Investigating the Influence of Reaction Conditions and the Properties of Ceria for the Valorisation of Glycerol

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Catalyst Preparation
2.3. Catalyst Testing
2.4. Calculations
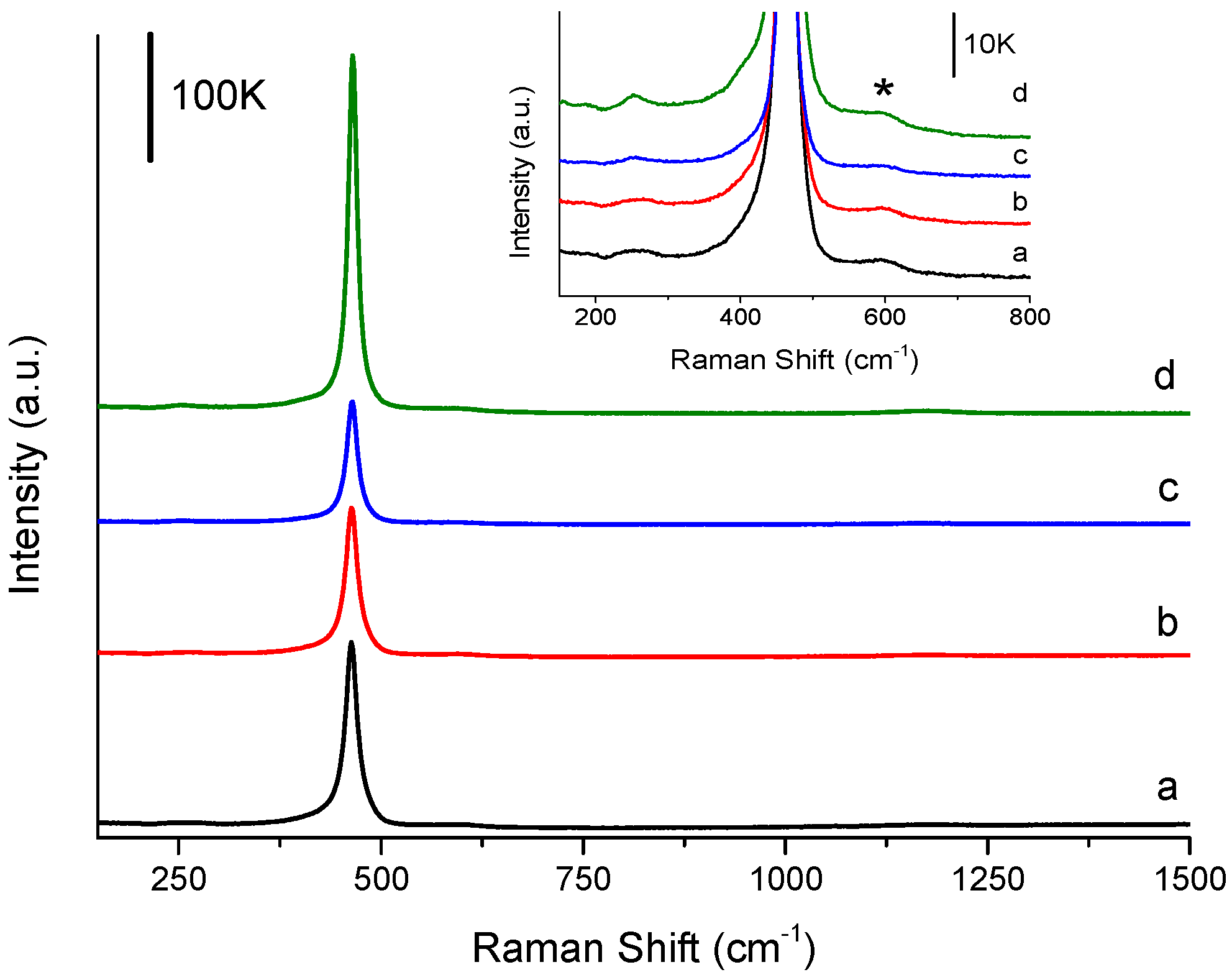
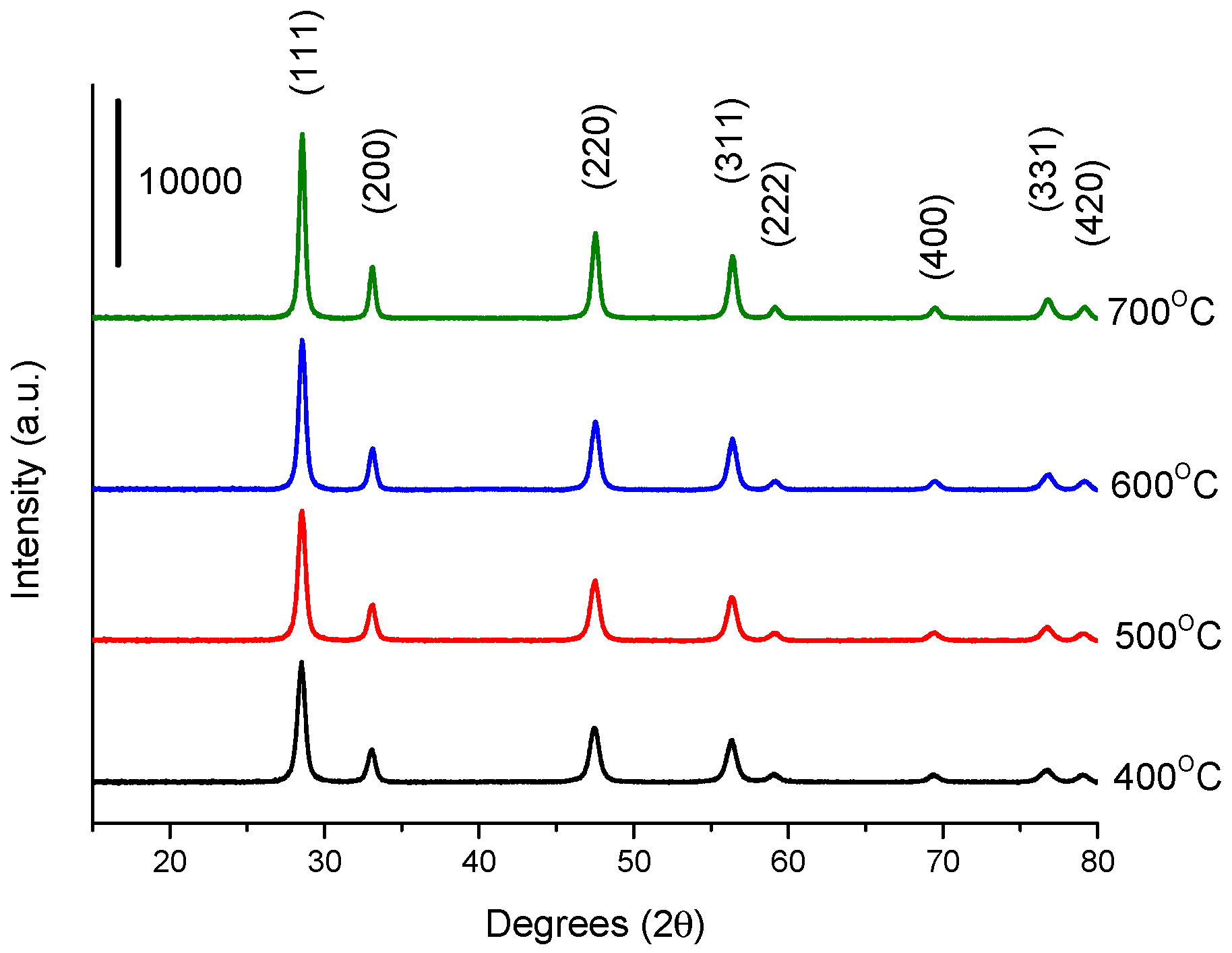
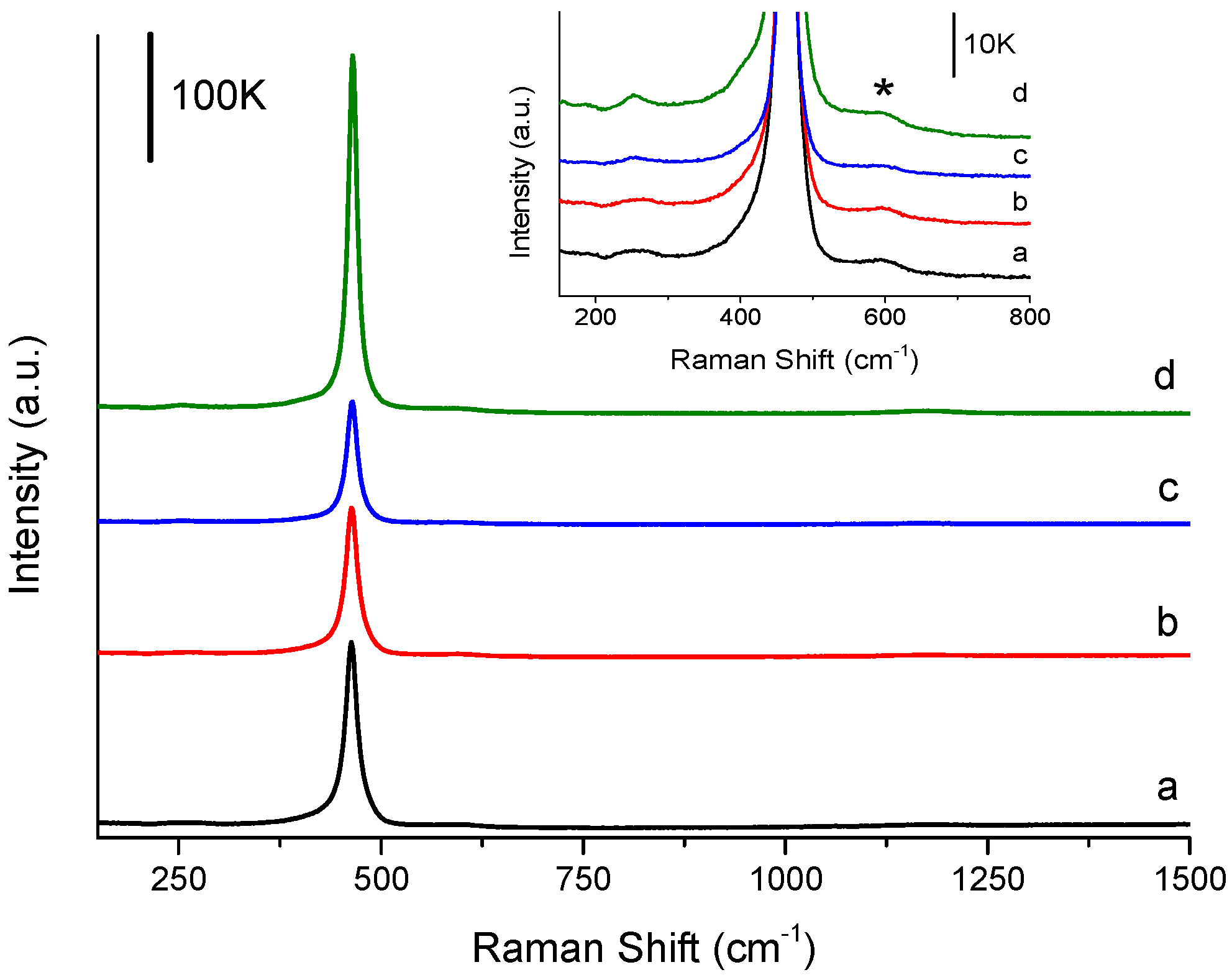
2.5. Catalyst Characterisation
3. Results and Discussion
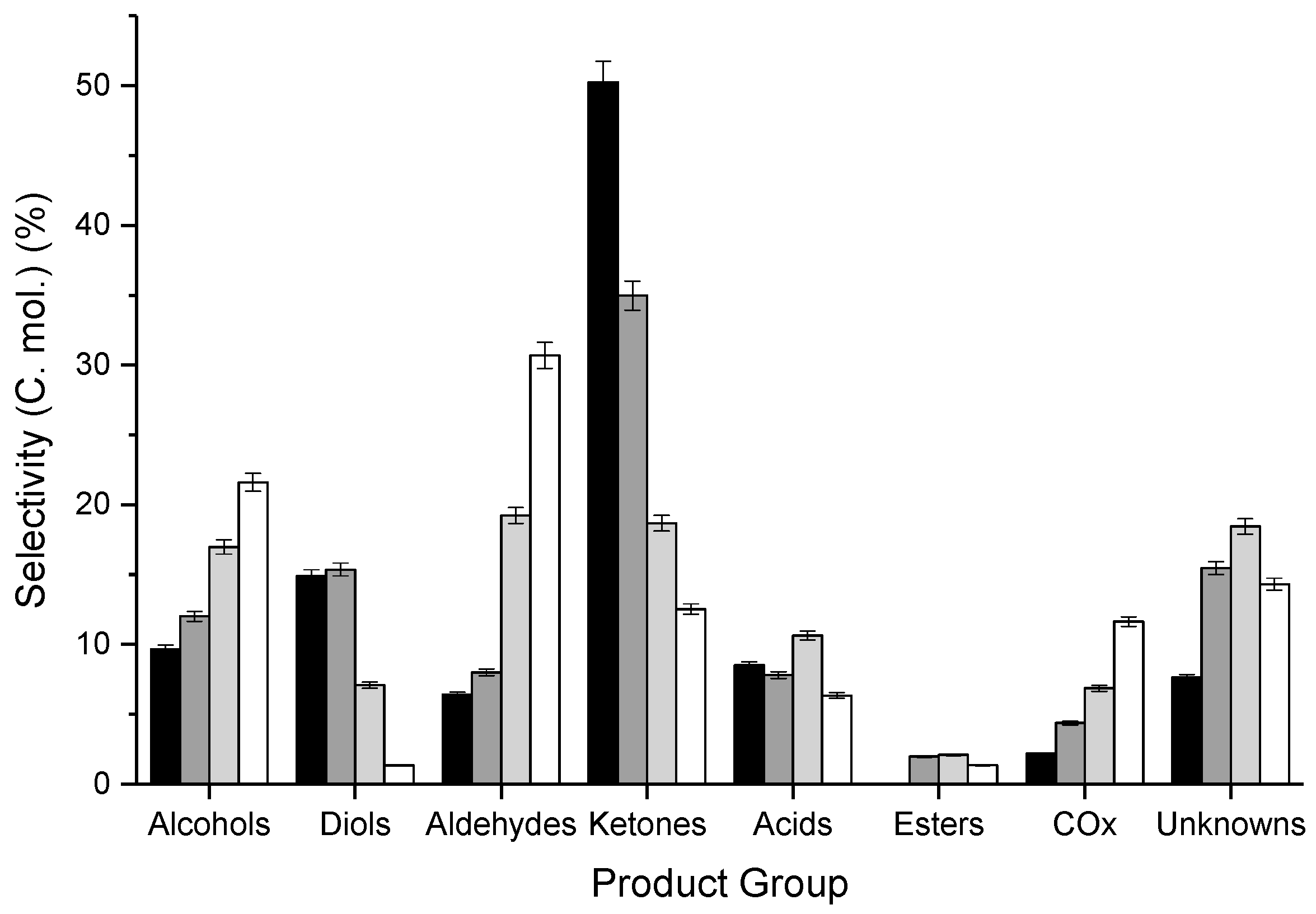
3.1. Influence of Contact Time and Reaction Temperature
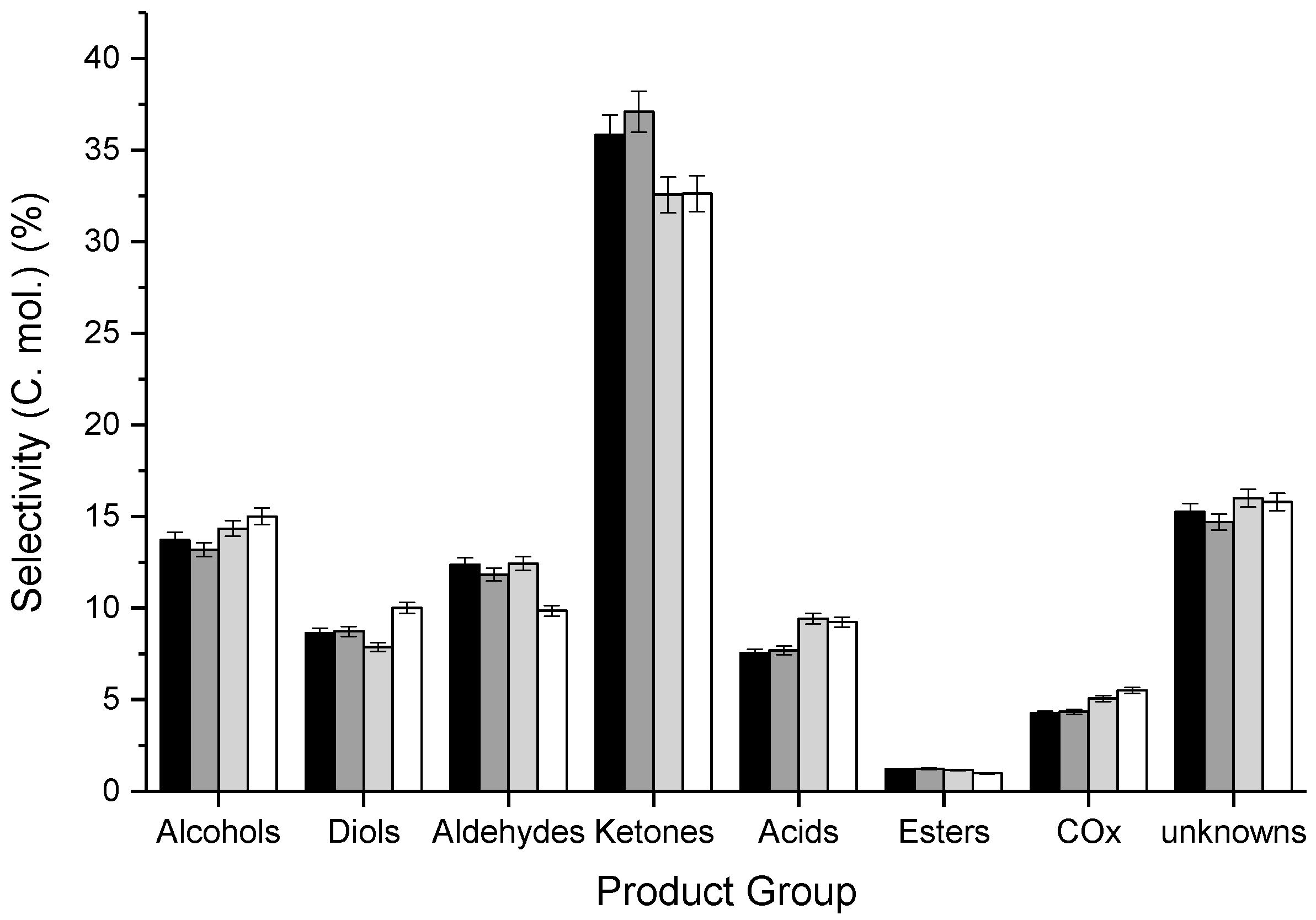
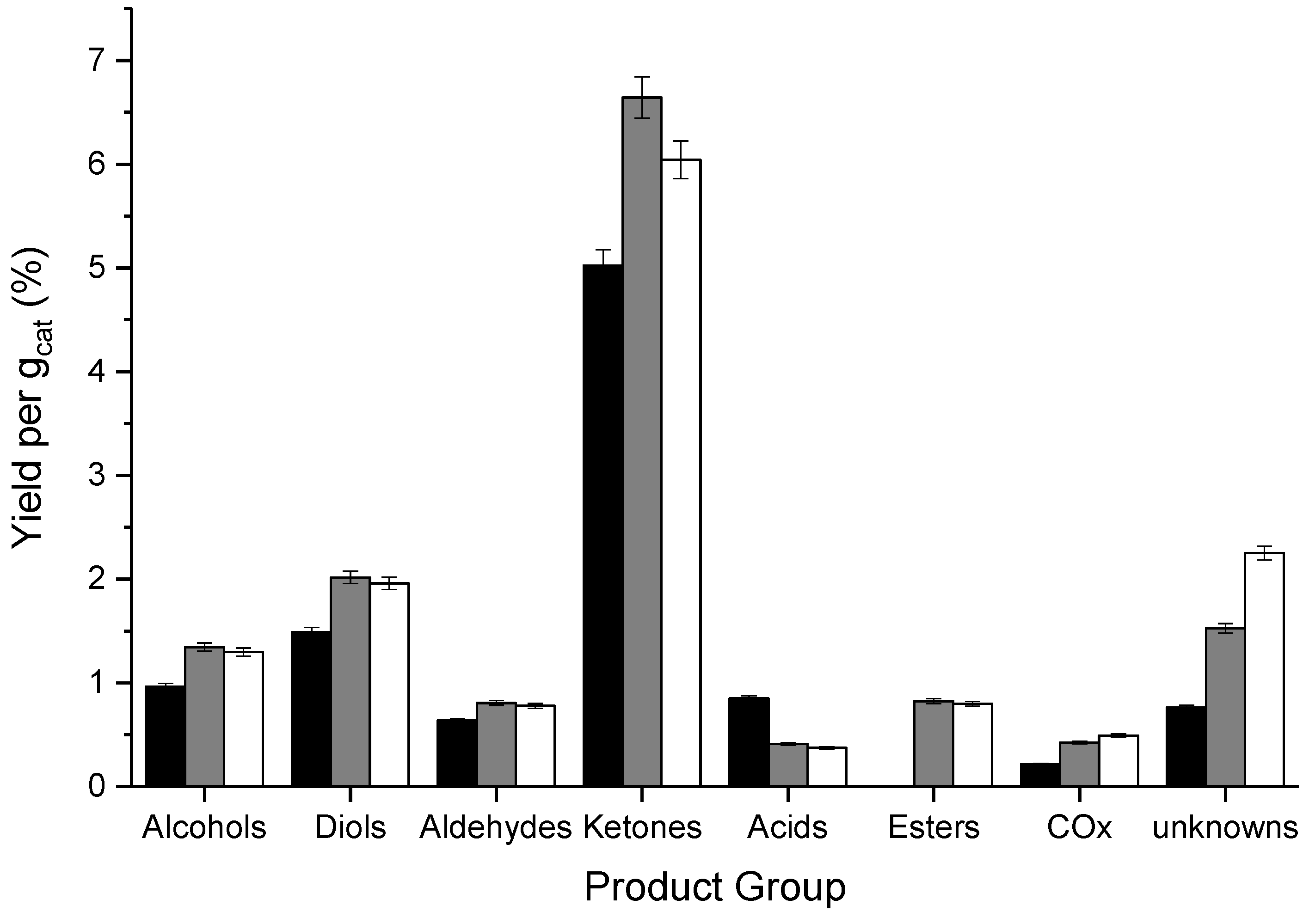
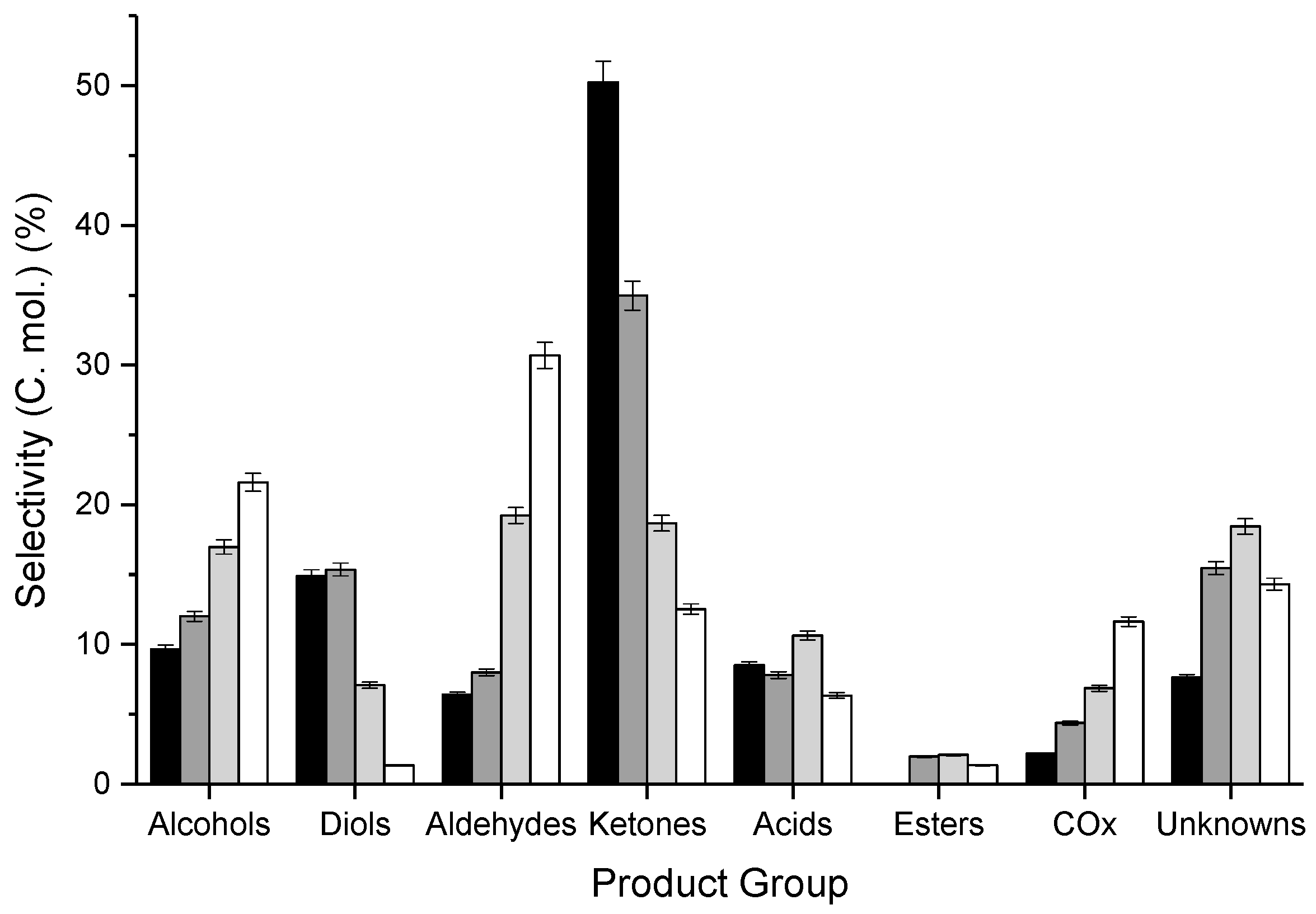
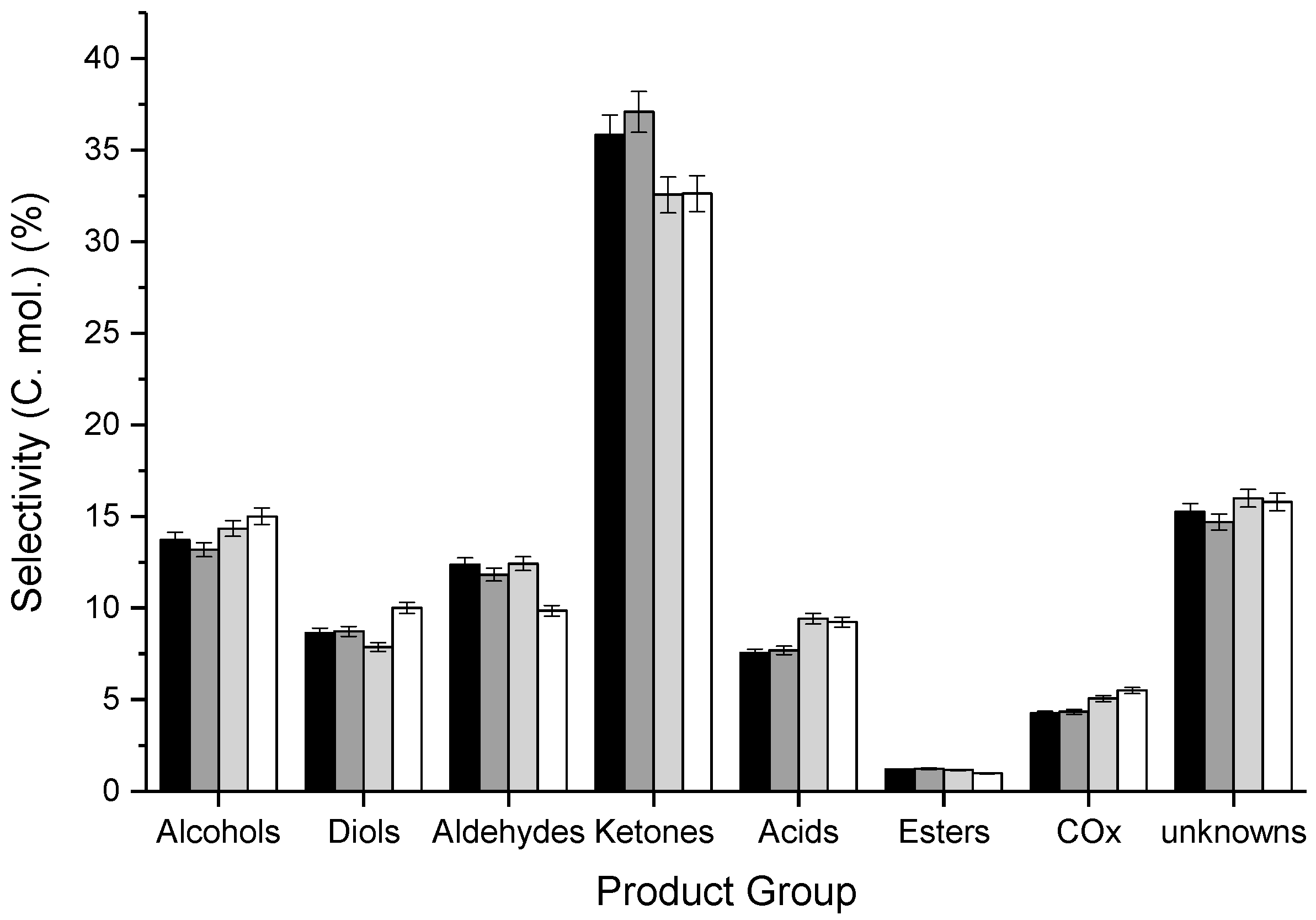
3.2. Influence of Catalyst Calcination Temperature
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gu, Y.; Jerome, F. Glycerol as a sustainable solvent for green chemistry. Green Chem. 2010, 12, 1127–1138. [Google Scholar] [CrossRef]
- Sun, D.; Yamada, Y.; Sato, S.; Ueda, W. Glycerol as a potential renewable raw material for acrylic acid production. Green Chem. 2017, 19, 3186–3213. [Google Scholar] [CrossRef]
- Otera, J. Transesterification. Chem. Rev. 1993, 93, 1449–1470. [Google Scholar] [CrossRef]
- Twigg, M.V.; Spencer, M.S. Deactivation of supported copper metal catalysts for hydrogenation reactions. Appl. Catal. A 2001, 212, 161–174. [Google Scholar] [CrossRef]
- Wainwright, M.S. Catalytic processes for methanol synthesis—Established and future. Stud. Surf. Sci. Catal. 1988, 36, 95–108. [Google Scholar]
