Atomistic Simulations of the Defect Chemistry and Self-Diffusion of Li-ion in LiAlO2


1
Department of Materials, Imperial College London, London SW7 2AZ, UK
2
Faculty of Engineering, Environment and Computing, Coventry University, Priory Street, Coventry CV1 5FB, UK
3
Solid State Physics Section, University of Athens, Panepistimiopolis Zografos, 157 84 Athens, Greece
4
Department of Mechanical Engineering, University of West Attica, 12210 Athens, Greece
*
Author to whom correspondence should be addressed.
Energies 2019, 12(15), 2895; https://doi.org/10.3390/en12152895
Submission received: 28 June 2019
/
Revised: 13 July 2019
/
Accepted: 25 July 2019
/
Published: 27 July 2019
Abstract
:Lithium aluminate, LiAlO2, is a material that is presently being considered as a tritium breeder material in fusion reactors and coating material in Li-conducting electrodes. Here, we employ atomistic simulation techniques to show that the lowest energy intrinsic defect process is the cation anti-site defect (1.10 eV per defect). This was followed closely by the lithium Frenkel defect (1.44 eV per defect), which ensures a high lithium content in the material and inclination for lithium diffusion from formation of vacancies. Li self-diffusion is three dimensional and exhibits a curved pathway with a migration barrier of 0.53 eV. We considered a variety of dopants with charges +1 (Na, K and Rb), +2 (Mg, Ca, Sr and Ba), +3 (Ga, Fe, Co, Ni, Mn, Sc, Y and La) and +4 (Si, Ge, Ti, Zr and Ce) on the Al site. Dopants Mg2+ and Ge4+ can facilitate the formation of Li interstitials and Li vacancies, respectively. Trivalent dopants Fe3+, Ni3+ and Mn3+ prefer to occupy the Al site with exoergic solution energies meaning that they are candidate dopants for the synthesis of Li (Al, M) O2 (M = Fe, Ni and Mn) compounds.

1. Introduction
The ever-increasing demand for energy generated, initiated further research into alternate methods of renewable energy production and storage methodologies for the power production. In that respect, Li-based ceramics are technologically important and considered by the community for applications ranging from nuclear fusion to batteries [1,2,3,4,5,6,7,8]. LiAlO2, a lithium based ceramic material, has been considered with the prospect of being used as a tritium breeder material in magnetic confined nuclear fusion reactors after promising initial research regarding the physical and thermodynamic properties of the material as well as the potential tritium breeding ratio (TBR) [8,9]. In addition, this material has great potential as a coating material in Li-conducting electrodes [10].
Previous studies have highlighted the importance of atomic scale calculation to gain insights on the defect processes (intrinsic defect processes, doping and diffusion) of oxides for energy applications [11,12,13,14,15]. Previous experimental and theoretical studies determined a wide range of activation energies for Li self-diffusion in γ-LiAlO2 (0.50–1.26 eV) [16,17,18,19,20,21,22]. Additionally, there are no systematic studies of the intrinsic defect processes and doping in this material. In the present study, we have employed atomistic simulations to investigate the structure, intrinsic defect processes, Li self-diffusion and the introduction of dopants.
2. Computational Methods
The present computational study was based on classical pair-wise potential calculations to describe LiAlO2 via the General Utility Lattice Program (GULP) code [23,24]. In the present approach, the total energy (lattice energy) is determined by long range (i.e., Coulombic) and short range [i.e., electron-electron repulsive and attractive intermolecular forces (van der Waals forces)]. The latter were modelled using Buckingham potentials (refer to the supplementary information). The van der Waals forces arising from the spontaneous formation of instantaneous dipoles are very important as the formation energy results are sensitive to those forces. The present modelling approach takes into the account of Van der Waals forces as a function of the interatomic distance (r) [23,24]. Two-body Buckingham potential mentioned in the supplementary information consists of two parts. The first part of the equation represents the Pauli repulsion (electron-electron) and the second part denotes the van der Waals interaction. Ionic positions and lattice parameters were relaxed using the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm [24]. Convergence criteria dictated that in relaxed configurations, forces on each atom was <0.001 eV/Å. To introduce point defects in the lattice we used the Mott-Littleton methodology [25] similarly to recent work [26,27,28]. It is established that although the present methodology may overestimate the defect formation enthalpies at the dilute limit, the trends will be unchanged [29]. In a thermodynamic perspective defect parameters can be described by comparing the real (i.e., defective) crystal to an isobaric or isochoric ideal (i.e., non-defective) crystal. Defect formation parameters can be interconnected through thermodynamic relations [30,31], with the present atomistic simulations corresponding to the isobaric parameters for the migration and formation processes [32,33,34].
3. Results
3.1. Crystal Structure, Intrinsic Defect Processes and Li Diffusion
The LiAlO2 crystal structure considered in the present study is the tetragonal γ-phase (Figure 1) with lattice parameters a = b = 5.1687 Å, c = 6.2679 Å, and α = β = γ = 90° [35]. This crystal structure consists of LiO4 and AlO4 tetrahedra. They are inter-connected via edge and corner sharing to form three dimensional channels. The calculated structural parameters are given in Table 1 and it can be seen that there is excellent agreement with experiment [35] validating the potentials (refer to Table S1 in the Supplementary Materials) used in this study.
Next, we considered the process of calculating the defect energy required for the key intrinsic defect processes such as Frenkel, Schottky and anti-site in LiAlO2. Frenkel and Schottky defect formation energies were calculated by combining individual point defects. Frenkel energy is the sum of vacancy and interstitial energies. Schottky energy was calculated by adding appropriate vacancy energies and substracting cohesive energy. Relative energetics of these defect processes are useful in predicting the electrochemical behaviour of LiAlO2. The reaction energies for the intrinsic defect processes can be represented by using the established Kröger-Vink notation [36].
