Copper-Tin Alloys for the Electrocatalytic Reduction of CO2 in an Imidazolium-Based Non-Aqueous Electrolyte
by

Robert L. Sacci
1,*, 
Stephanie Velardo
2,
Lu Xiong
2,
Daniel A. Lutterman
1,*,† and
Joel Rosenthal
2,* 1
Chemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
2
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA
*
Authors to whom correspondence should be addressed.
†
Knoxville Catholic High School, 9245 Fox Lonas Rd NW, Knoxville, TN 37923, USA.
Energies 2019, 12(16), 3132; https://doi.org/10.3390/en12163132
Submission received: 12 June 2019
/
Revised: 30 July 2019
/
Accepted: 7 August 2019
/
Published: 15 August 2019
(This article belongs to the Special Issue Carbon Capture, Storage and Utilization)
Abstract
:The ability to synthesize value-added chemicals directly from CO2 will be an important technological advancement for future generations. Using solar energy to drive thermodynamically uphill electrochemical reactions allows for near carbon-neutral processes that can convert CO2 into energy-rich carbon-based fuels. Here, we report on the use of inexpensive CuSn alloys to convert CO2 into CO in an acetonitrile/imidazolium-based electrolyte. Synergistic interactions between the CuSn catalyst and the imidazolium cation enables the electrocatalytic conversion of CO2 into CO at −1.65 V versus the standard calomel electrode (SCE). This catalyst system is characterized by overpotentials for CO2 reduction that are similar to more expensive Au- and Ag-based catalysts, and also shows that the efficacy of the CO2 reduction reaction can be tuned by varying the CuSn ratio.

1. Introduction
The development of cost-effective systems for the conversion of CO2 into value-added products and high-energy species such as CO and saturated hydrocarbons is of global importance [1,2]. In 2016, industrial, commercial, and private transportation alone generated over 5000 metric tons of atmospheric CO2 through the burning of fossil fuels [3]. Together with other CO2 emitting processes, fossil fuel combustion continues to drive climate change and exacerbates the natural greenhouse effect [4]. Without improved energy harvesting, storage, and CO2 sequestration technologies, this trend will only continue to increase [5]. Renewable electric energy sources such as solar or wind harvesting technologies can be utilized to power thermodynamically uphill energy storing electrochemical reactions [1,2,5]. Historically, coinage metals (Au, Ag, and Cu) have been utilized as electrocatalysts for the reduction of CO2 to CO and saturated hydrocarbons (methane, ethane, propane, etc.) via Equations (1) and (2), respectively [6,7].
Electrolysis using copper cathodes can also promote the conversion of CO2 into simple alcohols (i.e., methanol, ethanol, propanol, etc.) via the balanced process shown in Equation (3). In addition to the direct reduction of CO2 to hydrocarbons and oxygenates, a secondary process that converts H2O into H2 can be used in conjunction with the electrocatalytic CO production to produce syngas (a CO/H2 mixture), which is a primary feedstock that can be unconverted into liquid carbon-based fuels such as gasoline and diesel via Fischer-Tropsch processes [8].
It has been shown that the 2e−/2H+ CO2 reduction reaction (CO2RR) to generate CO is promoted by metallic cathodes in the presence of imidazolium-based ([Im]+) non-aqueous electrolytes [9,10,11,12,13]. The [Im]+ cation is believed to serve as a proton donor and also acts to stabilize reduced CO2 intermediates that are formed/adsorbed at the cathode/electrolyte interface [14], while promoting proton and electron transfer processes attendant to CO2 reduction [15]. In fact, CO2 electroreduction in [Im]+-based electrolytes has been shown to occur selectively and efficiently using inexpensive non-noble cathodes such as bismuth [16,17]. These metals inhibit the kinetics for the H2 evolution reaction (HER) by increasing the activation energy of the surface hydride formation [18,19]. In this manner, H2 Faradaic efficiency (FE) is dramatically suppressed (FEH2 < 2%), allowing the CO2RR reactivity to dominate on Bi [13,17,20]. Sn represents another post-transition metal that has been shown to facilitate the CO2RR in [Im]+-based electrolytes, although with a slightly higher overpotential than Bi [16].
In many systems, bimetallic and alloy catalysts have shown improvements over their base components for electrocatalysis applications. Since, as mentioned above, Sn has been shown to facilitate the CO2 reduction reaction (CO2RR) to generate CO from CO2 dissolved in [Im]+/MeCN-based electrolytes (j ~ 7.5 mA/cm at −1.95 V vs. SCE), we considered the ability of Sn-containing alloys to promote the CO2RR under similar conditions. In this study, we present the use of Cu6Sn5 and Cu6Sn10 thin film alloys as electrocatalysts for the CO2RR in [Im]+/MeCN-based electrolytes. This work complements recent studies by Haruyama and coworkers, which demonstrated that electrodeposited CuSn alloys can promote the reduction of CO2 to both CO and formate in aqueous electrolytes that do not contain [Im]+ promoters [21]. These authors reported that the selectivity of formate production was enhanced with increased Sn stoichiometry in the film. In this work, we compare the activity and selectivity of a pure phase CuSn alloy with one having excess Sn, and document the CO2RR performance of these systems in MeCN containing [BMIM]+ electrolytes.
2. Experimental
Reagents, solvents, and all other chemicals were purchased from Fisher, Alfa Aesar, TCI America, Sigma-Aldrich, Acros Organics, Matrix Scientific, or Cambridge Isotopes Laboratories. Electrochemical grade tetrabutylammonium hexafluorophosphate (TBAPF6) was obtained from TCI America and was purified by recrystallization from ethanol prior to use. Compressed carbon dioxide gas was supplied by Keen Compressed Gas Company.
Cu6Sn5 and Cu6Sn10 targets were prepared from stoichiometric mixtures of Sn (99.8%, Alfa Aesar, Haverhill, MA, USA) and Cu (99.5%, Sigma-Aldrich, St. Louis, MO, USA) that were ball milled with yttria-stabilized zirconia balls for 15 min. This powder was then pressed into a 50 mm diameter disk, annealed at 200 °C under Ar, and slowly cooled to ambient temperature. The Cu6Sn5 target was analyzed by X-ray diffraction (XRD) and was shown to correspond to the expected η-Cu6Sn5 (C2/c) monoclinic phase formed at low temperatures with lattice parameters a = 11.0262 (±0.0014) Å, b = 7.2745 (±0.0012) Å, and c = 9.846 (±0.0021) Å [22]. The Cu6Sn10 target was a mixture of Sn and the Cu6Sn5 monoclinic phase.
Thin film depositions were carried out using direct current (DC) magnetron sputtering of the above alloyed targets in argon plasma onto (2000-grit) polished nickel foils. Sputtering was conducted at 10 W, 20 mTorr, and with a 5 cm target–substrate distance. This resulted in a deposition rate of about 0.9 nm s−1 and yielded thin films with the structure of the high temperature hexagonal Cu6Sn5 (P63/mmc, ICDD 00-002-0713) phase [22] and Cu6Sn10 was a mixture of Sn (I4/mmm, ICDD 96-900-8571) and the η-Cu6Sn5 phase (see Results and Discussion). Film thicknesses were systematically varied between 0.1 and 0.5 mm.
2.1. Film Characterization
A Scintag PDS 2000 diffractometer equipped with Cu-Kα radiation and a nickel filter were used for XRD collection. Scattering angles of 10–100° were collected in 0.02°-steps with a count time of 5 s per step. Rietveld refinements were performed using the PANalytical HighScore Plus software (3.5, PANalytical B.V., Almelo, The Netherlands) package by indexing with powder diffraction files (ICDD and COD).
