Lyngbyastatins 8–10, Elastase Inhibitors with Cyclic Depsipeptide Scaffolds Isolated from the Marine Cyanobacterium Lyngbya semiplena


Abstract
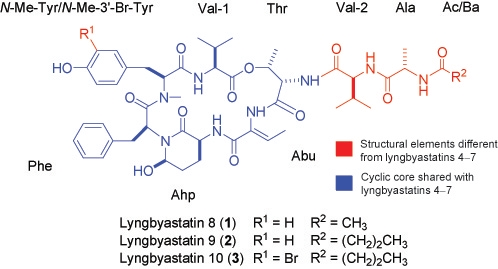
:
1. Introduction
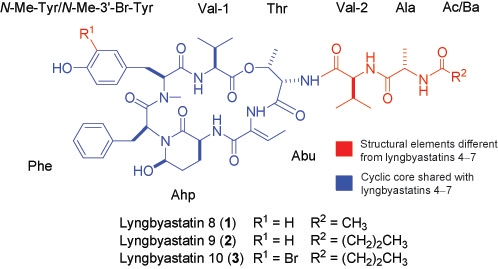
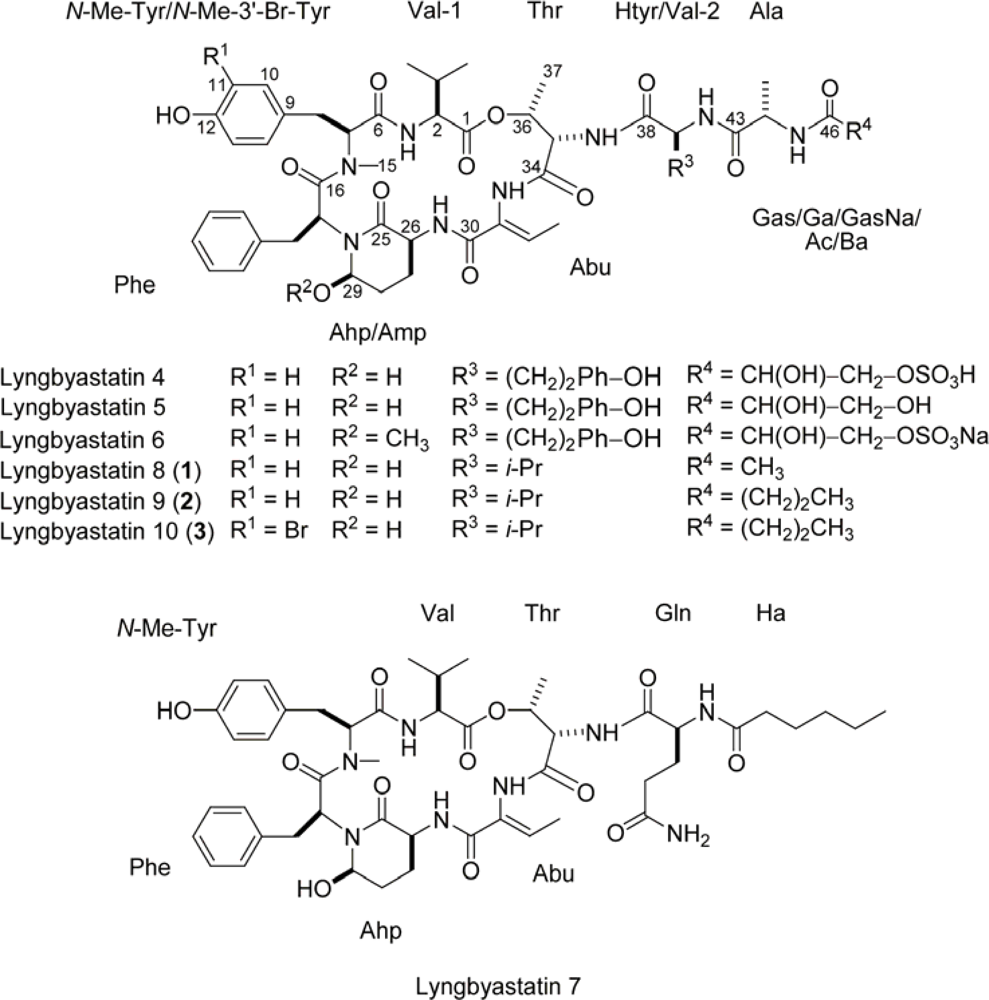
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Extraction and Isolation
3.3. Lyngbyastatin 8 (1)
3.4. Lyngbyastatin 9 (2)
3.5. Lyngbyastatin 10 (3)
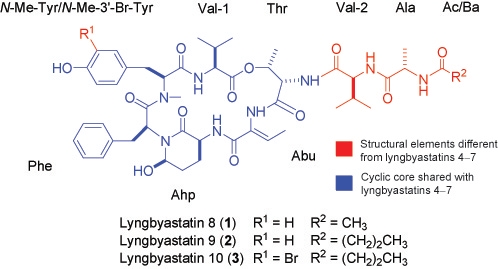
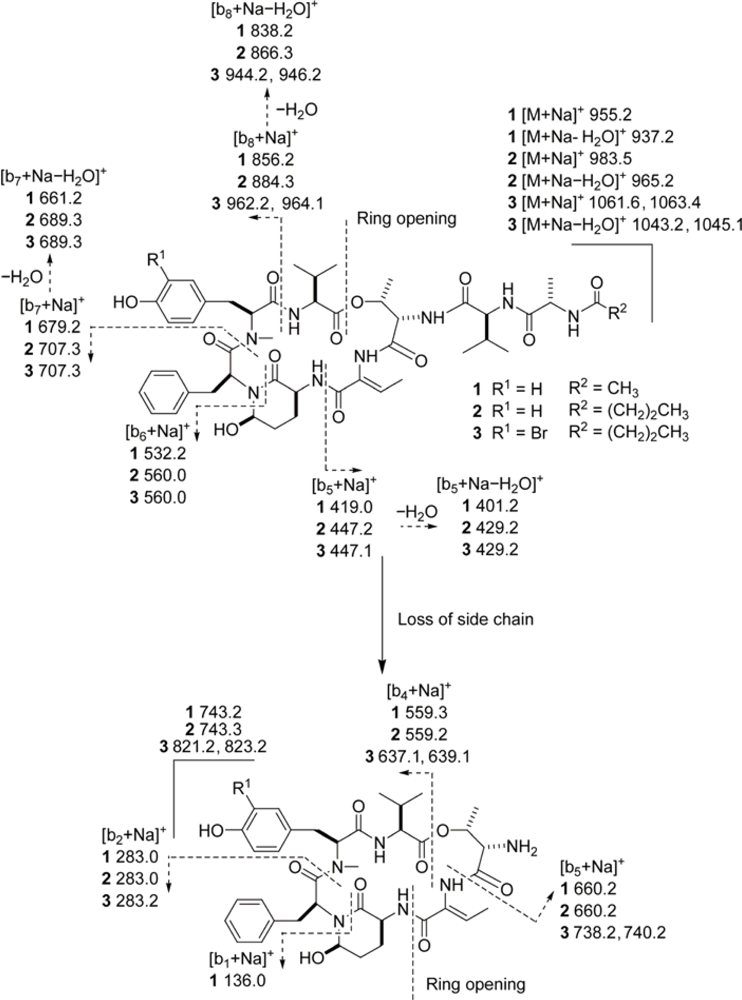
3.6. ESIMS Fragmentation
3.7. Marfey’s Analysis
3.8. Elastase Assays
Acknowledgments
Glossary of Terms
| Abu | 2-amino-2-butenoic acid; |
| Ahp | 3-amino-6-hydroxy-2-piperidone; |
| Amp | 3-amino-6-methoxy-2-piperidone; |
| Ba | butanoic acid; |
| CAD | collisionally activated decomposition; |
| CE | collision energy; |
| CEP | collision cell entrance potential; |
| CUR | curtain gas; |
| DP | declustering potential; |
| EP | entrance potential; |
| EPI | enhanced product ion; |
| FDLA | 1-fluoro-2,4-dinitro-5-leucinamide; |
| Ga | glyceric acid; |
| Gas | glyceric acid sulfate; |
| GS1 | gas 1; |
| GS2 | gas 2; |
| Ha | hexanoic acid; |
| HRESI/APCIMS | high resolution electrospray ionization/atmospheric pressure chemical ionization mass spectrometry (dual probe); |
| Htyr | homotyrosine; |
| IS | ionspray voltage; |
| TEM | temperature. |
- Supplementary Material Available: Figure S1, Tables S1–S3 and NMR spectra for compounds 1–3.