The reaction energies are reported in Figure 2. The lowest intrinsic defect energy process was calculated to be the cation mixing (anti-site) in which Li and Al exchange their atomic positions. This defect was noted in various oxide materials experimentally and theoretically [37,38,39,40,41,42,43,44,45,46,47]. The primary reasons for this defect include experimental conditions for the preparation of as-prepared compounds and cycling of as-prepared materials particularly in battery applications. Using the Pechini sol–gel process, Dominko et al. [48] synthesised a pure orthorhombic phase of Li2MnSiO4. Later, Politaev et al. [39] observed a small amount of cation exchange (Li-Mn) defect in their experimental preparation (solid-state reactions in hydrogen at 950–1150 °C) of monoclinic phase of Li2MnSiO4. Nyten et al. [38] observed a shift in the potential plateau between first and second cycles in Li2FeSiO4 and suggested that this is due to the structural rearrangement associated with the interchange of some of the Li and Fe ions. The second lowest reaction energy is calculated for the Frenkel defect and this process is higher only by 0.34 eV than the anti-site defect. This would signify the concentration of lithium vacancies and interstitials, inferring this material is suitable for use as a tritium breeder. Other defects exhibit high reaction energies meaning that they are not significant in this material.
Ion diffusion is a key issue in many energy related materials, including cathode materials for batteries. The current simulation approach has the ability to calculate Li-ion diffusion pathways together with activation energies. As experimental determination of Li-ion diffusion is quite challenging, simulation results would be useful to assist in the investigation of experimental data. The results reveal that there is a single Li local hop with the jump distance of 3.09 Å. The activation energy for this hop is calculated to be 0.53 eV. Long range diffusion pathway was constructed by connecting local Li hops. Figure 3a shows the three dimensional long range diffusion pathway with a non-linear pattern. This shows that Li-ion conduction in this material is high. In a previous theoretical study based on the molecular dynamics by Jacobs et al. [18], it is shown that Li-ion diffusion is three dimensional though the pathway is not clear enough from their calculated snapshots derived at different temperatures. Density functional theory simulations were also employed to calculate the Li-ion diffusivity in γ-LiAlO2 [21,22]. In those calculations, Li hopping mechanisms and activation energies for different Li-Li separations are reported. However, the information regarding the Li migration pathway (linear or non-linear) is unclear. Our simulation clearly presents the diffusion pathway that can be directly compared in experiments.
The activation energy calculated here (0.53 eV) is within the range of previous theoretical studies (0.5–0.63 eV) [18,21,22] and lower that previous experimental (NMR spectroscopy and conductivity measurements) work by Idris et al. (0.70 eV) [17]. More direct experimental work (e.g., secondary ion mass spectrometry, SIMS) would be required to experimentally pinpoint the activation energy of Li self-diffusion. As was previously demonstrated, simple atomistic models can yield reliable activation energies of diffusion as compared to experiment with mechanisms compatible with DFT [49].
The calculated activation energy exceeds 0.5 eV and it is therefore anticipated that LiAlO2 will only be a mediocre ion conductor. In Table 2 we compare the activation energies of migration of recently investigated Li, Na, Mg ion conducting oxides considered for battery applications. There is a good agreement between our calculations and experiments for some oxide materials. As mentioned earlier, experimental investigation of activation energy is often difficult. Current simulations, in addition to giving activation energy information, have the added bonus of elucidating diffusion pathways that can be useful in the interpretation of experimental data.
3.2. Solution of Dopants
This section details the processes of monovalent, divalent, trivalent and tetravalent doping into the lithium aluminate lattice. Monovalent dopants (R = Na, K and Rb) and divalent dopants (M = Mg, Ca, Sr and Ba) were considered on the Al site in order to increase the Li content in the form of Li interstitials in LiAlO2 according the following reaction equations:
This efficient engineering strategy can increase the lithium density in lithium-based ceramics and tune mechanical, electrical and optical properties of materials such as lithium aluminate to further increase the applicability for use as a tritium breeding material. Solution enthalpies calculated for monovalent dopants are highly endothermic (>4.5 eV) suggesting that the doping process is quite difficult (refer to Figure 4a). Solution enthalpy gradually increases with ionic radius. High solution enthalpies are due to the larger radii of cations than that of Al3+ (0.53 Å). In the case of divalent cations, Mg2+ is promising as the solution enthalpy of MgO is −5.66 eV (refer to Figure 4b). The highly exothermic solution enthalpy is due to the ionic radius of Mg2+ (0.57 Å) which is closer to the ionic radius of Al3+. The other three dopants exhibit endoergic solution enthalpies, as their ionic radii significantly deviate from the ionic radius of Al3+.
Next, we considered trivalent dopants (X = Ga, Fe, Co, Ni, Mn, Sc, Y and La) on the Al site according to the following reaction equation:
Figure 5 shows the solution enthalpies of X2O3. The results present nickel as the favourable isovalent dopant on the Al site, having the lowest solution enthalpy of −2.03 eV to integrate into the lattice. Solution enthalpies for both Fe3+ and Mn3+ are also negative implying that these two dopants are also worth examining experimentally. The favourable nature of potential candidate dopants (Fe3+, Ni3+ and Mn3+) can be due to their ionic radii which are closer to the ionic radius of Al3+ (0.53 Å). However, the dopant Co3+ exhibits a positive solution enthalpy though its ion radius is closer to that of Al3+ indicating other factors should be responsible. The dopants Ga3+ and Sc3+ exhibit almost zero solution enthalpies. Other dopants are not favourable as their solution enthalpies are endoergic. From Mn3+ to La3+ solution enthalpy gradually increases with ionic radius.