- Thompson, J.C.; He, B.B. Characterization of crude glycerol from biodiesel production from multiple feedstocks. Appl. Eng. Agric. 2006, 22, 261. [Google Scholar] [CrossRef]
- Katryniok, B.; Paul, S.; Capron, M.; Dumeignil, F. Towards the Sustainable Production of Acrolein by Glycerol Dehydration. ChemSusChem 2009, 2, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.-H.; Wang, H.-P.; Liang, Y.; Xu, B.-Q. Sustainable production of acrolein: Gas-phase dehydration of glycerol over 12-tungstophosphoric acid supported on ZrO2 and SiO2. Green Chem. 2008, 10, 1087–1093. [Google Scholar] [CrossRef]
- Wang, F.; Dubois, J.-L.; Ueda, W. Catalytic dehydration of glycerol over vanadium phosphate oxides in the presence of molecular oxygen. J. Catal. 2009, 268, 260–267. [Google Scholar] [CrossRef]
- Ott, L.; Bicker, M.; Vogel, H. Catalytic dehydration of glycerol in sub- and supercritical water: A new chemical process for acrolein production. Green Chem. 2006, 8, 214–220. [Google Scholar] [CrossRef]
- Haider, M.H.; Dummer, N.F.; Zhang, D.; Miedziak, P.; Davies, T.E.; Taylor, S.H.; Willock, D.J.; Knight, D.W.; Chadwick, D.; Hutchings, G.J. Rubidium- and caesium-doped silicotungstic acid catalysts supported on alumina for the catalytic dehydration of glycerol to acrolein. J. Catal. 2012, 286, 206–213. [Google Scholar] [CrossRef]