Scanning electron microscopy (SEM) images and energy dispersive X-ray (EDX) spectra were obtained using a JEOL JSM 7400F Scanning Electron Microscope. X-ray photoelectron spectroscopy (XPS) experiments were carried out using a VG ESCALAB 220I-XL spectrometer equipped with a non-monochromatic Al Kα source operating at 300 W. Initial XPS survey scans were collected at a pass energy of 100 eV using a step size of 1.0 eV. All high-resolution XPS spectra were recorded at a pass energy of 20 eV using a step size of 0.1 eV. Atomic ratios were determined using a Shirley-type baseline and standard relative sensitivity factors. Additional XPS characterization of the sputtered films was carried out using a PHI 3056 spectrometer with a non-monochromatic Mg Kα source operated at 15 kV and 350 W. Through comparison with the adventitious C 1s signal, the XPS spectra in Figure 1 will show a 0.9 eV shift to lower binding energies.
2.2. Electrochemistry
All electrochemistry was performed using either a CHI-620D potentiostat/galvanostat or a CHI-720D bipotentiostat. Cyclic voltammetry (CV) and linear-sweep voltammetry (LSV) experiments were carried out using standard three-electrode configurations. A section of platinum gauze (Sigma 99.9%) served as the auxillary electrode, and was separated from the working and reference electrodes by Nafion membranes. All potentials were measured against a Ag/AgCl reference electrode (1.0 M KCl, CH Instruments) and were converted into SCE (ESCE = EAg/AgCl + 44 mV). Cyclic voltammograms were recorded at 100 mV/s and were IR compensated. Unless otherwise noted, 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in MeCN was used as the supporting electrolyte for all electrochemical studies. The MeCN utilized for electrochemical experiments was not rigorously dried and contained adventitious levels of moisture.
Controlled potential electrolysis (CPE) experiments were performed in gas-tight two-compartment cells with a Nafion (NRE-212) membrane separating the anode and cathode compartments, as has been previously described [17]. Both the cathode and anode compartments of the electrolysis cell were filled with 20.0 mL of 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in MeCN, and were bubbled with CO2, Ar, or N2 for at least 30 min prior to performing the electrochemical experiments.
During all CPE experiments, the cathode solution was stirred vigorously while a constant supply of MeCN-saturated CO2 gas was fed into the headspace of the electrolysis cell at a rate of 5.0 cm3/min. The cathode compartment was vented into an autosampling loop of a gas chromatograph (GC) (SRI Instruments, SRI-8610C). GC acquisitions were initiated every 15 min by placing the autosampling loop in line with a packed HayeSep D column and a packed MoleSieve 13× column. Argon (Keen, 99.999%) was used as the GC carrier gas. Both GC columns connected directly to a thermal conductivity detector (TCD) to quantify hydrogen and a flame ionization detector (FID) equipped with a methanizer unit to quantify the production of reduced carbon species such as carbon monoxide. The partial current densities associated with electrolytic formation of CO and H2 were calculated from the GC peak area as follows:
In the above two equations, α and β represent conversion factors based on the calibration of the GC with standard samples of CO and H2, respectively, F = 9.65 × 104 C mol−1, p0 = 1 atm, R = 82.1 mL atm K−1 mol−1, and T = 298 K. Faradaic efficiencies for the formation of a given product were calculated by dividing the partial current densities by the total current. 1H NMR experiments were performed on aliquots of the electrolyte solution at the completion of each electrolysis run to determine whether nonvolatile CO2RR products had formed, and to monitor changes to the composition of the electrolyte solution. NMR spectra were recorded at 25 °C on a Bruker 600 MHz NMR spectrometer.
3. Results and Discussion
3.1. Catalyst Characterization
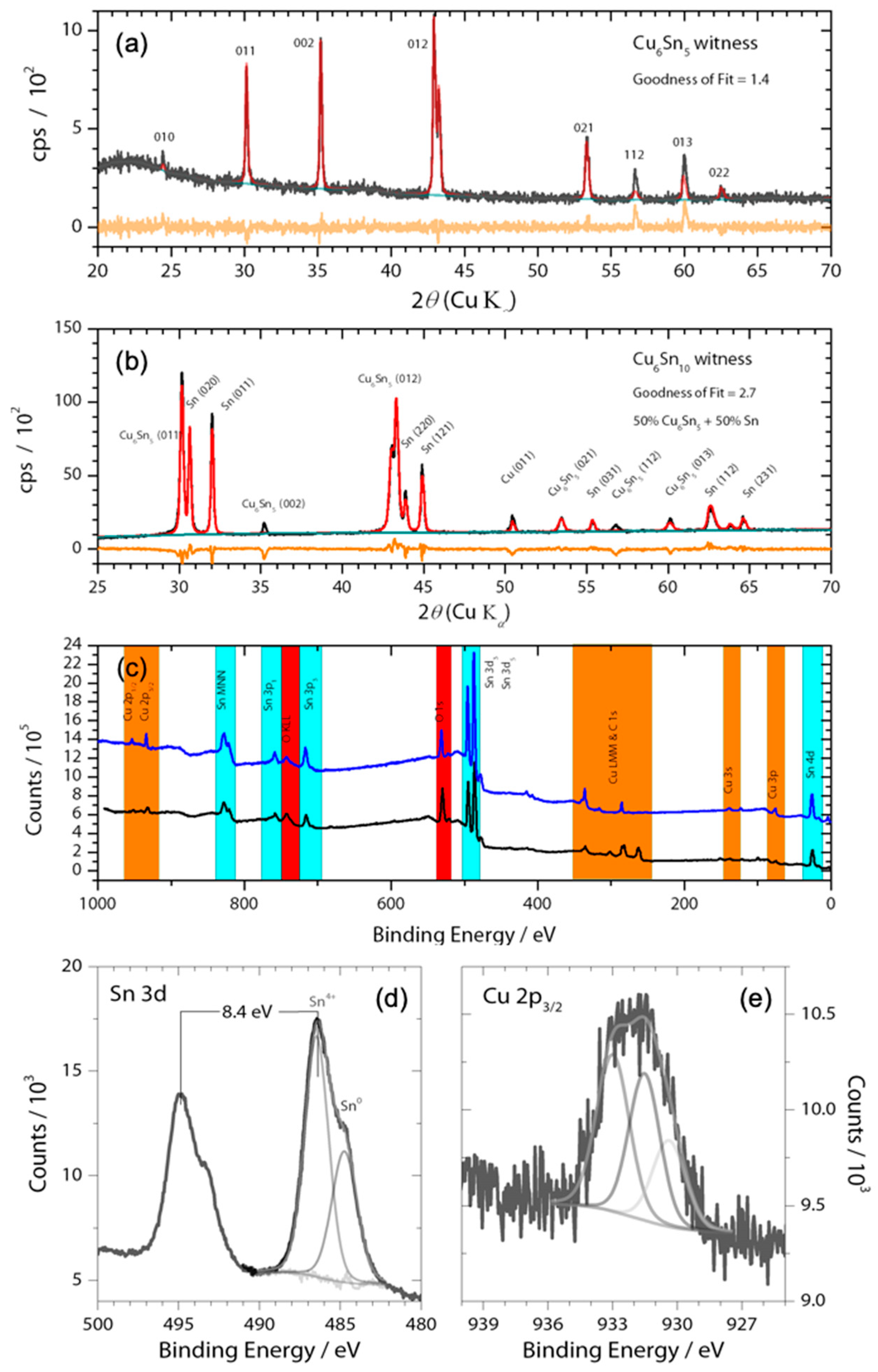
The CuSn alloys were deposited from Cu6Sn5 or Cu6Sn10 targets onto nickel foils. X-ray diffraction experiments showed that the thin film generated by sputtering the Cu6Sn5 target belonged to the P63/mmc space group (Figure 1a), which is indicative of hexagonal η-Cu6Sn5. By contrast, the thin film generated by sputtering the Cu6Sn10 target was shown to be a mixture of η-Cu6Sn5 and pure Sn phases. Given that thin films typically have a preferred orientation, the refinement of the diffraction pattern was kept qualitative for phase identification. The stoichiometry of these deposits was confirmed by XPS analysis (Figure 1); however, these experiments also showed evidence of the presence of a native Sn-oxide layer (Figure 1c,d) on the film surface that was at least 2-nm thick. The Cu 2p3/2 peak appears to be composed of three species. There appears to be Cu2O and CuO given the signal at 933.2 and 930.5 eV, respectively [23]. The Cu 2p3/2 peak in Cu6Sn5 has been found to be around 932.6 eV [24], which agrees with our value of 931.6 eV (recall 0.9 eV offset). The sputtering produced a rough and polycrystalline surface with sub-micron grains, as demonstrated by the SEM images shown in Figure 2. The presence of a pure Sn phase in Cu6Sn10 qualitatively appears to decrease the average particle size of this sputtered film, as evidenced by the SEM images of Figure 2.