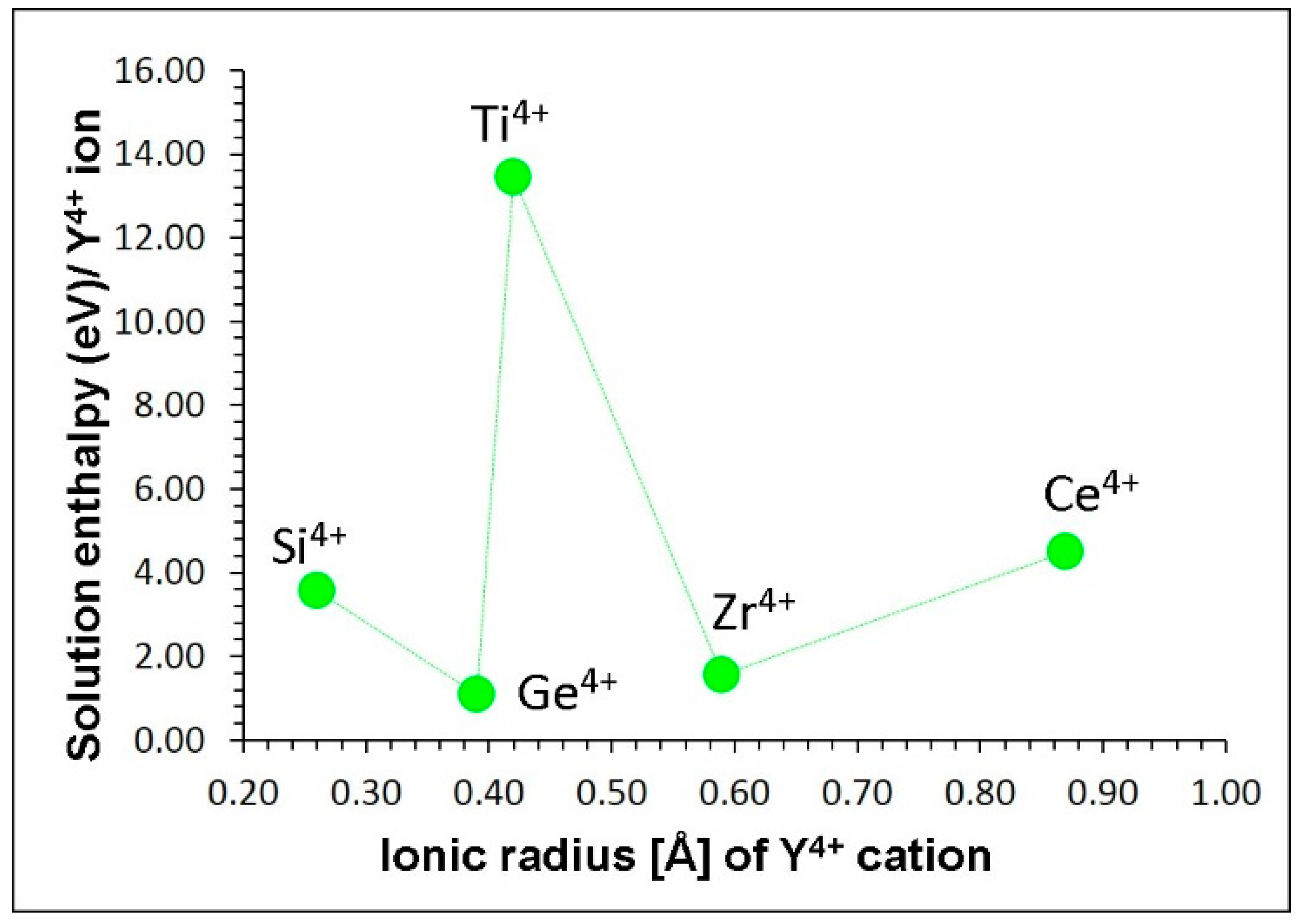
Finally, tetravalent dopants (Y = Si, Ge, Ti, Zr and Ce) were considered on the Al site. This doping process will introduce additional Li vacancies in LiAlO2 assisting vacancy mediated self-diffusion according to the following equation:
We report the solution energies in Figure 6. The results present germanium as the favourable tetravalent dopant on the aluminium site, having the lowest solution enthalpy of 1.08 eV to integrate into the lattice. The second most stable dopant is Zr with the solution enthalpy of 1.54 eV. The ionic radii of these two dopants are closer to the ionic radius of Al3+. However, the high positive solution enthalpy of Ti (~14 eV) is unclear though its ionic radius is somewhat closer to that of Al3+. Furthermore, this dopant cannot be doped under any condition. Both Si and Ce exhibit high solution enthalpies (~4 eV) due to the mismatching of their ionic radii with that of Al3+.
4. Conclusions
In this computational modelling investigation, we have employed static atomistic simulation techniques to study the defect processes (intrinsic disorder mechanisms and doping) and Li self-diffusion in LiAlO2. The lowest energy intrinsic defect process is the Li-Al anti-site implying that a small concentration of Li on the Al site and Al on the Li site will be present. The long range Li-ion diffusion is three-dimensional with an activation energy of migration of 0.53 eV. Numerous dopants were considered and it is calculated that the solution of GeO2 will increase the concentration of the Li vacancies that are required to act as vehicles of diffusion. Furthermore, Mg2+ on the Al site would increase the concentration of Li interstitials that are necessary for the enhancement in the capacity of NaAlO2. It is anticipated that the present atomistic simulation work will encourage the further experimental investigation of LiAlO2.