- Magatani, Y.; Okumura, K.; Dubois, J.-L.; Devaux, J.-F. Catalyst and process for preparing acrolein and/or acrylic acid by dehydration reaction of glycerin. Patent WO2011033689A1, 24 March 2011. [Google Scholar]
- Omata, K.; Matsumoto, K.; Murayama, T.; Ueda, W. Direct oxidative transformation of glycerol to acrylic acid over Nb-based complex metal oxide catalysts. Catal. Today 2016, 259, 205–212. [Google Scholar] [CrossRef]
- Sato, S.; Akiyama, M.; Takahashi, R.; Hara, T.; Inui, K.; Yokota, M. Vapor-phase reaction of polyols over copper catalysts. Appl. Catal. A Gen. 2008, 347, 186–191. [Google Scholar] [CrossRef]
- Chiu, C.-W.; Dasari, M.A.; Suppes, G.J.; Sutterlin, W.R. Dehydration of glycerol to acetol via catalytic reactive distillation. AICHE J. 2006, 52, 3543–3548. [Google Scholar] [CrossRef]
- Sonnati, M.O.; Amigoni, S.; Taffin de Givenchy, E.P.; Darmanin, T.; Choulet, O.; Guittard, F. Glycerol carbonate as a versatile building block for tomorrow: Synthesis, reactivity, properties and applications. Green Chem. 2013, 15, 283–306. [Google Scholar] [CrossRef]
- Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F. Improved utilisation of renewable resources: New important derivatives of glycerol. Green Chem. 2008, 10, 13–30. [Google Scholar] [CrossRef]
- Tomishige, K.; Nakagawa, Y.; Tamura, M. Selective hydrogenolysis and hydrogenation using metal catalysts directly modified with metal oxide species. Green Chem. 2017, 19, 2876–2924. [Google Scholar] [CrossRef]
