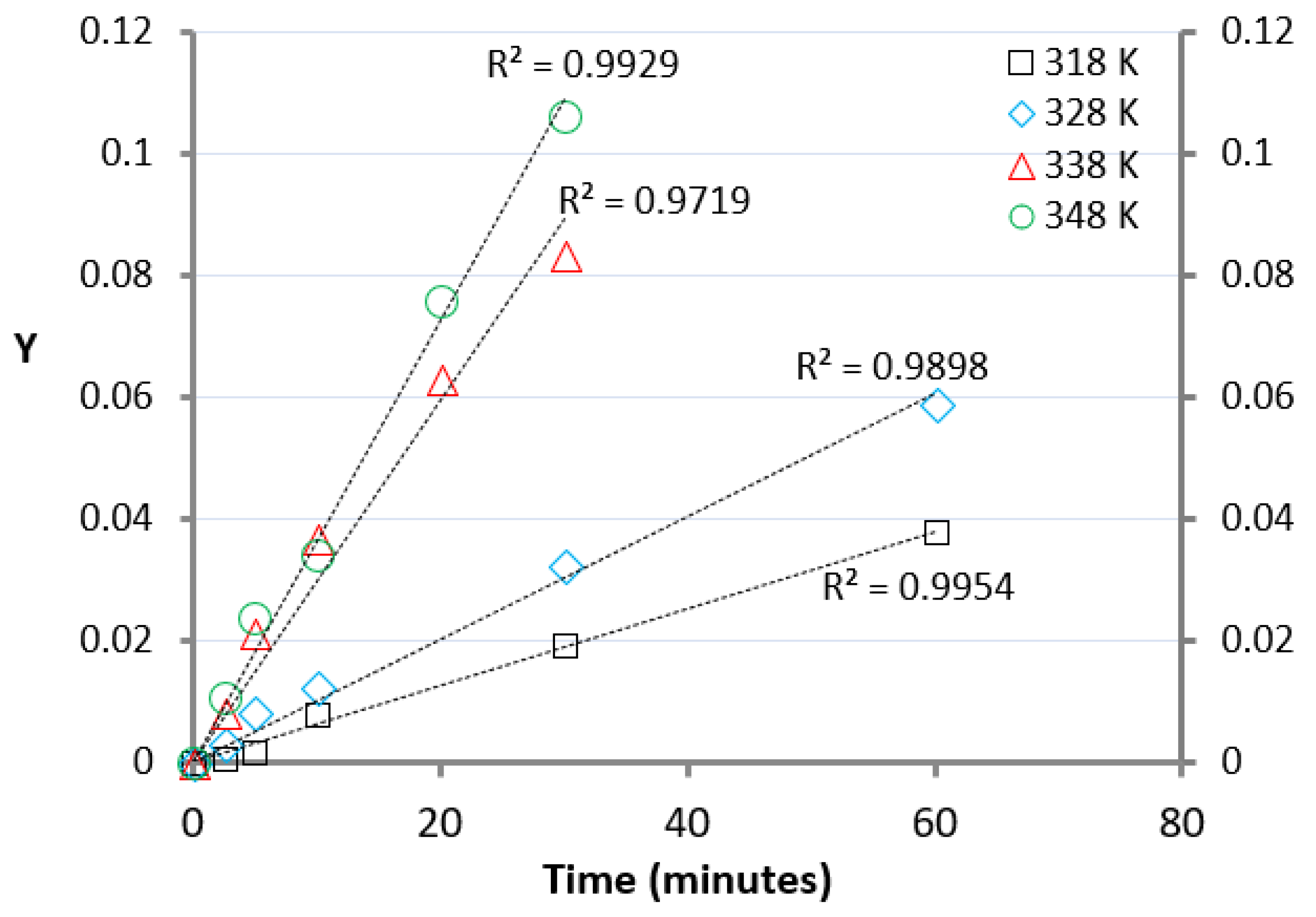
Figure 3.
Arrhenius and van’t Hoff plots for EL synthesis through direct esterification of the respective acid and ethanol.
Figure 3.
Arrhenius and van’t Hoff plots for EL synthesis through direct esterification of the respective acid and ethanol.