3.2. Catalyst Activity
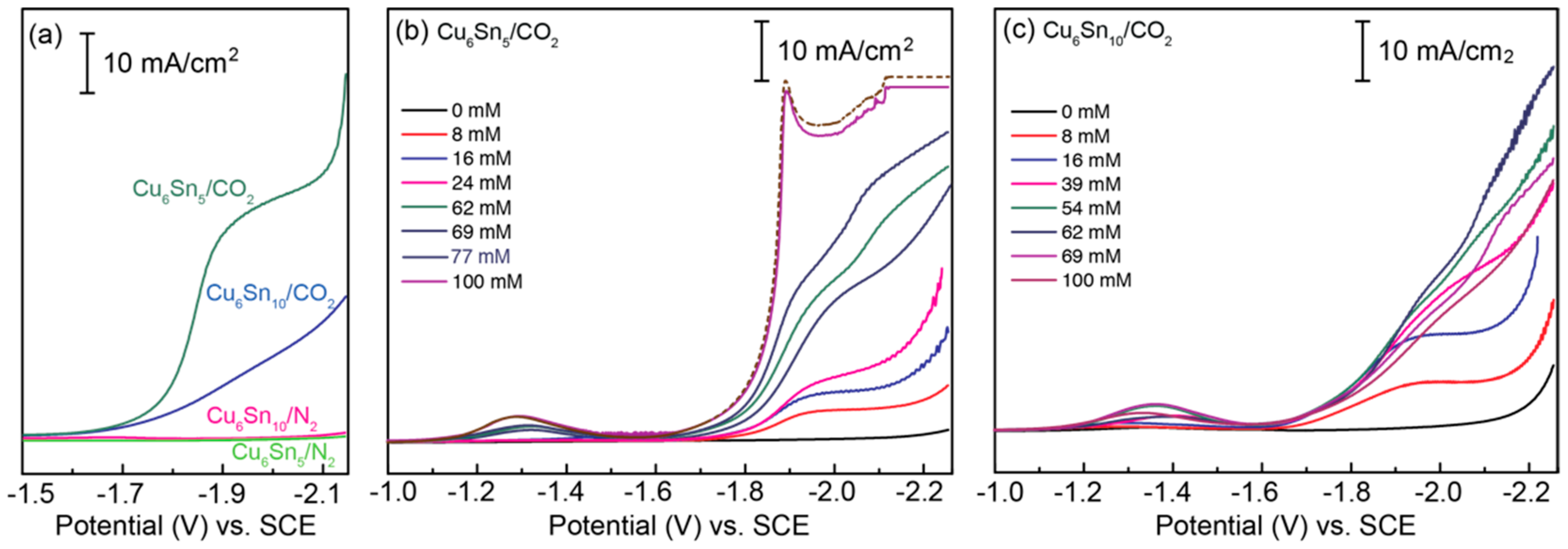
Figure 3a shows linear sweep voltammograms recorded for the CuSn alloys submerged in electrolyte solutions containing 0.1M TBAPF6 and 70 mM of the ionic liquid (IL) additive [BMIM]OTf, under an atmosphere of either N2 or CO2. Notably, no current response is observed for either CuSn film in the absence of CO2, consistent with the voltammetric response being correlated with CO2RR activity. Panels (b) and (c) of Figure 3 demonstrate the effect of varying the concentration of [BMIM]OTf on the CO2RR activity of both CuSn alloys. Notably, in the absence of the [BMIM]+-based IL, no CO2 activation takes place, indicating that the [Im]+ cation is critical to the observed CO2RR and voltammetric responses. The addition of [BMIM]OTf to the CO2-saturated solution clearly shows CO2RR activity with an onset potential of E ~ −1.65 V vs. SCE for both the Cu6Sn5 and Cu6Sn10 films, but only in the presence of CO2 (Figure 3a). The need for both CO2 and [BMIM]+ to observe the current responses of Figure 3 is consistent with the imidazolium-assisted activation of CO2 by the CuSn films, as opposed to direct [BMIM]+ reduction [13,20].
Alteration of the Cu/Sn ratio of the thin-film catalysts impacts their activity toward CO2 activation; for Cu6Sn5, the maximum current is reached at ~100 mM [BMIM]OTf, and that of Cu6Sn10 is reached at a concentration of ~60 mM (Figure 3). The CO2RR current increases exponentially with the potential for Cu6Sn5, while for the electrode with excess Sn, the current increases linearly between −1.65 and −1.9 V, suggesting different limiting kinetics for the two systems, even at the relatively fast sweep rates (ν = 100 mV/s) used to collect the voltammetry data shown in Figure 3.
Controlled potential electrolysis (CPE) of CO2 saturated solutions of MeCN containing 0.1 M TBAPF6 and [BMIM]OTf at −1.95 V versus SCE, were carried out for both the Cu6Sn5 and Cu6Sn10 cathodes. Table 1 summarizes how the CPE selectivities for the reduction of CO2 to CO (FECO) are influenced by the amount of Sn in the alloys. The two alloys promote the evolution of CO with FECO = 34% and 41% for Cu6Sn5 and Cu6Sn10, respectively. In both cases, only minimal H2 production is observed (FEH2 < 0.1%). The suppression of H2 evolution observed for the CuSn alloys in the presence of [BMIM]+ is akin to that which has been observed for Bi and Sn cathodes in the presence of nonaqueous [Im]+-based electrolytes [16,20]. The proton equivalents needed for both the CO2RR and H2 evolution catalysis likely come from adventitious water in the MeCN-based electrolyte, and the weakly acidic [BMIM]+ cations (vide infra).
We have previously reported CPE with bare supporting Ni-electrodes in MeCN containing 0.1 M TBAPF6 and [BMIM]OTf at −1.95 V versus SCE, and demonstrated that no CO2RR takes place under these conditions [16,17]. Accordingly, the CO evolution reported in Table 1 must be promoted by the CuSn alloys. We note that the electrocatalytic formation of multicarbon products was not observed by NMR, consistent with prior studies of the CO2RR on CuSn alloys under aqueous conditions [21]. The current densities for the CO2RR are lower than those reported for electrodeposited non-noble metal cathodes such as Bi and Sn, which may be due to the electrodeposited platforms having much higher specific surface areas [16] compared with the evaporated CuSn films studied here. This difference in surface area (and therefore activity) is to be expected, as vapor depositions typically produce a smoother surface than those generated using electrodeposition methods. As such, we expect that CuSn alloys may demonstrate improved kinetics for the CO2RR if synthesized through controlled co-electrodeposition methods or if used in nanoparticle form [25].