- Bienholz, A.; Schwab, F.; Claus, P. Hydrogenolysis of glycerol over a highly active CuO/ZnO catalyst prepared by an oxalate gel method: Influence of solvent and reaction temperature on catalyst deactivation. Green Chem. 2010, 12, 290–295. [Google Scholar] [CrossRef]
- Furikado, I.; Miyazawa, T.; Koso, S.; Shimao, A.; Kunimori, K.; Tomishige, K. Catalytic performance of Rh/SiO2 in glycerol reaction under hydrogen. Green Chem. 2007, 9, 582–588. [Google Scholar] [CrossRef]
- Yin, A.-Y.; Guo, X.-Y.; Dai, W.-L.; Fan, K.-N. The synthesis of propylene glycol and ethylene glycol from glycerol using Raney Ni as a versatile catalyst. Green Chem. 2009, 11, 1514–1516. [Google Scholar] [CrossRef]
- Maglinao, R.L.; He, B.B. Catalytic Thermochemical Conversion of Glycerol to Simple and Polyhydric Alcohols Using Raney Nickel Catalyst. Ind. Eng. Chem. Res. 2011, 50, 6028–6033. [Google Scholar] [CrossRef]
- Dasari, M.A.; Kiatsimkul, P.-P.; Sutterlin, W.R.; Suppes, G.J. Low-pressure hydrogenolysis of glycerol to propylene glycol. Appl. Catal. A Gen. 2005, 281, 225–231. [Google Scholar] [CrossRef]
- Velasquez, M.; Santamaria, A.; Batiot-Dupeyrat, C. Selective conversion of glycerol to hydroxyacetone in gas phase over La2CuO4 catalyst. Appl. Catal. B Environ. 2014, 160–161, 606–613. [Google Scholar] [CrossRef]
- Nimlos, M.R.; Blanksby, S.J.; Qian, X.; Himmel, M.E.; Johnson, D.K. Mechanisms of Glycerol Dehydration. J. Phys. Chem. A 2006, 110, 6145–6156. [Google Scholar] [CrossRef] [PubMed]