Figure 4.
Direct esterification of Levulinic Acid (LA) at different catalyst concentrations. Reaction conditions: molar ratio ethanol:LA = 1; AlCl3∙6H2O from 1 to 5%mol, temperature = 348 K, time = 8 h.
Figure 4.
Direct esterification of Levulinic Acid (LA) at different catalyst concentrations. Reaction conditions: molar ratio ethanol:LA = 1; AlCl3∙6H2O from 1 to 5%mol, temperature = 348 K, time = 8 h.
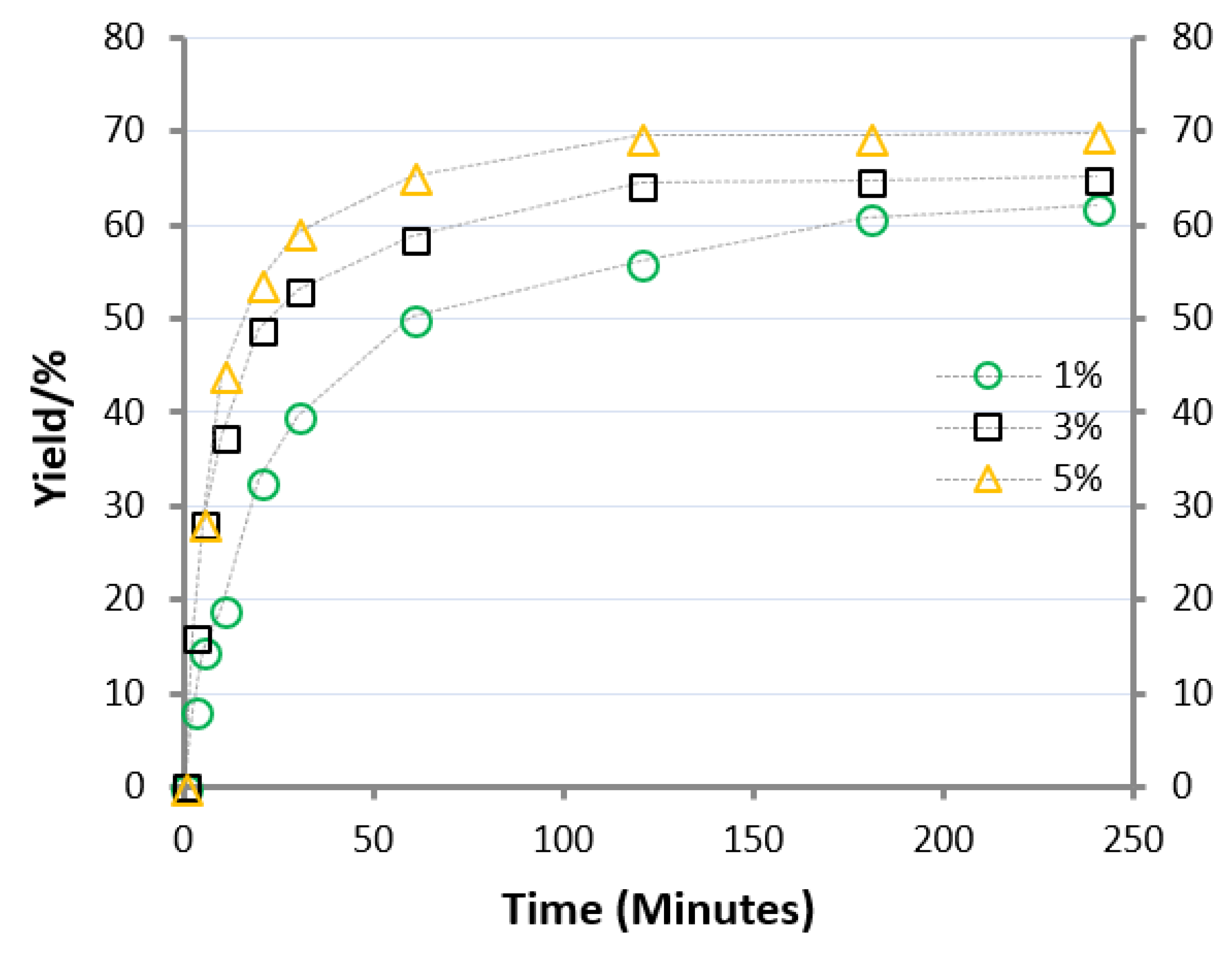
Figure 5.