References and Notes
- Tan, LT. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–979. [Google Scholar]
- Kwan, JC; Rocca, JR; Abboud, KA; Paul, VJ; Luesch, H. Total structure determination of grassypeptolide, a new marine cytotoxin. Org Lett 2008, 10, 789–792. [Google Scholar]
- Luesch, H; Yoshida, WY; Moore, RE; Paul, VJ; Corbett, TH. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium. Lyngbya majuscula J Am Chem Soc 2001, 123, 5418–5423. [Google Scholar]
- Taori, K; Paul, VJ; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J Am Chem Soc 2008, 130, 1806–1807. [Google Scholar]
- Kwan, JC; Eksioglu, EA; Liu, C; Paul, VJ; Luesch, H. Grassystatins A–C from marine cyanobacteria, potent cathepsin E inhibitors that reduce antigen presentation. J Med Chem 2009, 52, 5732–5747. [Google Scholar]
- Matthew, S; Ross, C; Rocca, JR; Paul, VJ; Luesch, H. Lyngbyastatin 4, a dolastatin 13 analogue with elastase and chymotrypsin inhibitory activity from the marine cyanobacterium Lyngbya confervoides. J Nat Prod 2007, 70, 124–127. [Google Scholar]
- Taori, K; Matthew, S; Rocca, JR; Paul, VJ; Luesch, H. Lyngbyastatins 5–7, potent elastase inhibitors from Floridian marine cyanobacteria, Lyngbya spp. J Nat Prod 2007, 70, 1593–1600. [Google Scholar]
- Yamaki, H; Sitachitta, N; Sano, T; Kaya, K. Two new chymotrypsin inhibitors isolated from the cyanobacterium Microcystis aeruginosa NIES-88. J Nat Prod 2005, 68, 14–18. [Google Scholar]
- Ploutno, A; Shoshan, M; Carmeli, S. Three novel protease inhibitors from a natural bloom of the cyanobacterium Microcystis aeruginosa. J Nat Prod 2002, 65, 973–978. [Google Scholar]
- Itou, Y; Ishida, K; Shin, HJ; Murakami, M. Oscillapeptins A to F, serine protease inhibitors from the three strains of Oscillatoria agardhii. Tetrahedron 1999, 55, 6871–6882. [Google Scholar]
- Nakanishi, I; Kinoshita, T; Sato, A; Tada, T. Structure of porcine pancreatic elastase complexed with FR901277, a novel macrocyclic inhibitor of elastases, at 1.6 Å resolution. Biopolymers 2000, 53, 434–445. [Google Scholar]