Supplementary Materials
The following are available online at https://www.mdpi.com/1996-1073/12/15/2895/s1, Table S1: Interatomic potential parameters used in the atomistic simulations of LiAlO2.
Author Contributions
Computation N.K. and J.D.; Writing, N.K. and A.C.; Analysis N.K., A.C., E.N.S. and Y.P.
Funding
This research was financially supported by the European Union’s H2020 Programme under Grant Agreement no 824072–HARVESTORE.
Acknowledgments
We acknowledge Coventry University and Imperial College London for providing computing facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, M.S. Lithium Batteries and Cathode Materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar] [CrossRef] [PubMed]
- Recham, N.; Chotard, J.N.; Dupont, L.; Delacourt, C.; Walker, W.; Armand, M.; Tarascon, J.M. A 3.6 V lithium-based fluorosulphate insertion positive electrode for lithium-ion batteries. Nat. Mater. 2009, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Masquelier, C.; Croguennec, L. Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chem. Rev. 2013, 113, 6552–6591. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Fisher, C.A.J. Lithium and sodium battery cathode materials: computational insights into voltage, diffusion and nanostructural properties. Chem. Soc. Rev. 2014, 43, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Jay, E.E.; Rushton, M.J.D.; Chroneos, A.; Grimes, R.W.; Kilner, J.A. Genetics of superionic conductivity in lithium lanthanum titanates. Phys. Chem. Chem. Phys. 2015, 17, 178–183. [Google Scholar] [CrossRef]
- Ma, S.G.; Gao, T.; Li, S.C.; Ma, X.J.; Shen, Y.H.; Lu, T.C. Theoretical investigations on the a-LiAlO2 properties via first-principles calculation. Fusion Eng. Des. 2016, 113, 324–330. [Google Scholar] [CrossRef]
- Paudel, H.P.; Lee, Y.L.; Senor, D.J.; Duan, Y. Tritium diffusion pathways in γ-LiAlO2 pellets used in TPBAR: A first-principles density functional theory investigation. J. Phys. Chem. C 2018, 122, 9755–9765. [Google Scholar] [CrossRef]
- Park, J.S.; Meng, X.B.; Elam, J.W.; Hao, S.Q.; Wolverton, C.; Kim, C.; Cabana, J. Ultrathin lithium-ion conducting coatings for increased interfacial stability in high voltage lithium-ion batteries. Chem. Mater. 2014, 26, 3128–3134. [Google Scholar] [CrossRef]
- Kuganathan, N.; Islam, M.S. Li2MnSiO4 Lithium Battery Material: Atomic-Scale Study of Defects, Lithium Mobility, and Trivalent Dopants. Chem. Mater. 2009, 21, 5196–5202. [Google Scholar] [CrossRef]
- Rupasov, D.; Chroneos, A.; Parfitt, D.; Kilner, J.A.; Grimes, R.W.; Istomin, S.Y.; Antipov, E.V. Oxygen diffusion in Sr0.75Y0.25CoO2.625: A molecular dynamics study. Phys. Rev. B 2009, 79, 172102. [Google Scholar] [CrossRef]
- Seymour, I.D.; Chroneos, A.; Kilner, J.A.; Grimes, R.W. Defect processes in orthorhombic LnBaCo2O5.5 double perovskites. Phys. Chem. Chem. Phys. 2011, 13, 15305–15310. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.A.J.; Kuganathan, N.; Islam, M.S. Defect chemistry and lithium-ion migration in polymorphs of the cathode material Li2MnSiO4. J. Mater. Chem. A 2013, 1, 4207–4214. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defect Chemistry and Na-Ion Diffusion in Na3Fe2(PO4)3 Cathode Material. Materials 2019, 12, 1348. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Alvani, C.; Casadio, S.; Mancini, M.; Nannetti, C. In-situ tritium release (CORELLI-2 experiment) and ex-reactor ionic conductivity of substoichiometric LiAlO2 breeder ceramics. J. Nucl. Mater. 1995, 224, 236–244. [Google Scholar] [CrossRef]
- Indris, S.; Heitjans, P.; Uecker, R.; Rolling, B. Li ion dynamics in a LiAlO2 single crystal studied by 7Li NMR spectroscopy and conductivity measurements. J. Phys. Chem. C 2012, 116, 14243–14247. [Google Scholar] [CrossRef]