- Antal, M.J.; Mok, W.S.L.; Roy, J.C.; Raissi, A.T.; Anderson, D.G.M. Pyrolytic sources of hydrocarbons from biomass. J. Anal. Appl. Pyrolysis 1985, 8, 291–303. [Google Scholar] [CrossRef]
- Chai, S.-H.; Wang, H.-P.; Liang, Y.; Xu, B.-Q. Sustainable production of acrolein: Investigation of solid acid-base catalysts for gas-phase dehydration of glycerol. Green Chem. 2007, 9, 1130–1136. [Google Scholar] [CrossRef]
- Haider, M.H.; Dummer, N.F.; Knight, D.W.; Jenkins, R.L.; Howard, M.; Moulijn, J.; Taylor, S.H.; Hutchings, G.J. Efficient green methanol synthesis from glycerol. Nat. Chem. 2015, 7, 1028–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.R.; Smith, P.J.; Mugford, K.S.; Douthwaite, M.; Dummer, N.F.; Willock, D.J.; Howard, M.; Knight, D.W.; Taylor, S.H.; Hutchings, G.J. New insights for the valorisation of glycerol over MgO catalysts in the gas-phase. Catal. Sci. Technol. 2019, 9, 1464–1475. [Google Scholar] [CrossRef]
- Chen, J.; Cai, Q.; Lu, L.; Leng, F.; Wang, S. Upgrading of the Acid-Rich Fraction of Bio-oil by Catalytic Hydrogenation-Esterification. ACS Sustain. Chem. Eng. 2017, 5, 1073–1081. [Google Scholar] [CrossRef]
- Idriss, H.; Diagne, C.; Hindermann, J.P.; Kiennemann, A.; Barteau, M.A. Reactions of Acetaldehyde on CeO2 and CeO2-Supported Catalysts. J. Catal. 1995, 155, 219–237. [Google Scholar] [CrossRef]