- Matern, U; Schleberger, C; Jelakovic, S; Weckesser, J; Schulz, GE. Binding structure of elastase inhibitor scyptolin A. Chem Biol 2003, 10, 997–1001. [Google Scholar]
- Lee, AY; Smitka, TA; Bonjouklian, R; Clardy, J. Atomic structure of the trypsin–A90720A complex: a unified approach to structure and function. Chem Biol 1994, 1, 113–117. [Google Scholar]
- Taori, K; Paul, VJ; Luesch, H. Kempopeptins A and B, serine protease inhibitors with different selectivity profiles from a marine cyanobacterium, Lyngbya sp. J Nat Prod 2008, 71, 1625–1629. [Google Scholar]
- Tremblay, GM; Janelle, MF; Bourbonnais, Y. Anti-inflammatory activity of neutrophil elastase inhibitors. Curr Opin Investig Drugs 2003, 4, 556–565. [Google Scholar]
- von Elert, E; Oberer, L; Merkel, P; Huhn, T; Blom, JF. Cyanopeptolin 954, a chlorine-containing cymotrypsin inhibitor of Microcystis aeruginosa NIVA Cya 43. J Nat Prod 2005, 68, 1324–1327. [Google Scholar]
- Mayumi, T; Kato, H; Kawasaki, Y; Harada, K-I. Formation of diagnostic product ions from cyanobacterial cyclic peptides by the two-bond fission mechanism using ion trap liquid chromatography/multi-stage mass spectrometry. Rapid Commun Mass Spectom 2007, 21, 1025–1033. [Google Scholar]
- Claridge, TDW. High-Resolution NMR Techniques in Organic Chemistry; Elsevier: San Diego, CA, USA, 1999. [Google Scholar]
- Araya-Maturana, R; Delgado-Castro, T; Cardona, W; Weiss-López, BE. Use of long-range C–H (nJ n > 3) heteronuclear multiple bond connectivity in the assignment of the 13C NMR spectra of complex organic molecules. Curr Org Chem 2001, 5, 253–263. [Google Scholar]
- Araya-Maturana, R; Pessoa-Mahana, H; Weiss-López, B. Very long-range correlations (nJC,H n > 3) in HMBC spectra. Nat Prod Commun 2008. [Google Scholar]
- Marfey, P. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res Commun 1984, 49, 591–596. [Google Scholar]