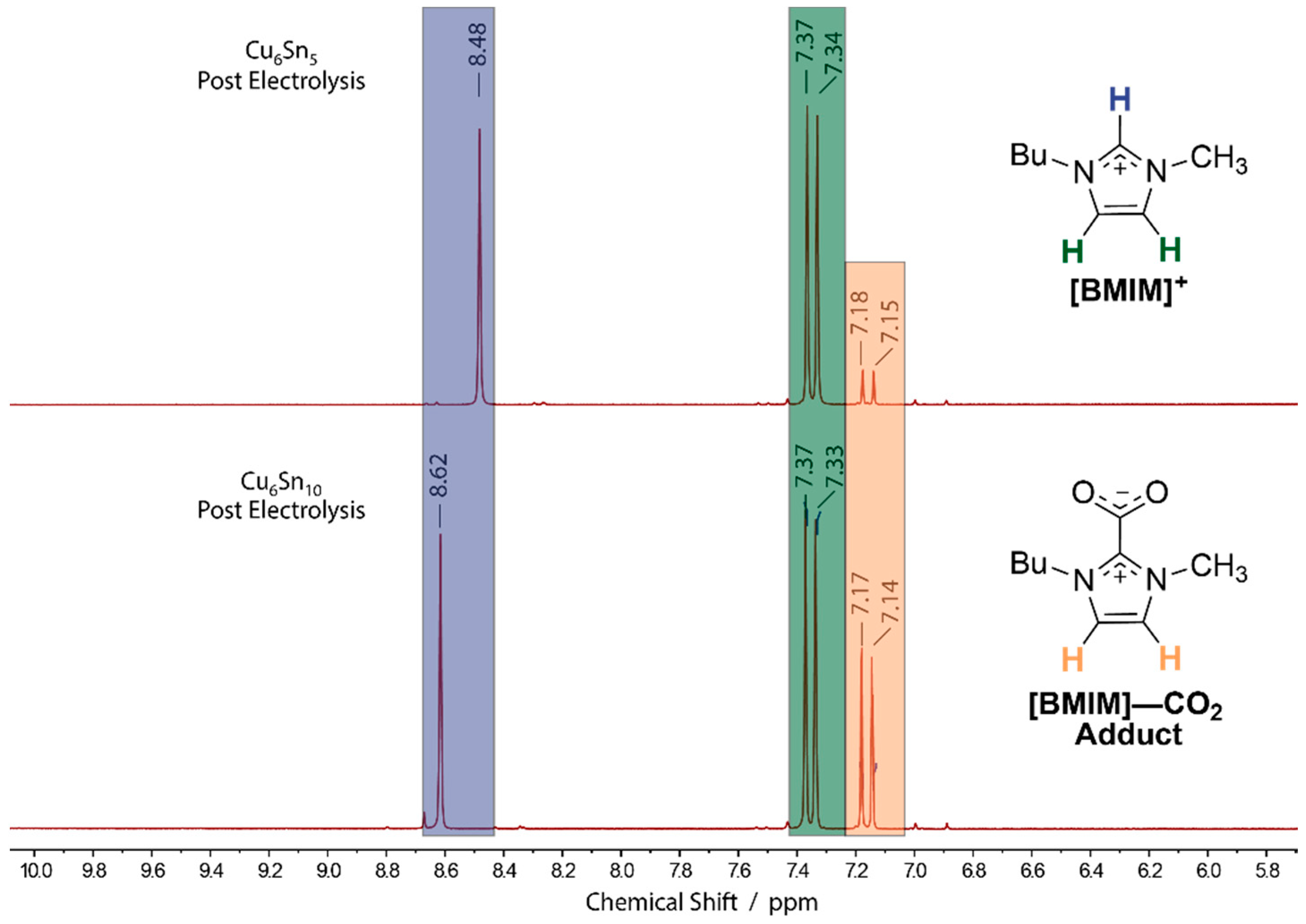
The formation of nonvolatile CO2RR products was analyzed by 1H NMR spectroscopy. Neither formate, formaldehyde, nor methanol, which are all common nonvolatile C1 products formed from reduction of CO2 were detected in the electrolyte solution following CPE experiments using either of the CuSn alloys as electrocatalysts. However, as shown in Figure 4, a product formed from the [BMIM]+ cation was observed by 1H NMR. In particular, the analysis of the post electrolysis solution showed both distinct peaks for the starting [BMIM]+ cation as well as peaks corresponding to the zwitterionic [BMIM]–CO2 adduct, as characterized by the diagnostic signals at 7.16 ppm in the 1H NMR spectra [26,27,28]. The structure of the [BMIM]–CO2 adduct is shown in Figure 5. We note that other than the starting [BMIM]+ and TBA+ cations, the [BMIM]–CO2 adduct is the major solution-phase product following CPE and CO2RR for both the C6Sn5 and C6Sn10 alloys, suggesting that the majority of the charge passed during the CPE experiments may be directed toward the activation of the [BMIM]+ as opposed to CO2. Importantly, we note that this reactivity is distinct from that observed for Bi and Sn in MeCN-based electrolytes containing [Im]+-based cations [13,16,17].
The NMR spectra show that significantly less of the [BMIM]–CO2 adduct is formed upon CPE with the Cu6Sn5 alloy compared with the analogous experiments employing the C6Sn10 thin film, as judged by the relative intensities of the adduct peaks at ~7.16 ppm versus those for the pristine [BMIM]+ cation at ~7.35 ppm, on both panels of Figure 4. We note that the position of the C(2)–H 1H NMR resonance of [BMIM]+ varies between ~8.4–8.6 ppm for the two post-CPE spectra shown in Figure 5. This variance is likely due to hydrogen-bonding interactions between the anionic oxygen atoms of the electrochemically generated [BMIM]–CO2 adduct and the acidic C(2)–H proton residue of [BMIM]+ in the solution [28]. Consistent with this rationale ([BMIM]–CO2•••[BMIM]+) is the observation that the C(2)–H residue of [BMIM]+ appears further downfield (Figure 4, bottom) with increasing concentrations of the [BMIM]–CO2 adduct. The formation of the [BMIM]–CO2 adduct is undesirable because of its ability to bind strongly to acidic protons and catalyst surfaces [15,28]. As such, the observation (vide supra) that Cu6Sn10 displays a lower apparent rate order and current density for the CO2RR is consistent with an enhanced production of [BMIM]–CO2 with the more Sn-rich catalyst film.
3.3. Adsorption Pre-Catalysis Peak
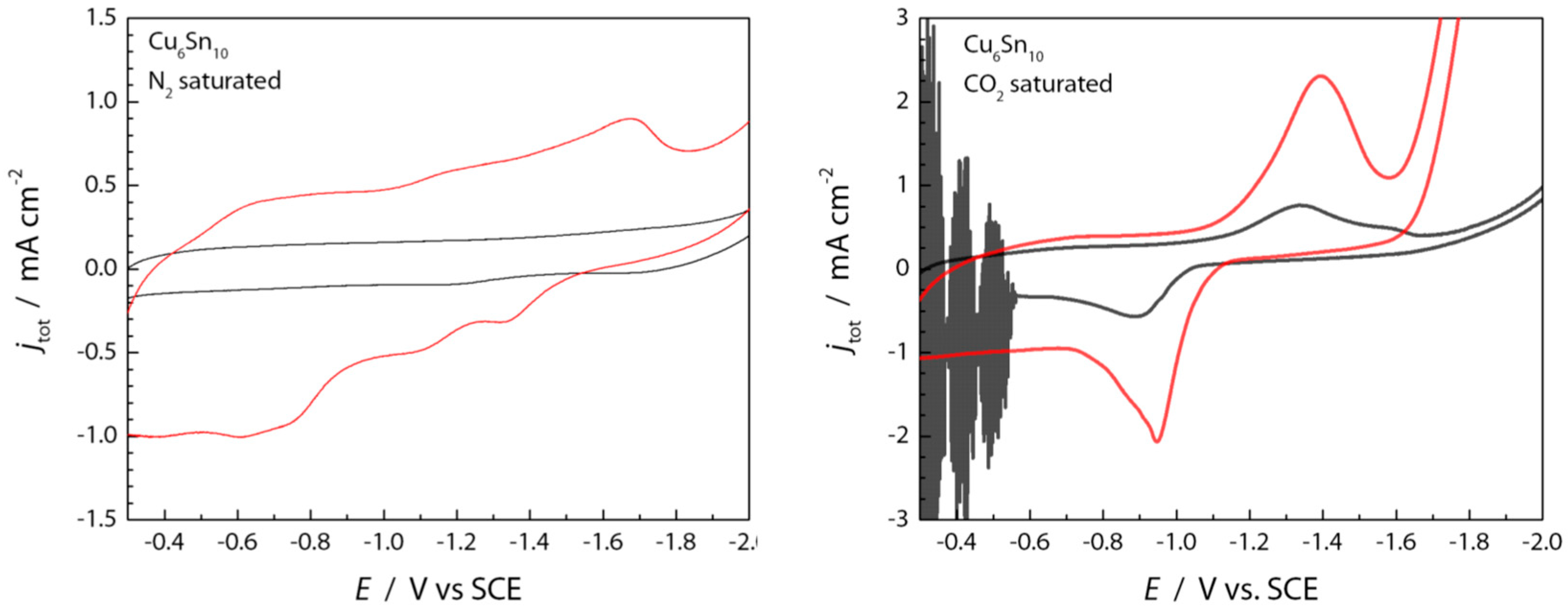
There is a clear pre-catalysis wave in the LSV traces shown in Figure 3, centered around −1.4 V. Further inspection of the double layer regions by cyclic voltammetry (Figure 5) shows a clear quasi-reversible peak that is only observed in CO2-saturated electrolyte solutions (Figure 5, right). The addition of 100 mM [BMIM]+ to the N2-saturated MeCN containing 0.1 M TBAPF6 (Figure 5, left) causes an increase of charge stored within the double layer region [29,30], which may be presumed to be due to the adsorption of [BMIM]+ at the electrode/electrolyte interface of the negatively polarized CuSn film [31,32,33]. Similar phenomena have been observed for Au electrodes [34,35,36,37], and it has been demonstrated that [Im]+ cations prefer to π-stack and lie flat on electrode surfaces, thereby supporting more capacitive charge build-up than an inner layer containing large TBA+ cations [38,39,40].