- Chen, T.L.; Mullins, D.R. Adsorption and Reaction of Acetaldehyde over CeOX(111) Thin Films. J. Phys. Chem. C 2011, 115, 3385–3392. [Google Scholar] [CrossRef]
- Calaza, F.C.; Xu, Y.; Mullins, D.R.; Overbury, S.H. Oxygen Vacancy-Assisted Coupling and Enolization of Acetaldehyde on CeO2(111). J. Am. Chem. Soc. 2012, 134, 18034–18045. [Google Scholar] [CrossRef] [PubMed]
- Mamontov, E.; Egami, T.; Brezny, R.; Koranne, M.; Tyagi, S. Lattice Defects and Oxygen Storage Capacity of Nanocrystalline Ceria and Ceria-Zirconia. J. Phys. Chem. B 2000, 104, 11110–11116. [Google Scholar] [CrossRef]
- Qiao, Z.-A.; Wu, Z.; Dai, S. Shape-Controlled Ceria-based Nanostructures for Catalysis Applications. ChemSusChem 2013, 6, 1821–1833. [Google Scholar] [CrossRef]
- McBride, J.R.; Hass, K.C.; Poindexter, B.D.; Weber, W.H. Raman and X-ray studies of Ce1−xRExO2−y, where RE = La, Pr, Nd, Eu, Gd, and Tb. J. Appl. Phys. 1994, 76, 2435–2441. [Google Scholar] [CrossRef]
- Hua, G.; Zhang, L.; Fei, G.; Fang, M. Enhanced catalytic activity induced by defects in mesoporous ceria nanotubes. J. Mater. Chem. 2012, 22, 6851–6855. [Google Scholar] [CrossRef]
- Nolan, M.; Parker, S.C.; Watson, G.W. Reduction of NO2 on Ceria Surfaces. J. Phys. Chem. B 2006, 110, 2256–2262. [Google Scholar] [CrossRef]
- Guo, M.; Lu, J.; Wu, Y.; Wang, Y.; Luo, M. UV and Visible Raman Studies of Oxygen Vacancies in Rare-Earth-Doped Ceria. Langmuir 2011, 27, 3872–3877. [Google Scholar] [CrossRef]