- Brey, WW; Edison, AS; Nast, RE; Rocca, JR; Saha, S; Withers, RS. Design, construction, and validation of a 1-mm triple-resonance high-temperature-superconducting probe for NMR. J Magn Reson 2006, 179, 290–293. [Google Scholar]
- Matthew, S; Ross, C; Paul, VJ; Luesch, H. Pompanopeptins A and B, new cyclic peptides from the marine cyanobacterium. Lyngbya confervoides Tetrahedron 2008, 64, 4081–4089. [Google Scholar]
- Unable to obtain IR due to lack of sample.
- The retention time of N-Me-d-Tyr-l-FDLA was deduced from that of its enantiomer, N-Me-l-Tyr-d-FDLA.
- The retention time of N-Me-3′-Br-d-Tyr-l-FDLA was deduced from that of its enantiomer N-Me-3′-Br-l-Tyr-d-FDLA.
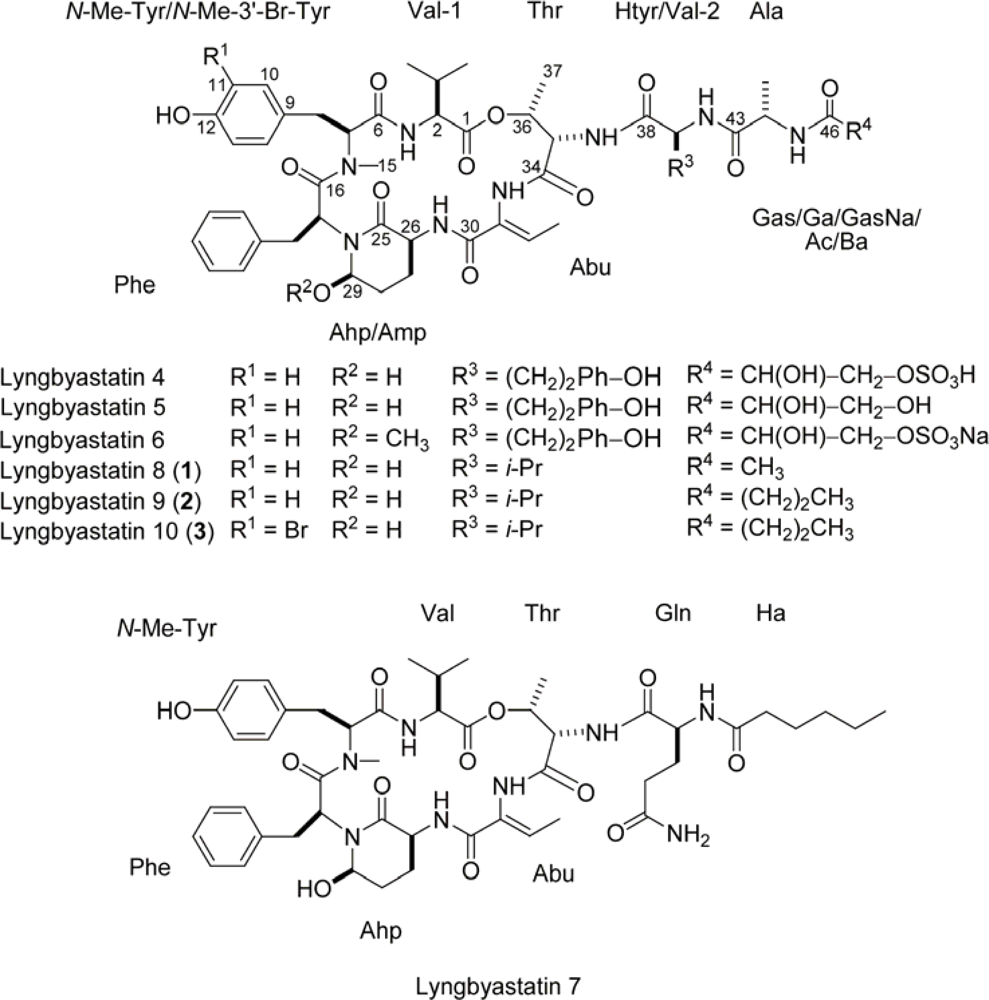
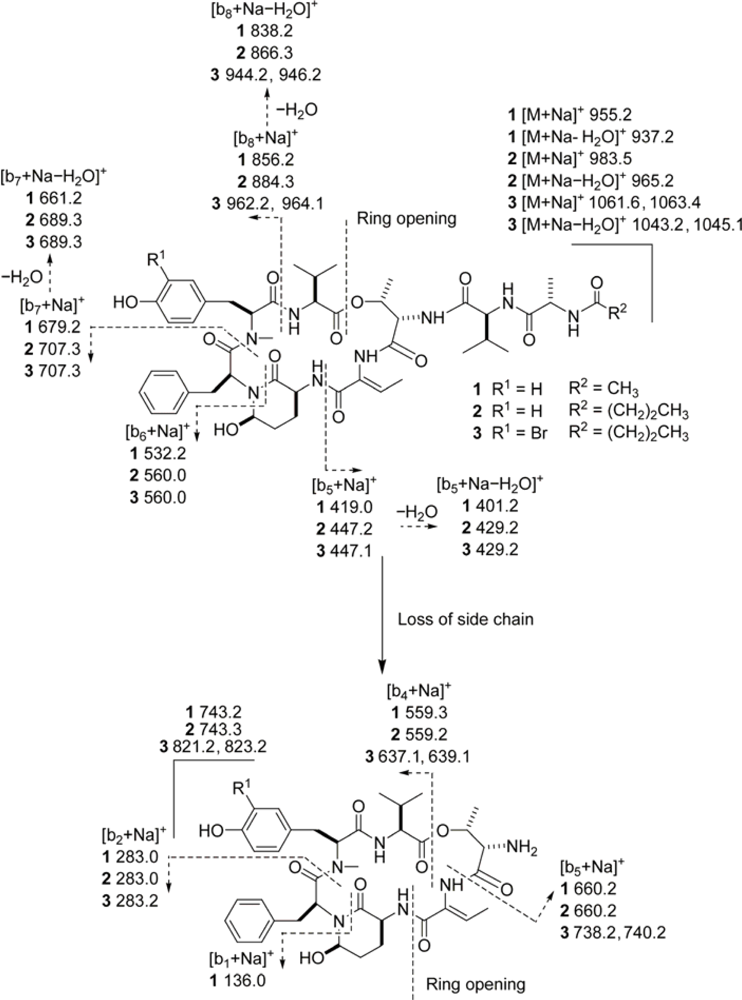

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C/H No. | Lyngbyastatin 8 (1) | Lyngbyastatin 9 (2) | Lyngbyastatin 10 (3) | |||
|---|---|---|---|---|---|---|
| δH, mult. (J in Hz) | δC, mult.a | δH, mult. (J in Hz) | δC, mult.a | δH, mult. (J in Hz) | δC, mult.a | |
| 1 | c | c | c | |||
| 2 | 4.64, br | 55.9, d | 4.62, m | 55.9, d | 4.60, m | 56.2, d |
| 3 | 2.05, m | 30.4, d | 2.05, m | 30.3, d | 2.03, m | 30.4, d |
| 4 | 0.86, d (6.6) | 19.0, q | 0.85d | 18.9, q | 0.86, d (7.0) | 19.0, q |
| 5 | 0.74, d (6.6) | 17.2, q | 0.74, d (6.6) | 17.1, q | 0.74, d (7.0) | 17.2, q |
| NH | 7.50, br d (6.3) | 7.52, br d (6.8) | 7.52, br d (7.3) | |||
| 6 | c | c | c | |||
| 7 | 4.87, dd (11.8, 1.9) | 60.6, d | 4.87, dd (12.0, 0) | 60.5, d | 4.86, dd (12.0, 2.4) | 60.6, d |
| 8a | 3.08, dd (−13.4, 1.9) | 32.5, t | 3.08, dd (−13.0, 0) | 32.4, t | 3.08, dd (−13.6, 2.4) | 32.2, t |
| 8b | 2.69, dd (−13.4, 11.8) | 2.69, dd (−13.0, 12.2) | 2.72, dd (−13.6, 12..0) | |||
| 9 | 127.3, s | 127.2, s | c | |||
| 10 | 6.97, d (8.0) | 130.3, d | 6.97, d (7.6) | 130.2, d | 7.25, s | 133.4, d |
| 11 | 6.76, d (8.0) | 115.2, d | 6.76, d (7.6) | 115.1, d | c | |
| 12 | 156.2, s | 156.0, s | c | |||
| 13 | 6.76, d (8.0) | 115.2, d | 6.76, d (7.6) | 115.1, d | 6.93, d (8.2) | 116.5, d |