- Jacobs, J.P.; San Miguel, M.A.; Alvarez, L.J.; Giral, P.B. Lithium diffusion in γ-LiAlO2, a molecular dynamics simulation. J. Nucl. Mater. 1996, 232, 131–137. [Google Scholar] [CrossRef]
- Hu, Q.; Lei, L.; Jiang, X.; Feng, Z.C.; Tang, M.; He, D. Li ion diffusion in LiAlO2 investigated by Raman spectroscopy. Solid State Sci. 2014, 37, 103–107. [Google Scholar] [CrossRef]
- Wohlmuth, D.; Epp, V.; Bottke, P.; Hanzu, I.; Bitschnau, B.; Letofsky-Papst, I.; Kriechbaum, M.; Amenitsch, H.; Hofer, F.; Wilkening, M. Order vs. disorder-a huge increase in ionic conductivity of nanocrystalline LiAlO2 embedded in an amorphous-like matrix of lithium aluminate. J. Mater. Chem. A 2014, 2, 20295–20306. [Google Scholar] [CrossRef]
- Islam, M.M.; Bredow, T. Interstitial lithium diffusion pathways in γ-LiAlO2: A computational study. J. Phys. Chem. Lett. 2015, 6, 4622–4626. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, F.; Fjellvåg, H.; Vajeeston, P. A first principle comparative study of the ionic diffusivity in LiAlO2 and NaAlO2 polymorphs for solid-state battery applications. Phys. Chem. Chem. Phys. 2018, 20, 9824–9832. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Mott, N.F.; Littleton, M.J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 1938, 34, 485–499. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Chroneos, A. Lithium diffusion in Li5FeO4. Sci. Rep. 2018, 8, 5832. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Kelaidis, N.; Chroneos, A. Defects, Lithium Mobility and Tetravalent Dopants in the Li3NbO4 Cathode Material. Sci. Rep. 2019, 9, 2192. [Google Scholar] [CrossRef]
- Kuganathan, N.; Sgourou, E.N.; Panayiotatos, Y.; Chroneos, A. Defect Process, Dopant Behaviour and Li Ion Mobility in the Li2MnO3 Cathode Material. Energies 2019, 12, 1329. [Google Scholar] [CrossRef]
- Grimes, R.W.; Busker, G.; McCoy, M.A.; Chroneos, A.; Kilner, J.A.; Chen, S.P. The Effect of Ion Size on Solution Mechanism and Defect Cluster Geometry. Bunsen Ges. Phys. Chem. 1997, 101, 1204–1210. [Google Scholar] [CrossRef]
- Varotsos, P.; Alexopoulos, K. Thermodynamics of Point Defects and Their Relation with the Bulk Properties; Amelinckx, S., Gevers, R., Nihoul, J., Eds.; Elsevier: Amsterdam, The Netherlands, 1986. [Google Scholar]
- Varotsos, P. Comparison of models that interconnect point defect parameters in solids with bulk properties. J. Appl. Phys. 2007, 101, 123503. [Google Scholar] [CrossRef]
- Chroneos, A.; Vovk, R.V. Modeling self-diffusion in UO2 and ThO2 by connecting point defect parameters with bulk properties. Solid State Ion. 2015, 274, 1–3. [Google Scholar] [CrossRef]
- Cooper, M.W.D.; Grimes, R.W.; Fitzpatrick, M.E.; Chroneos, A. Modeling oxygen self-diffusion in UO2 under pressure. Solid State Ion. 2015, 282, 26–30. [Google Scholar] [CrossRef]
- Sarlis, N.V.; Skordas, E.S. Pressure and temperature dependence of the oxygen self-diffusion activation volume in UO2 by a thermodynamic model. Solid State Ion. 2016, 290, 121–123. [Google Scholar] [CrossRef]
- Marezio, M. The crystal structure and anomalous dispersion of γ-LiAlO2. Acta Crystallogr. 1965, 19, 396–400. [Google Scholar] [CrossRef]
- Kröger, F.A.; Vink, H.J. Relations between the Concentrations of Imperfections in Crystalline Solids. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: Cambridge, MA, USA, 1956; Volume 3, pp. 307–435. [Google Scholar]
- Armstrong, A.R.; Kuganathan, N.; Islam, M.S.; Bruce, P.G. Structure and Lithium Transport Pathways in Li2FeSiO4 Cathodes for Lithium Batteries. J. Am. Chem. Soc. 2011, 133, 13031–13035. [Google Scholar] [CrossRef] [PubMed]
- Nytén, A.; Abouimrane, A.; Armand, M.; Gustafsson, T.; Thomas, J.O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochem. Commun. 2005, 7, 156–160. [Google Scholar] [CrossRef]