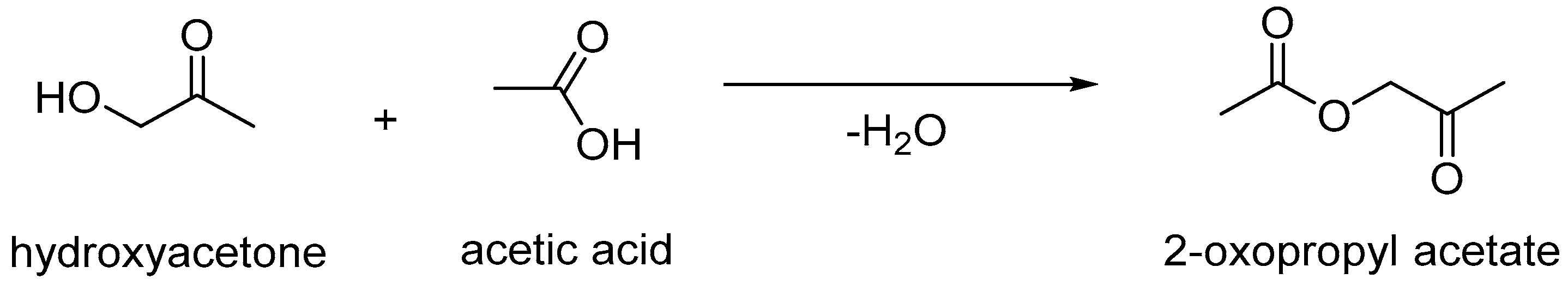


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
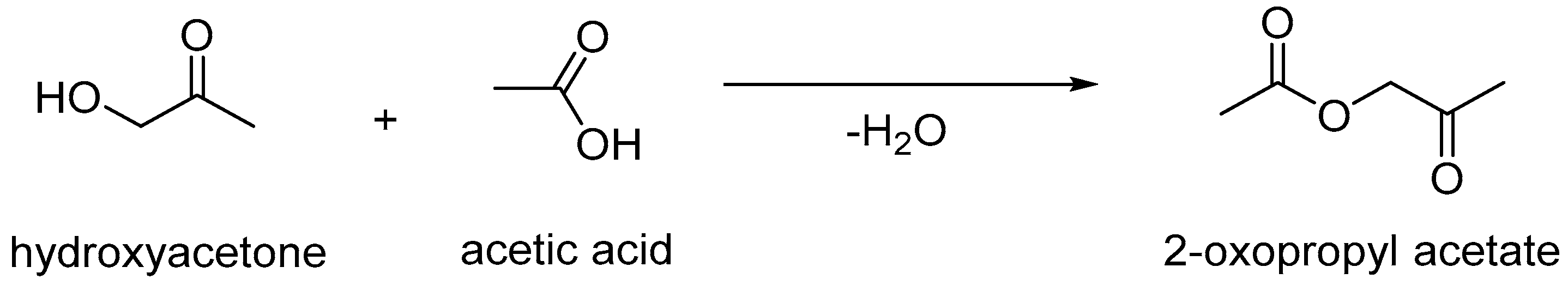{kind=link}
| Time (min) | A 1 (%) | B 2 (%) |
|---|---|---|
| 0.0 | 98 | 2 |
| 1.0 | 98 | 2 |
| 15.0 | 2 | 98 |
| 17.0 | 2 | 98 |
| 18.0 | 98 | 2 |
| 20.0 | 98 | 2 |
| RT a/°C | CGLYb/% | Mol. Balance c/% | Yield/% e | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BCd | BH | BO | Alc. | Diols | Ald. | Ket. | Ac. | Est. | COx | Unk. | ||
| 320 | 21 | 100 | 97 | 95 | 2.0 | 3.1 | 3.1 | 10.6 | 1.8 | 0.0 | 0.5 | 1.6 |
| 360 | 84 | 87 | 78 | 72 | 10.1 | 12.9 | 12.9 | 29.4 | 6.6 | 1.7 | 3.7 | 13.0 |
| 400 | 98 | 80 | 68 | 62 | 16.6 | 6.9 | 6.9 | 18.3 | 10.4 | 2.0 | 6.7 | 18.1 |
| 440 | 100 | 77 | 62 | 57 | 21.6 | 1.3 | 1.3 | 12.5 | 6.3 | 1.3 | 11.6 | 14.3 |
| Calcination Temperature/°C | BET Surface Area a/m2 g−1 | Crystallite Size b/nm | Defect Density c/% | Defect Density per Surface Area c/×10−3 % m−2 | FWHM d of F2g Mode |
|---|---|---|---|---|---|
| 400 | 38 | 14 | 0.58 | 1.5 | 17.96 |
| 500 | 34 | 15 | 0.83 | 2.4 | 18.03 |
| 600 | 27 | 16 | 0.11 | 0.4 | 16.61 |
| 700 | 22 | 19 | 0.05 | 0.3 | 13.35 |
| CT/°C | Catalyst Mass/g | Ar Flow Rate/mL min−1 | CGLYa/% | Mol. Balance/% b | Yield d/% | STYMeOHe/g1 h−1 kg−1 | Carbon Deposition/mgCoke g−1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BCc | BH | BO | Alc. | Diols | Ald. | Ket. | Ac. | COx | Est. | Unk. | ||||||