| 14 | 6.97, d (8.0) | 130.3, d | 6.97, d (7.6) | 130.2, d | 6.97, d (8.2) | 130.0, d |
| 15 | 2.74, s | 30.2, q | 2.74, s | 30.1, q | 2.75, s | 30.3, q |
| OH | 9.38, s | 9.41, s | 8.48, s | |||
| 16 | 170.6, s | 170.3, s | c | |||
| 17 | 4.71, dd (12.1, 3.1) | 50.0, d | 4.71, dd (12.1, 3.0) | 49.9, d | 4.70, dd (11.7, 3.2) | 50.2, d |
| 18a | 2.86, dd (−12.8, 12.1) | 35.0, t | 2.85, dd (−12.8, 12.1) | 35.0, t | 2.88, dd (−13.7, 11.7) | 35.2, t |
| 18b | 1.80, dd (−12.8, 3.1) | 1.80, dd (−12.8, 3.0) | 1.87, dd (−13.7, 3.2) | |||
| 19 | 136.8, s | 136.5, s | c | |||
| 20/24 | 6.82, d (7.2) | 129.3, d | 6.82, d (7.3) | 129.2, d | 6.78, d (7.4) | 129.5, d |
| 21/23 | 7.18, m | 127.7, d | 7.18, m | 127.6, d | 7.16, dd (7.4, 7.3) | 128.0, d |
| 22 | 7.14, m | 126.1, d | 7.14, m | 126.0, d | 7.13, t (7.3) | 126.4, d |
| 25 | c | 168.5, s | c | |||
| 26 | 3.77, ddd (12.4, 9.2, 2.4) | 47.9, d | 3.77, ddd (14.1, 10.6, 2.3) | 47.9, d | 3.78, ddd (11.6, 8.8, 2.2) | 48.2, d |
| 27a | 2.40, dddd (−12.4, 12.4, 11.7, 2.7) | 21.7, t | 2.40, dddd (14.1, −12.4, 11.3, 4.4) | 21.6, t | 2.40, m | 21.8, t |
| 27b | 1.56, m | 1.56, m | 1.56, m | |||
| 28a | 1.70, br d (11.7) | 29.1, t | 1.71, br d (11.3) | 29.0, t | 1.71, br d (12.4) | 29.2, t |
| 28b | 1.56, m | 1.55, m | 1.57, m | |||
| 29 | 5.06, s | 73.5, d | 5.06, s | 73.4, s | 5.07, s | 73.7, d |
| NH | 7.21, d (9.2) | 7.21, br | 7.21, br | |||
| OH | 6.10, s | 6.11, br | 3.15, se | |||
| 30 | c | 162.7, s | c | |||
| 31 | 130.1, s | 129.7, s | c | |||
| 32 | 6.49, q (6.8) | 131.6, d | 6.49, q (6.8) | 131.5, d | 6.50, q | 132.1, d |
| 33 | 1.47, d (6.8) | 12.9, q | 1.47, d (6.8) | 12.8, q | 1.47, d | 12.9, q |
| NH | 6.56, s | 6.58, s | ||||
| 34 | c | c | c | |||
| 35 | 4.62, m | 55.1, d | 4.63, m | 55.0, d | 4.61, m | 55.7, d |
| 36 | 5.53, br | 71.4, d | 5.53, br | 71.4, d | 5.50, br | c |
| 37 | 1.21, d (6.2) | 17.6, q | 1.21, d (6.2) | 17.6, q | 1.21, d (6.7) | 17.8, q |
| NH | 7.91, br | 7.92, br | 7.92, br | |||