As shown in the right panel of Figure 5, the presence of [BMIM]+ appears to increase the cathodic (at −1.35 V) and anodic (at −0.90 V) waves associated with the precatalytic CO2 binding wave. Imidazolium cations are known to coordinate strongly with CO2 through π-π interactions, hydrogen bonding, and formation of CO2 adducts [28]. As is also shown in the right panel of Figure 5 (black trace), a quasi-reversible wave is observed for CV traces recorded for the CuSn film in MeCN containing 0.1M TBAPF6 under CO2. This wave arises at potentials more positive than those required for CO2RR catalysis, suggesting that the current response originates from the electrosorption of CO2. The current response associated with this wave increases in the presence of 0.1 M [BMIM]+ (Figure 5, right, red trace), suggesting that [BMIM]+ is assisting in the CO2 adsorption/activation, potentially via cooperative interactions between [BMIM]+, CO2, and the negatively polarized CuSn film [17,41].
4. Conclusions
Electrochemical reduction of CO2 to fuels using Cu-based cathodes and powered by renewable sources of electric current have attracted recent attention [42]. To date, however, CuSn alloys have been largely ignored as cathode materials for CO2 reduction; however, these alloys are commonly available and inexpensive to produce, making them attractive material for the development of CO2RR platforms. In this work, we have shown that sputtering CuSn thin film alloys produces submicron-sized particulates on Ni substrates. The CO2RR activities of Cu6Sn5 and C6Sn10 films were ascertained by voltammetry and CPE experiments. When used in conjunction with imidazolium-based nonaqueous electrolytes, the CuSn films show activity for the reduction of CO2 to CO at E = −1.6 V, and display modest Faradaic efficiencies for this transformation at an applied potential of E = −1.95 V. The CuSn alloy containing the higher ratio of Sn displayed lower efficiency and slower kinetics for the conversion of CO2 to CO compared with Cu6Sn5. CPE experiments conducted using both CuSn materials in the presence of CO2 and the [BMIM]+-containing electrolyte lead to deprotonation of the imidazolium and formation of the corresponding [BMIM]–CO2 adduct, as judged by 1H NMR experiments. Interestingly, the C6Sn10 material showed higher levels of [BMIM]–CO2 generation, while displaying lower efficiency and activity for CO production, suggesting that the formation of the imidazolium–CO2 adduct inhibits the CO2RR with CuSn-based cathodes. We note that this deleterious reaction mode involving the [BMIM]+ electrolyte is not an issue for CuSn alloys employed for the CO2RR with aqueous electrolytes that do not contain imidazolium cations. Accordingly, unlike other non-noble metal cathodes we have studied, CuSn alloys may be most useful for the CO2RR when restricted to aqueous electrolyzer designs.
When both of the CuSn cathode materials are not utilized under aqueous conditions, the presence of the [BMIM]+ promoter in the CO-saturated MeCN-based electrolyte is critical to the observed CO2RR activity. In addition to providing the proton equivalents required to drive the 2e−/2H+ conversion of CO2 to CO, the [BMIM]+ ion also appears to promote the electrosorption of CO2 at the CuSn/electrolyte interface, as judged by voltammetric experiments. As such, the interaction of the imidazolium electrolyte with the polarized CuSn cathodes and CO2 is likely critical to improving the kinetics of the electrocatalysis, since the formation of surface-adsorbed CO2 intermediates is energetically unfavorable and often rate-limiting for electrochemical CO2RR platforms [41].
In future work, advanced spectroscopic methods will be useful tools as we seek to better understand the precise interplay between the adsorbed [Im]+ (and other electrolyte cations) and the cathode surface, and how the electrode/electrolyte interfacial structure affects CO2 adsorption and reduction [39]. Recent work demonstrating that the interplay between adsorbed [Im]+ cations and other weakly acidic organic cations can be used to significantly enhance CO2 reduction, as well as tune CO2RR product distributions [17,29]. As such, further improvements in this aspect of the CO2RR require the continued lowering of CO2 adsorption/activation energy using electrolyte promoters, and the leveraging of [cation]+•••cathode interactions may be especially critical in this regard.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript and contributed equally.
Funding
The experiments and authors were supported as part of the Fluid Interface Reactions, Structures and Transport (FIRST) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (BES-DOE).
Acknowledgments
The experiments and authors were supported as part of the Fluid Interface Reactions, Structures and Transport (FIRST) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (BES-DOE). This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| [BMIM]+ | n-butyl-N′-methyl imidazolium |
| CPE | controlled potential electrolysis |
| DC | direct current |
| FE | Faradaic efficiency |
| GC | gas chromatography |
| NMR | nuclear magnetic resonance |
| TBA+ | tetrabutylammonium |
| OTf | triflate |
| SCE | standard calomel electrode |
| XPS | X-ray photoelectron spectroscopy |
References
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, T.R.; Dogutan, D.K.; Reece, S.Y.; Surendranath, Y.; Teets, T.S.; Nocera, D.G. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem. Rev. 2010, 110, 6474–6502. [Google Scholar] [CrossRef] [PubMed]
- US Energy Information Administration. Monthly Energy Review—May 2018; U.S. Energy Information Administration: Washington, DC, USA, 2018.