| 400 | 0.4998 | 15 | 83 | 82 (83) | 75 | 67 | 7.9 | 6.0 | 8.5 | 24.7 | 5.2 | 2.9 | 0.7 | 11.3 | 84.3 | 17 |
| 500 | 0.5497 | 15 | 83 | 80 (81) | 72 | 66 | 7.4 | 5.9 | 7.9 | 24.9 | 5.2 | 2.9 | 0.7 | 10.7 | 80.0 | 21 |
| 600 | 0.6995 | 20 | 80 | 84 (86) | 76 | 70 | 8.1 | 5.3 | 8.4 | 25.2 | 6.4 | 3.4 | 0.6 | 11.7 | 59.3 | 19 |
| 700 | 0.8508 | 25 | 84 | 81 (84) | 74 | 67 | 7.7 | 6.9 | 6.8 | 22.4 | 6.3 | 3.8 | 0.5 | 11.5 | 51.1 | 22 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, P.J.; Smith, L.; Dummer, N.F.; Douthwaite, M.; Willock, D.J.; Howard, M.; Knight, D.W.; Taylor, S.H.; Hutchings, G.J. Investigating the Influence of Reaction Conditions and the Properties of Ceria for the Valorisation of Glycerol. Energies 2019, 12, 1359. https://doi.org/10.3390/en12071359
Smith PJ, Smith L, Dummer NF, Douthwaite M, Willock DJ, Howard M, Knight DW, Taylor SH, Hutchings GJ. Investigating the Influence of Reaction Conditions and the Properties of Ceria for the Valorisation of Glycerol. Energies. 2019; 12(7):1359. https://doi.org/10.3390/en12071359
Chicago/Turabian StyleSmith, Paul J., Louise Smith, Nicholas F. Dummer, Mark Douthwaite, David J. Willock, Mark Howard, David W. Knight, Stuart H. Taylor, and Graham J. Hutchings. 2019. "Investigating the Influence of Reaction Conditions and the Properties of Ceria for the Valorisation of Glycerol" Energies 12, no. 7: 1359. https://doi.org/10.3390/en12071359