| 38 | c | 171.7, s | c | |||
| 39 | 4.36, m | 57.0, d | 4.37, m | 56.8, d | 4.36, dd (8.8, 6.1) | 57.1, d |
| 40 | 2.05, m | 30.4, d | 2.05, m | 30.3, d | 2.03, m | 30.4, d |
| 41 | 0.85, d (6.2) | 19.0, q | 0.84d | 13.3, q | 0.85, d (6.1) | 19.0, q |
| 42 | 0.80, d (6.4) | 17.5, q | 0.80, d (6.5) | 17.4, q | 0.80, d (6.6) | 17.5, q |
| NH | 7.77, br | 7.69, br | 7.68, br | |||
| 43 | 172.5, s | 172.3, s | c | |||
| 44 | 4.33, dq (7.4, 6.7) | 47.8, d | 4.34, dq (7.2, 6.8) | 47.7, d | 4.32, dq (7.6, 7.0) | 47.9, d |
| 45 | 1.17, d (6.7) | 17.8, q | 1.18, d (6.8) | 17.6, q | 1.18, d (7.0) | 17.8, q |
| NH | 8.08, d (7.4) | 8.04, d (7.2) | 8.01, d (7.6) | |||
| 46 | 169.0, s | 171.8, s | c | |||
| 47 | 1.82, s | 22.2, q | 2.07, m (2H) | 36.7, t | 2.07, m (2H) | 36.8, t |
| 48 | 1.48, m (2H) | 18.3, t | 1.48, m (2H) | 18.5, t | ||
| 49 | 0.83d | 13.3, q | 0.83, t (7.3) | 13.4, q | ||
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kwan, J.C.; Taori, K.; Paul, V.J.; Luesch, H. Lyngbyastatins 8–10, Elastase Inhibitors with Cyclic Depsipeptide Scaffolds Isolated from the Marine Cyanobacterium Lyngbya semiplena. Mar. Drugs 2009, 7, 528-538. https://doi.org/10.3390/md7040528
Kwan JC, Taori K, Paul VJ, Luesch H. Lyngbyastatins 8–10, Elastase Inhibitors with Cyclic Depsipeptide Scaffolds Isolated from the Marine Cyanobacterium Lyngbya semiplena. Marine Drugs. 2009; 7(4):528-538. https://doi.org/10.3390/md7040528
Chicago/Turabian StyleKwan, Jason C., Kanchan Taori, Valerie J. Paul, and Hendrik Luesch. 2009. "Lyngbyastatins 8–10, Elastase Inhibitors with Cyclic Depsipeptide Scaffolds Isolated from the Marine Cyanobacterium Lyngbya semiplena" Marine Drugs 7, no. 4: 528-538. https://doi.org/10.3390/md7040528
APA StyleKwan, J. C., Taori, K., Paul, V. J., & Luesch, H. (2009). Lyngbyastatins 8–10, Elastase Inhibitors with Cyclic Depsipeptide Scaffolds Isolated from the Marine Cyanobacterium Lyngbya semiplena. Marine Drugs, 7(4), 528-538. https://doi.org/10.3390/md7040528