- Karl, T.R.; Trenberth, K.E. Modern global climate change. Science 2003, 302, 1719–1723. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Prakash, G.K.S.; Goeppert, A. Anthropogenic chemical carbon cycle for a sustainable future. J. Am. Chem. Soc. 2011, 133, 12881–12898. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels. technological use of CO2. Chem. Rev. 2013, 11, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.R.; Siang, Y.B. Recent advances in understanding mechanisms for the electrochemical reduction of carbon dioxide. Curr. Opin. Electrochem. 2018, 8, 126–134. [Google Scholar]
- Rosenthal, J. Progress Toward the Electrocatalytic Production of Liquid Fuels from Carbon Dioxide; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 299–338. [Google Scholar]
- Rosen, B.A.; Salehi-Khojin, A.; Thorson, M.R.; Zhu, W.; Whipple, D.T.; Kenis, P.J.A.; Masel, R.I. Ionic liquid-mediated selective conversion of CO2 to CO at low overpotentials. Science 2011, 334, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chi, M.; Veith, G.M.; Zhang, P.; Lutterman, D.A.; Rosenthal, J.; Overbury, S.H.; Dai, S.; Zhu, H. Rational design of Bi nanoparticles for efficient electrochemical CO2 Reduction: The elucidation of size and surface condition effects. ACS Catal. 2016, 6, 6255–6264. [Google Scholar] [CrossRef]
- Sun, L.; Ramesha, G.K.; Kamat, P.V.; Brennecke, J.F. Switching the reaction course of electrochemical CO2 reduction with ionic liquids. Langmuir 2014, 30, 6302–6308. [Google Scholar] [CrossRef]
- Tanner, E.E.L.; Batchelor-Mcauley, C.; Compton, R.G. Carbon dioxide reduction in room-temperature ionic liquids: the effect of the choice of electrode material, cation, and anion. J. Phys. Chem. C 2016, 120, 26442–26447. [Google Scholar] [CrossRef]
- Medina-Ramos, J.; DiMeglio, J.L.; Rosenthal, J. Efficient reduction of CO2 To CO with high current density using in situ or ex situ prepared Bi-based materials. J. Am. Chem. Soc 2014, 136, 8361–8367. [Google Scholar] [CrossRef] [PubMed]
- Winikoff, S.G.; DiMeglio, J.L.; Chemburkar, A.; Medina-Ramos, J.; Rosenthal, J.; Neurock, M. Cooperativity between bismuth electrodes and ionic liquid electrolytes promotes efficient electrocatalytic reduction of CO2 to CO. Nat. Commun. 2018. submitted. [Google Scholar]
- Matsubara, Y.; Grills, D.C.; Kuwahara, Y. Thermodynamic aspects of electrocatalytic CO2 reduction in acetonitrile and with an ionic liquid as solvent or electrolyte. ACS Catal. 2015, 5, 6440–6452. [Google Scholar] [CrossRef]
- Medina-Ramos, J.; Pupillo, R.C.; Keane, T.P.; DiMeglio, J.L.; Rosenthal, J. Efficient conversion of CO2 to CO using tin and other inexpensive and easily prepared post-transition metal catalysts. J. Am. Chem. Soc. 2015, 137, 5021–5027. [Google Scholar] [CrossRef] [PubMed]
- Atifi, A.; Boyce, D.W.; DiMeglio, J.L.; Rosenthal, J. Directing the outcome of CO2 reduction at bismuth cathodes using varied ionic liquid promoters. ACS Catal. 2018, 8, 2857–2863. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Motoo, S. The electrochemical behavior of ad-atoms and their effect on hydrogen evolution: Part I. Order-disorder rearrangement of copper ad-atoms on platinum. J. Electroanal. Chem. 1976, 72, 165–175. [Google Scholar] [CrossRef]
- Furuya, N.; Motoo, S. The electrochemical behavior of ad-atoms and their effect on hydrogen evolution: Part IV. Tin and lead ad-atoms on platinum. J. Electroanal. Chem. 1979, 98, 195–202. [Google Scholar] [CrossRef]
- DiMeglio, J.L.; Rosenthal, J. Selective conversion of CO2 to CO with high efficiency using an inexpensive bismuth-based electrocatalyst. J. Am. Chem. Soc 2013, 135, 8798–8801. [Google Scholar] [CrossRef]
- Morimoto, M.; Takatsuji, Y.; Yamasaki, R.; Hashimoto, H.; Nakata, I.; Sakakura, T.; Haruyama, T. Electrodeposited Cu-Sn alloy for electrochemical CO2 reduction to CO/HCOO−. Electrocatalysis 2018, 9, 323–332. [Google Scholar] [CrossRef]
- Baggetto, L.; Jumas, J.-C.; Górka, J.; Bridges, C.A.; Veith, G.M. Predictions of particle size and lattice diffusion pathway requirements for sodium-ion anodes using H-Cu6Sn5 thin films as a model system. Phys. Chem. Chem. Phys. 2013, 15, 10885–10894. [Google Scholar] [CrossRef]
- Moulder, J.F. Handbook of X-ray Photoelectron Spectroscopy; Physical Electronics: Eden Prairie, MN, USA, 1995. [Google Scholar]
- Naille, S.; Dedryvère, R.; Martinez, H.; Leroy, S.; Lippens, P.E.; Jumas, J.C.; Gonbeau, D. XPS study of electrode/electrolyte interfaces of H-Cu6Sn5 electrodes in li-ion batteries. J. Power Sources 2007, 174, 1086–1090. [Google Scholar] [CrossRef]
- Gawande, M.B.; Goswami, A.; Felpin, F.-X.; Asefa, T.; Huang, X.; Silva, R.; Zou, X.; Zboril, R.; Varma, R.S. Cu and Cu-based nanoparticles: Synthesis and applications in catalysis. Chem. Rev. 2016, 116, 3722–3811. [Google Scholar] [CrossRef] [PubMed]
- Cabaço, M.I.; Besnard, M.; Chávez, F.V.; Pinaud, N.; Sebastião, P.J.; Coutinho, J.A.P.; Danten, Y. Understanding chemical reactions of CO2 and its isoelectronic molecules with 1-butyl-3-methylimidazolium acetate by changing the nature of the cation: The case of CS2 in 1-butyl-1-methylpyrrolidinium acetate studied by NMR spectroscopy and density functional theory calculations. J. Chem. Phys. 2014, 140, 244307. [Google Scholar] [PubMed]