- Politaev, V.V.; Petrenko, A.A.; Nalbandyan, V.B.; Medvedev, B.S.; Shvetsova, E.S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 2007, 180, 1045–1050. [Google Scholar] [CrossRef]
- Liu, H.; Choe, M.J.; Enrique, R.A.; Orvañanos, B.; Zhou, L.; Liu, T.; Thornton, K.; Grey, C.P. Effects of Antisite Defects on Li Diffusion in LiFePO4 Revealed by Li Isotope Exchange. J. Phys. Chem. C 2017, 121, 12025–12036. [Google Scholar] [CrossRef]
- Kempaiah Devaraju, M.; Duc Truong, Q.; Hyodo, H.; Sasaki, Y.; Honma, I. Synthesis, characterization and observation of antisite defects in LiNiPO4 nanomaterials. Sci. Rep. 2015, 5, 11041. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Anurakavan, S.; Iyngaran, P.; Chroneos, A. Li3SbO4 lithium-ion battery material: Defects, lithium ion diffusion and tetravalent dopants. Mater. Chem. Phys. 2019, 225, 34–41. [Google Scholar] [CrossRef]
- Kordatos, A.; Kuganathan, N.; Kelaidis, N.; Iyngaran, P.; Chroneos, A. Defects and lithium migration in Li2CuO2. Sci. Rep. 2018, 8, 6754. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Ganeshalingam, S.; Chroneos, A. Defects, Dopants and Lithium Mobility in Li9V3(P2O7)3(PO4)2. Sci. Rep. 2018, 8, 8140. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Li2SnO3 as a Cathode Material for Lithium-ion Batteries: Defects, Lithium Ion Diffusion and Dopants. Sci. Rep. 2018, 8, 12621. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Defect Chemistry and Li-ion Diffusion in Li2RuO3. Sci. Rep. 2019, 9, 550. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Tsoukalas, L.H.; Chroneos, A. Defects, dopants and Li-ion diffusion in Li2SiO3. Solid State Ion. 2019, 335, 61–66. [Google Scholar] [CrossRef]
- Dominko, R.; Bele, M.; Gaberšček, M.; Meden, A.; Remškar, M.; Jamnik, J. Structure and electrochemical performance of Li2MnSiO4 and Li2FeSiO4 as potential Li-battery cathode materials. Electrochem. Commun. 2006, 8, 217–222. [Google Scholar] [CrossRef]
- Kushima, A.; Parfitt, D.; Chroneos, A.; Yildiz, B.; Kilner, J.A.; Grimes, R.W. Interstitialcy diffusion of oxygen in tetragonal La2CoO4+δ. Phys. Chem. Chem. Phys. 2011, 13, 2242–2249. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defects and dopant properties of Li3V2(PO4)3. Sci. Rep. 2019, 9, 333. [Google Scholar] [CrossRef]
- Cahill, L.S.; Chapman, R.P.; Britten, J.F.; Goward, G.R. 7Li NMR and Two-Dimensional Exchange Study of Lithium Dynamics in Monoclinic Li3V2(PO4)3. J. Phys. Chem. B 2006, 110, 7171–7177. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Fitzpatrick, M.E.; Vovk, R.V.; Chroneos, A. Defect process and lithium diffusion in Li2TiO3. Solid State Ion. 2018, 327, 93–98. [Google Scholar] [CrossRef]
- Ruprecht, B.; Wilkening, M.; Uecker, R.; Heitjans, P. Extremely slow Li ion dynamics in monoclinic Li2TiO3—Probing macroscopic jump diffusion via 7Li NMR stimulated echoes. Phys. Chem. Chem. Phys. 2012, 14, 11974–11980. [Google Scholar] [CrossRef] [PubMed]
- Fehr, T.; Schmidbauer, E. Electrical conductivity of Li2TiO3 ceramics. Solid State Ion. 2007, 178, 35–41. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defects, Dopants and Sodium Mobility in Na2MnSiO4. Sci. Rep. 2018, 8, 14669. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Chroneos, A. Na3V(PO4)2 cathode material for Na ion batteries: Defects, dopants and Na diffusion. Solid State Ion. 2019, 336, 75–79. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Vovk, R.; Chroneos, A. Defects, dopants and Mg diffusion in MgTiO3. Sci. Rep. 2019, 9, 4394. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Crystal structure of LiAlO2 (space group P41212).

Figure 2.
Energetics of intrinsic defect process calculated in tertragonal LiAlO2.
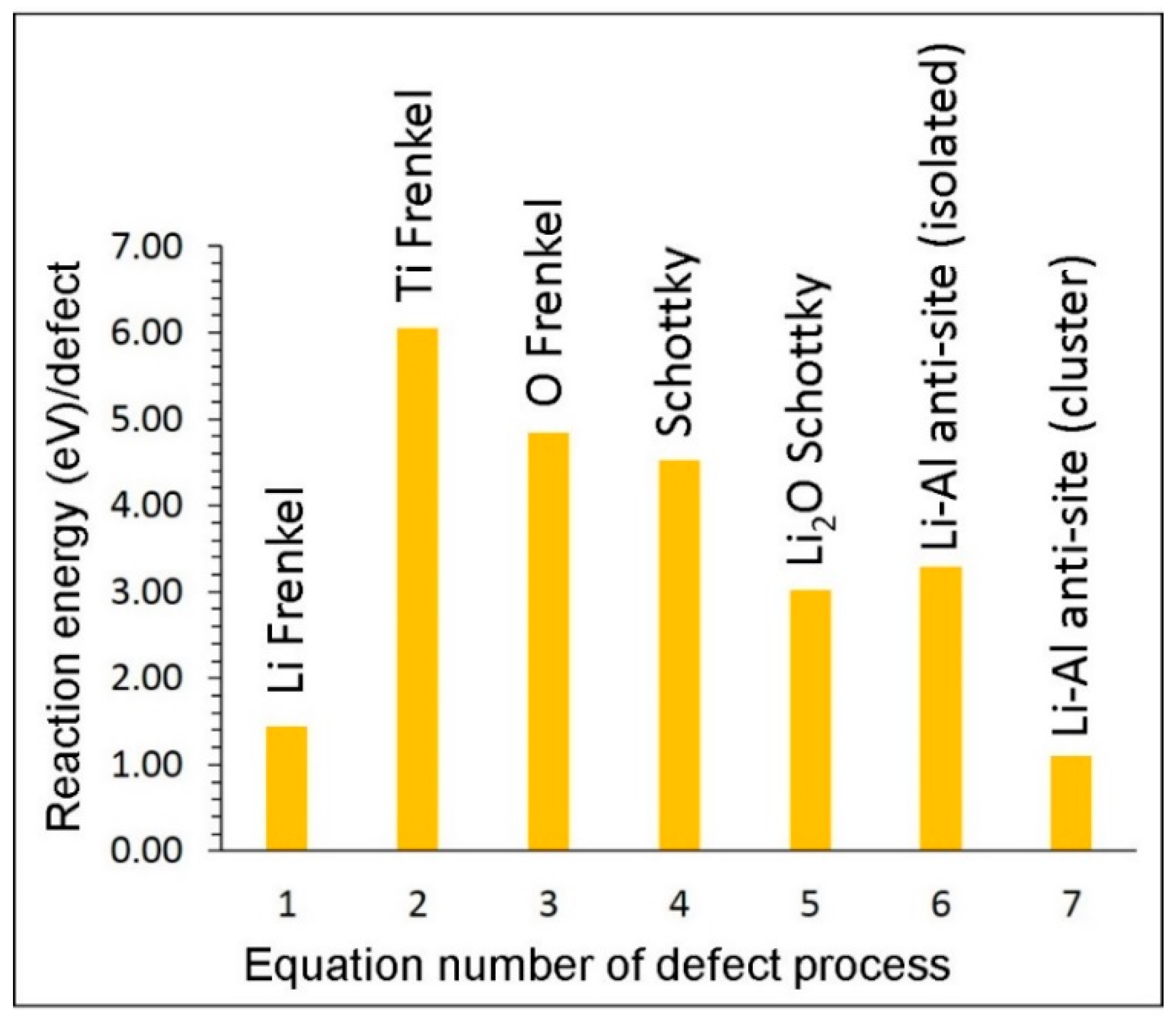
Figure 3.