Final reaction mixtures were obtained using an increasing amount of AlCl3∙6H2O in catalysis. Reaction conditions: molar ratio ethanol:LA = 1; AlCl3∙6H2O from 1 to 5%mol, temperature = 343 K, time = 8 h.
Figure 5.
Final reaction mixtures were obtained using an increasing amount of AlCl3∙6H2O in catalysis. Reaction conditions: molar ratio ethanol:LA = 1; AlCl3∙6H2O from 1 to 5%mol, temperature = 343 K, time = 8 h.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
k1, k−1 and Keq determined at 318, 328, 338 and 348 K.
| T | k1 | k−1 | Keq |
|---|---|---|---|
| 318 | 0.01004 | 0.004242 | 2.3669 |
| 328 | 0.016015 | 0.005791 | 2.7656 |
| 338 | 0.0393 | 0.012649 | 3.107 |
| 348 | 0.05733 | 0.017269 | 3.3199 |
Table 2.
Ea1, Ea−1, ΔH° and ΔS° evaluated for the direct-esterification reaction between LA and ethanol, catalyzed by AlCl3·6H2O.
Table 2.
Ea1, Ea−1, ΔH° and ΔS° evaluated for the direct-esterification reaction between LA and ethanol, catalyzed by AlCl3·6H2O.
| Ea1 (kJ K−1 mol−1) | Ea−1 (kJ K−1 mol−1) | ΔH° (kJ mol−1) | ΔS° (J K−1 mol−1) |
|---|---|---|---|
| 56.4 | 45.9 | 10.5 | 40.18 |
Table 3.
Compositions of organic and aqueous phases were obtained by using 3 and 5% AlCl3·6H2O in direct esterification.
Table 3.
Compositions of organic and aqueous phases were obtained by using 3 and 5% AlCl3·6H2O in direct esterification.
| 3% AlCl3·6H2O | 5% AlCl3·6H2O | ||||||
|---|---|---|---|---|---|---|---|
| Q | Composition | Distribution | Q | Composition | Distribution | ||
| (g) | (%wt) | (%wt) | (g) | (%wt) | (%wt) | ||
| Organic Phase | AL | 6.4 | 24.24 | 90.52 | 5.76 | 22.73 | 94.40 |
| EL | 16.3 | 61.74 | 99.76 | 17.20 | 67.88 | 98.48 | |
| EtOH | 2.5 | 9.47 | 89.29 | 2.18 | 8.60 | 89.32 | |
| H2O | 1.2 | 4.55 | 60.00 | 0.20 | 0.79 | 9.16 | |
| AlCl3·6H2O | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | |
| Aqueous Phase | AL | 0.67 | 22.41 | 9.48 | 0.34 | 6.73 | 5.60 |
| EL | 0.04 | 1.34 | 0.24 | 0.27 | 5.24 | 1.52 | |
| EtOH | 0.3 | 10.03 | 10.71 | 0.26 | 5.13 | 10.68 | |
| H2O | 0.8 | 26.76 | 40.00 | 1.98 | 39.06 | 90.84 | |
| AlCl3·6H2O | 1.18 | 39.46 | 100.00 | 2.23 | 43.83 | 100.00 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pastore, C.; D’Ambrosio, V. Intensification of Processes for the Production of Ethyl Levulinate Using AlCl3·6H2O. Energies 2021, 14, 1273. https://doi.org/10.3390/en14051273
AMA Style
Pastore C, D’Ambrosio V. Intensification of Processes for the Production of Ethyl Levulinate Using AlCl3·6H2O. Energies. 2021; 14(5):1273. https://doi.org/10.3390/en14051273
Chicago/Turabian StylePastore, Carlo, and Valeria D’Ambrosio. 2021. "Intensification of Processes for the Production of Ethyl Levulinate Using AlCl3·6H2O" Energies 14, no. 5: 1273. https://doi.org/10.3390/en14051273
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.