- Hanc-Scherer, F.A.; Montiel, M.A.; Montiel, V.; Herrero, E.; Sánchez-Sánchez, C.M. Surface structured platinum electrodes for the electrochemical reduction of carbon dioxide in imidazolium based ionic liquids. Phys. Chem. Chem. Phys. 2015, 17, 23909–23916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J.L.; Unali, G.; Perosa, A. A “By-productless” cellulose foaming agent for use in imidazolium ionic liquids. Chem. Commun. 2011, 47, 2970–2972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Han, Y.; Wang, Y.; Ye, S.; Yan, T. Comparing the differential capacitance of two ionic liquid electrolytes: Effects of specific adsorption. Electrochem. Commun. 2014, 38, 44–46. [Google Scholar] [CrossRef]
- Gore, T.R.; Bond, T.; Zhang, W.; Scott, R.W.J.; Burgess, I.J. Hysteresis in the measurement of double-layer capacitance at the gold–ionic liquid interface. Electrochem. Commun. 2010, 12, 1340–1343. [Google Scholar] [CrossRef]
- Moganty, S.S.; Baltus, R.E.; Roy, D. Electrochemical windows and impedance characteristics of [Bmim+][] and [Bdmim+][] ionic liquids at the surfaces of Au, Pt, Ta and glassy carbon electrodes. Chem. Phys. Lett. 2009, 483, 90–94. [Google Scholar] [CrossRef]
- Fedorov, M.V.; Kornyshev, A.A. Ionic liquids at electrified interfaces. Chem. Rev. 2014, 114, 2978–3036. [Google Scholar] [CrossRef]
- Kirchner, K.; Kirchner, T.; Ivaništšev, V.; Fedorov, M.V. Electrical double layer in ionic liquids: Structural transitions from multilayer to monolayer structure at the interface. Electrochim. Acta 2013, 110, 762–771. [Google Scholar] [CrossRef]
- Black, J.M.; Walters, D.; Labuda, A.; Feng, G.; Hillesheim, P.C.; Dai, S.; Cummings, P.T.; Kalinin, S.V.; Proksch, R.; Balke, N. Bias-dependent molecular-level structure of electrical double layer in ionic liquid on graphite. Nano Lett. 2013, 13, 5954–5960. [Google Scholar] [CrossRef] [PubMed]
- Uysal, A.; Zhou, H.; Feng, G.; Lee, S.S.; Li, S.; Cummings, P.T.; Fulvio, P.F.; Dai, S.; McDonough, J.K.; Gogotsi, Y.; et al. Interfacial ionic “liquids”: Connecting static and dynamic structures. J. Phys. Condens. Matter 2014, 27. [Google Scholar] [CrossRef] [PubMed]
- Lauw, Y.; Horne, M.D.; Rodopoulos, T.; Lockett, V.; Akgun, B.; Hamilton, W.A.; Nelson, A.R.J. Structure of [C 4mpyr][NTf 2] room-temperature ionic liquid at charged gold interfaces. Langmuir 2012, 28, 7374–7381. [Google Scholar] [CrossRef] [PubMed]
- García Rey, N.; Dlott, D.D. Structural transition in an ionic liquid controls CO2 electrochemical reduction. J. Phys. Chem. C 2015, 119, 20892–20899. [Google Scholar] [CrossRef]
- Siinor, L.; Siimenson, C.; Ivaništšev, V.; Lust, K.; Lust, E. Influence of cation chemical composition and structure on the double layer capacitance for Bi(111)|room temperature ionic liquid interface. J. Electroanal. Chem. 2012, 668, 30–36. [Google Scholar] [CrossRef]
- Yanson, A.I.; Rodriguez, P.; Garcia-Araez, N.; Mom, R.V.; Tichelaar, F.D.; Koper, M.T.M. Cathodic corrosion: A quick, clean, and versatile method for the synthesis of metallic nanoparticles. Angew. Chem. Int. Ed. 2011, 50, 6346–6350. [Google Scholar] [CrossRef]
- Hersbach, T.J.P.; Yanson, A.I.; Koper, M.T.M. Anisotropic etching of platinum electrodes at the onset of cathodic corrosion. Nat. Commun. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Tian, Z.; Priest, C.; Chen, L. Recent progress in the theoretical investigation of electrocatalytic reduction of CO2. Adv. Theory Simul. 2018, 1, 1800004–1800023. [Google Scholar] [CrossRef]
- Kunene, T.; Xiong, L.; Rosenthal, J. Solar-powered synthesis of hydrocarbons from carbon dioxide and water. Proc. Natl. Acad. Sci. USA 2019, 116, 9693–9695. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
(a,b) X-ray diffractogram of witness Cu6Sn5 and Cu6Sn10 thin films along with Rietveld refinement using Highscore Plus and ICDD; black lines are raw data, red lines are the fit data, and orange lines the residual; (c) Survey XPS (Mg source) spectrum of the thin films on nickel foil current collectors showing photoelectron emissions from only O, adventitious C, Cu, Sn, and Ni substrate. High resolution XPS spectra of (d) Sn 3d and (e) Cu 2p3/2 along with peak fittings highlights the presence of oxides within the Cu5Sn6 film. Note: Sn2+ may also be present in the Sn 3d XPS spectrum, as Sn4+ and Sn2+ have similar binding energies that differ by only ~0.6 eV. No corrections to binding energy were made.
Figure 1.
(a,b) X-ray diffractogram of witness Cu6Sn5 and Cu6Sn10 thin films along with Rietveld refinement using Highscore Plus and ICDD; black lines are raw data, red lines are the fit data, and orange lines the residual; (c) Survey XPS (Mg source) spectrum of the thin films on nickel foil current collectors showing photoelectron emissions from only O, adventitious C, Cu, Sn, and Ni substrate. High resolution XPS spectra of (d) Sn 3d and (e) Cu 2p3/2 along with peak fittings highlights the presence of oxides within the Cu5Sn6 film. Note: Sn2+ may also be present in the Sn 3d XPS spectrum, as Sn4+ and Sn2+ have similar binding energies that differ by only ~0.6 eV. No corrections to binding energy were made.
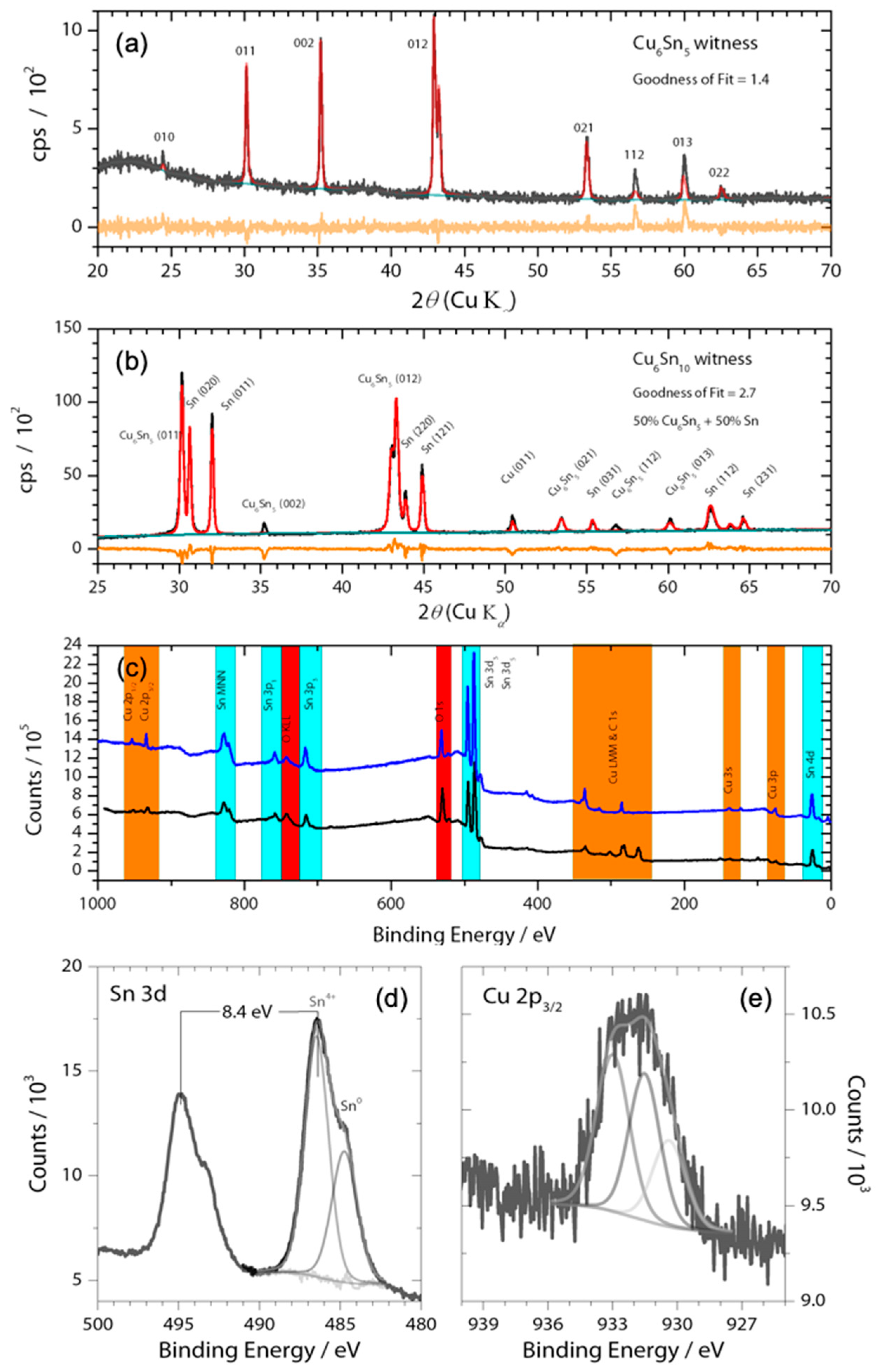
Figure 2.
Scanning electron micrograph (SEM) images recorded for evaporated thin films of (a) Cu6Sn5 and (b) Cu6Sn10. The scale bars correspond to a length of 1.0 μm.
Figure 2.
Scanning electron micrograph (SEM) images recorded for evaporated thin films of (a) Cu6Sn5 and (b) Cu6Sn10. The scale bars correspond to a length of 1.0 μm.

Figure 3.