(a) Calculated long range three dimensional Li-ion diffusion pathways and (b) energy profile diagram for the local Li hop involved in the long range diffusion.
Figure 3.
(a) Calculated long range three dimensional Li-ion diffusion pathways and (b) energy profile diagram for the local Li hop involved in the long range diffusion.
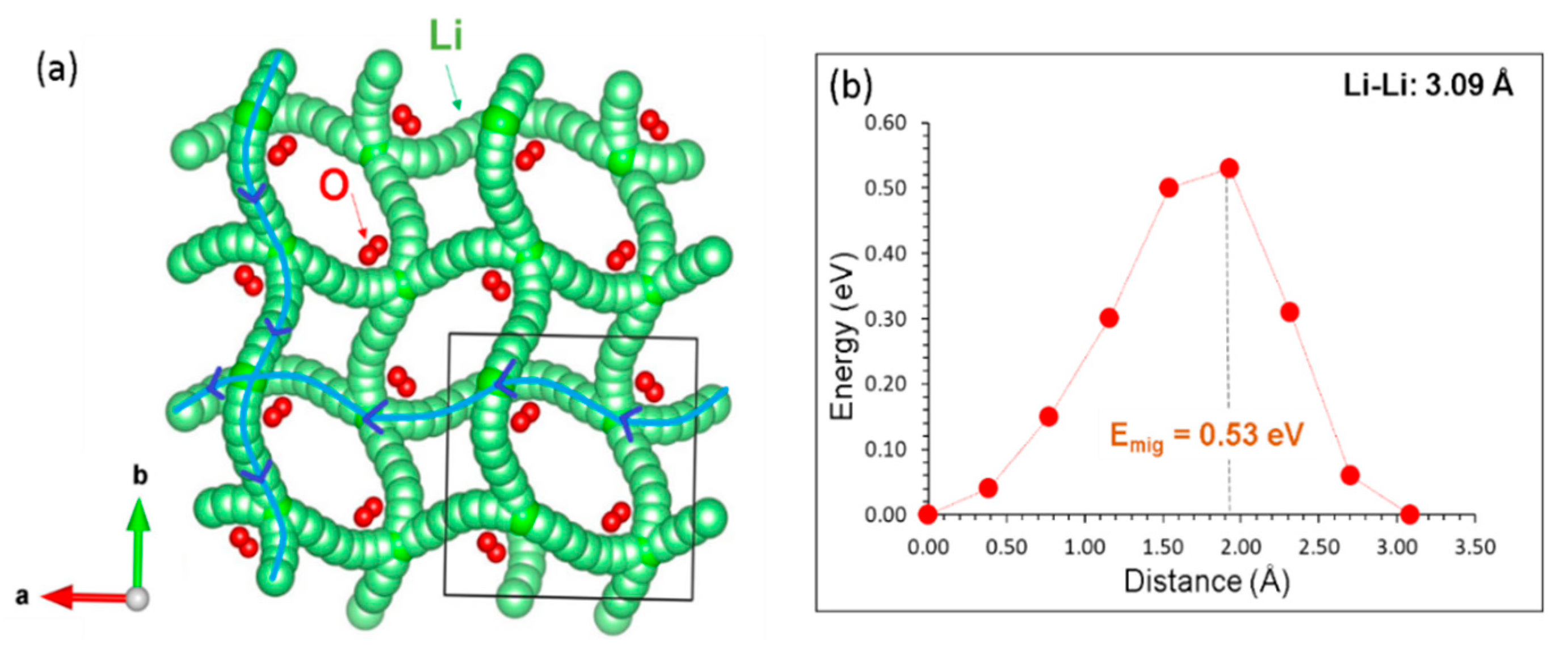
Figure 4.
Solution enthalpies of (a) R2O (R = Na, K and Rb) with respect to the R+ ionic radius and (b) MO (M = Mg, Ca, Sr and Ba) with respect to the M2+ ionic radius in LiAlO2.
Figure 4.
Solution enthalpies of (a) R2O (R = Na, K and Rb) with respect to the R+ ionic radius and (b) MO (M = Mg, Ca, Sr and Ba) with respect to the M2+ ionic radius in LiAlO2.

Figure 5.
Solution enthalpies of X2O3 (X = Ga, Fe, Co, Ni, Mn, Sc, Y and La) with respect to the X3+ ionic radius in LiAlO2.
Figure 5.
Solution enthalpies of X2O3 (X = Ga, Fe, Co, Ni, Mn, Sc, Y and La) with respect to the X3+ ionic radius in LiAlO2.
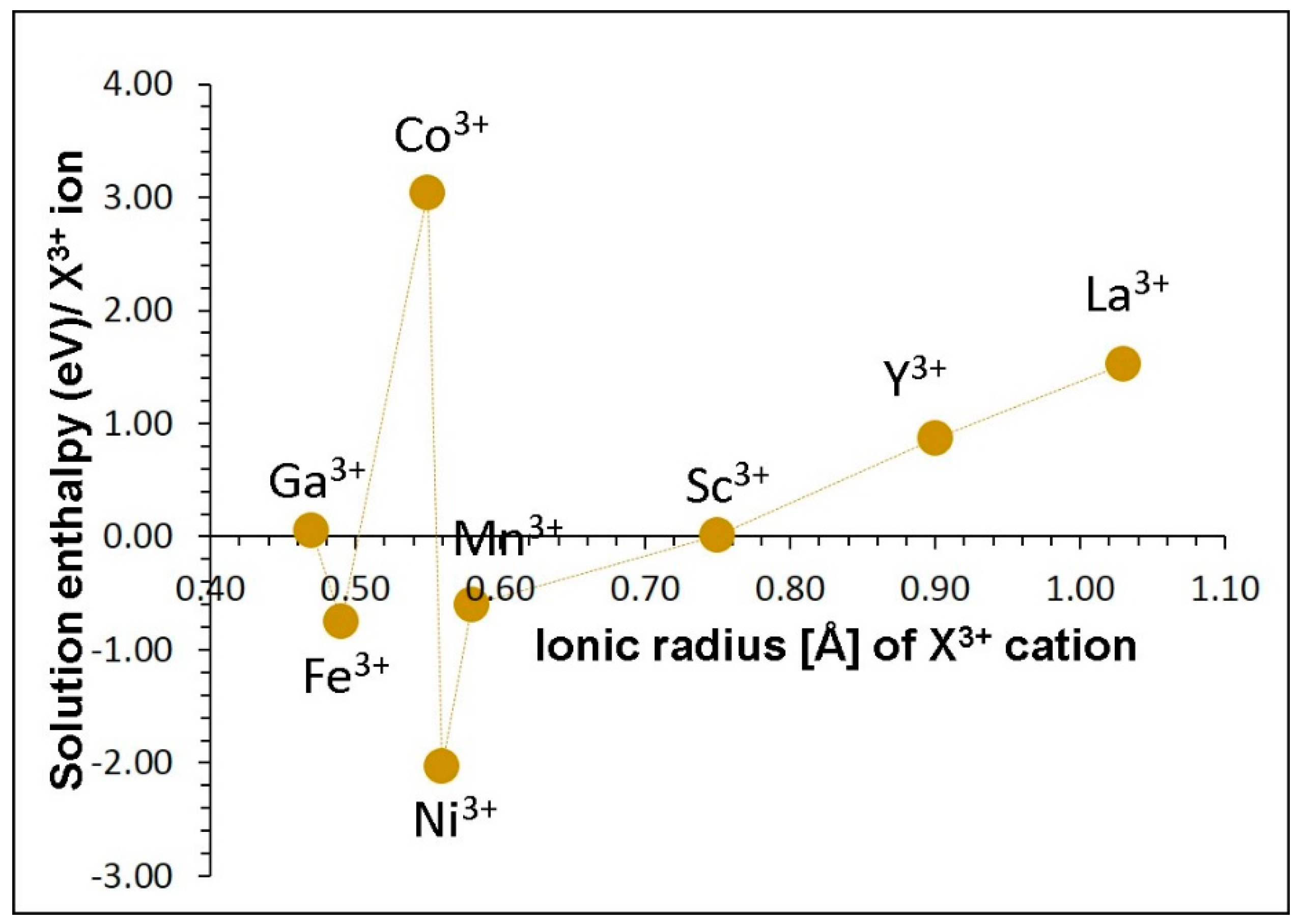
Figure 6.
Solution enthalpies of YO2 (Y = Si, Ti, Ge, Zr and Ce) with respect to the Y4+ ionic radius in LiAlO2.
Figure 6.
Solution enthalpies of YO2 (Y = Si, Ti, Ge, Zr and Ce) with respect to the Y4+ ionic radius in LiAlO2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated structural parameters for LiAlO2 (space group P41212) as compared to the experimental determinations [35].
Table 1.
Calculated structural parameters for LiAlO2 (space group P41212) as compared to the experimental determinations [35].
| Parameter | Exp | Calc | |∆|(%) |
|---|---|---|---|
| a = b (Å) | 5.1687 | 5.1403 | 0.55 |
| c (Å) | 6.2679 | 6.3860 | 1.88 |
| α = β = γ (°) | 90.00 | 90.00 | 0.00 |
| Volume [V = a × b × c (Å3)] | 167.4498 | 168.7370 | 0.77 |
Table 2.
Activation energies (eV) of ion self-diffusion (Li, Na, Mg) in recently investigated oxides considered for battery applications.
Table 2.
Activation energies (eV) of ion self-diffusion (Li, Na, Mg) in recently investigated oxides considered for battery applications.
| Material | Calculation | Experiment |
|---|---|---|
| LiAlO2 | 0.53 (this study) | 0.70–1.23 [17] |
| Li3V2(PO4)3 | 0.46–0.87 [50] | 0.73–0.83 [51] |
| Li2TiO3 | 0.51–0.72 [52] | 0.47–0.80 [53], 0.60–0.90 [54] |
| Na2MnSiO4 | 0.81 [55] | - |
| Na3V(PO4)2 | 0.59 [56] | - |
| Na3Fe2(PO4)3 | 0.45 [15] | - |
| MgTiO3 | 0.88 [57] | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kuganathan, N.; Dark, J.; Sgourou, E.N.; Panayiotatos, Y.; Chroneos, A. Atomistic Simulations of the Defect Chemistry and Self-Diffusion of Li-ion in LiAlO2. Energies 2019, 12, 2895. https://doi.org/10.3390/en12152895
AMA Style
Kuganathan N, Dark J, Sgourou EN, Panayiotatos Y, Chroneos A. Atomistic Simulations of the Defect Chemistry and Self-Diffusion of Li-ion in LiAlO2. Energies. 2019; 12(15):2895. https://doi.org/10.3390/en12152895
Chicago/Turabian StyleKuganathan, N., J. Dark, E.N. Sgourou, Y. Panayiotatos, and A. Chroneos. 2019. "Atomistic Simulations of the Defect Chemistry and Self-Diffusion of Li-ion in LiAlO2" Energies 12, no. 15: 2895. https://doi.org/10.3390/en12152895
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.