Linear sweep voltammograms (LSVs) recorded for (a) CuSn thin films in MeCN containing 0.1 M TBAPF6 and 70 mM [BMIM]OTf under either CO2 or N2; (b) Cu6Sn5 in MeCN containing 0.1 M TBAPF6 and varying concentrations of [BMIM]OTf under CO2; (c) Cu6Sn10 in MeCN containing 0.1 M TBAPF6 and varying concentrations of [BMIM]OTf under CO2. Notably, no appreciable current response is observed for either CuSn alloy in the absence of CO2 or [BMIM]OTf. All LSV data were recorded at a sweep rate of ν = 100 mV/s. Note: LSVs recorded for Cu6Sn5 in the presence of more than 100 mM [BMIM]OTf do not change appreciably. A representative LSV obtained for Cu6Sn5 with more than 100 mM of [BMIM]OTf is represented by the dashed brown trace in panel (b).
Figure 3.
Linear sweep voltammograms (LSVs) recorded for (a) CuSn thin films in MeCN containing 0.1 M TBAPF6 and 70 mM [BMIM]OTf under either CO2 or N2; (b) Cu6Sn5 in MeCN containing 0.1 M TBAPF6 and varying concentrations of [BMIM]OTf under CO2; (c) Cu6Sn10 in MeCN containing 0.1 M TBAPF6 and varying concentrations of [BMIM]OTf under CO2. Notably, no appreciable current response is observed for either CuSn alloy in the absence of CO2 or [BMIM]OTf. All LSV data were recorded at a sweep rate of ν = 100 mV/s. Note: LSVs recorded for Cu6Sn5 in the presence of more than 100 mM [BMIM]OTf do not change appreciably. A representative LSV obtained for Cu6Sn5 with more than 100 mM of [BMIM]OTf is represented by the dashed brown trace in panel (b).
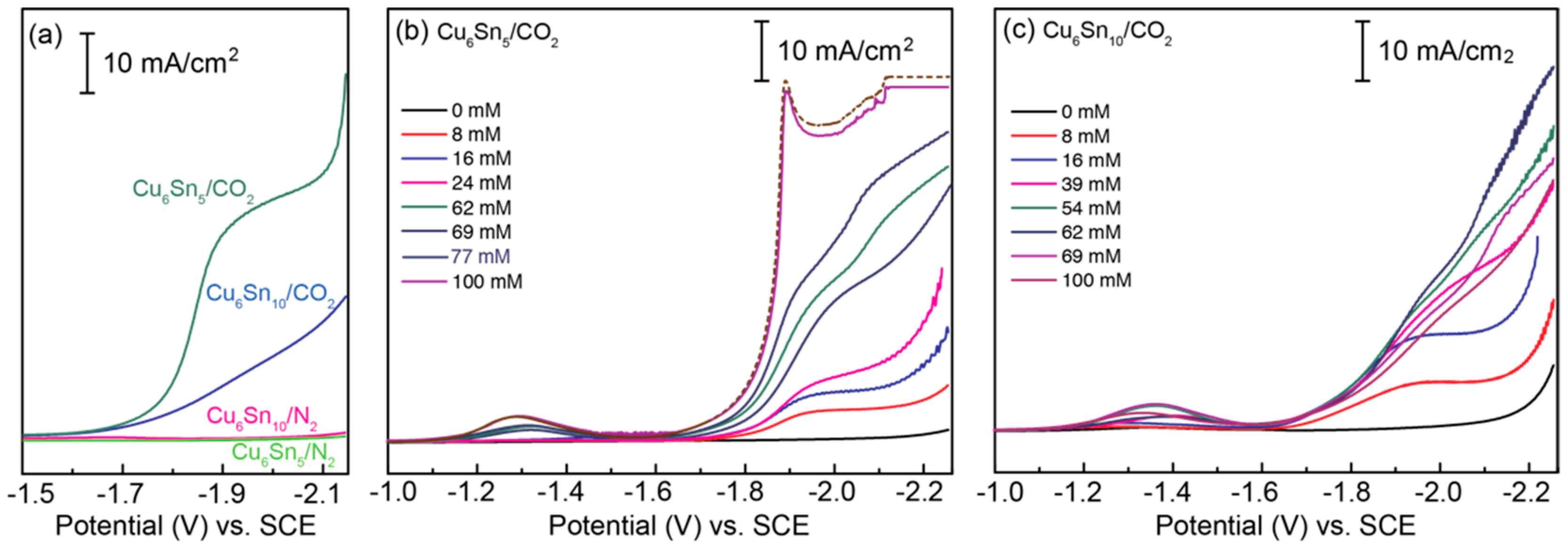
Figure 4.
1H NMR of the electrolyte solutions following a 3-h controlled potential electrolysis (CPE) at −1.95 V (SCE) using either (top) Cu6Sn5 or (bottom) Cu6Sn10 cathodes. CPE experiments employed CO2-saturated MeCN solutions containing 0.1 M TBAPF6 and [BMIM]OTf. Protonic resonances from the [BMIM]+ and [BMIM]–CO2 adduct are distinguished by color (blue, green, and orange).
Figure 4.
1H NMR of the electrolyte solutions following a 3-h controlled potential electrolysis (CPE) at −1.95 V (SCE) using either (top) Cu6Sn5 or (bottom) Cu6Sn10 cathodes. CPE experiments employed CO2-saturated MeCN solutions containing 0.1 M TBAPF6 and [BMIM]OTf. Protonic resonances from the [BMIM]+ and [BMIM]–CO2 adduct are distinguished by color (blue, green, and orange).

Figure 5.
Cyclic voltammograms (CV) highlighting the electrochemical double layer region (N2 sat) and the quasi-reversible pre-catalytic peak (CO2 sat) with (red) and without (black) [BMIM]OTf present. CV scans were initiated to negative potentials.
Figure 5.
Cyclic voltammograms (CV) highlighting the electrochemical double layer region (N2 sat) and the quasi-reversible pre-catalytic peak (CO2 sat) with (red) and without (black) [BMIM]OTf present. CV scans were initiated to negative potentials.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Steady state Faradaic efficiencies (FE) and current density (jtot) for controlled potential electrolysis at −1.95 V (SCE) using CuSn Alloys in MeCN-based electrolytes. Electrolysis experiments were conducted for 2.5 h. Table rows for experiments conducted in the absence of [BMIM]OTf are highlighted in blue.
Table 1.
Steady state Faradaic efficiencies (FE) and current density (jtot) for controlled potential electrolysis at −1.95 V (SCE) using CuSn Alloys in MeCN-based electrolytes. Electrolysis experiments were conducted for 2.5 h. Table rows for experiments conducted in the absence of [BMIM]OTf are highlighted in blue.
| Film | Electrolyte | FECO | FEH2 | jtot (mA·cm−2) |
|---|---|---|---|---|
| Cu6Sn5 | TBAPF6 | 41.1 ± 3.2% | 4.4 ± 0.3% | 0.44 ± 0.1 |
| TBAPF6 + [BMIM]OTf | 34.4 ± 2.7% | <0.1% | 3.9 ± 0.2 | |
| Cu6Sn10 | TBAPF6 | 22.5 ± 3.8% | 3.9 ± 0.4% | 0.6 ± 0.1 |
| TBAPF6 + [BMIM]OTf | 30.5 ± 3.1% | <0.1% | 2.8 ± 0.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sacci, R.L.; Velardo, S.; Xiong, L.; Lutterman, D.A.; Rosenthal, J. Copper-Tin Alloys for the Electrocatalytic Reduction of CO2 in an Imidazolium-Based Non-Aqueous Electrolyte. Energies 2019, 12, 3132. https://doi.org/10.3390/en12163132
AMA Style
Sacci RL, Velardo S, Xiong L, Lutterman DA, Rosenthal J. Copper-Tin Alloys for the Electrocatalytic Reduction of CO2 in an Imidazolium-Based Non-Aqueous Electrolyte. Energies. 2019; 12(16):3132. https://doi.org/10.3390/en12163132
Chicago/Turabian StyleSacci, Robert L., Stephanie Velardo, Lu Xiong, Daniel A. Lutterman, and Joel Rosenthal. 2019. "Copper-Tin Alloys for the Electrocatalytic Reduction of CO2 in an Imidazolium-Based Non-Aqueous Electrolyte" Energies 12, no. 16: 3132. https://doi.org/10.3390/en12163132
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.