
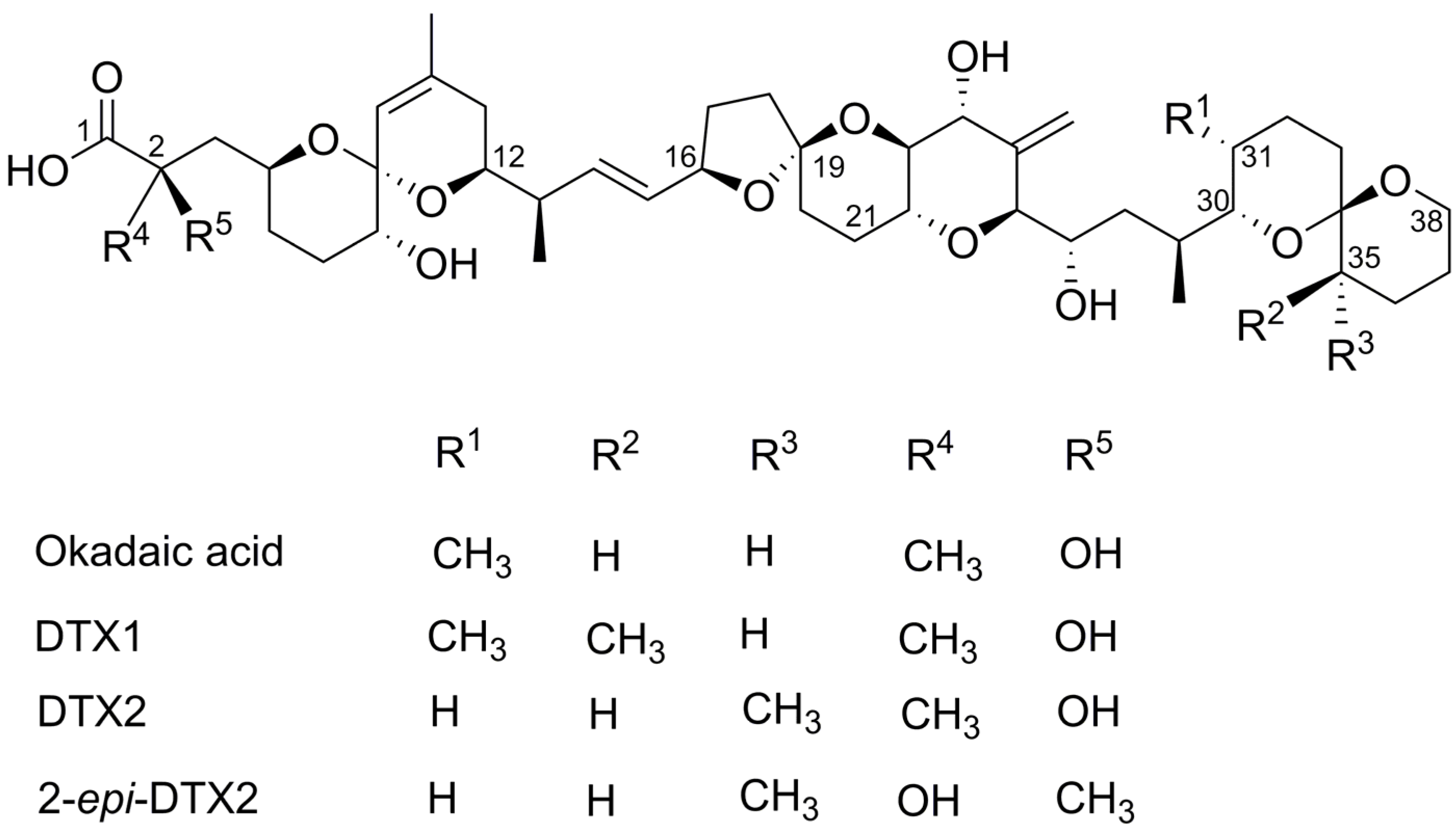
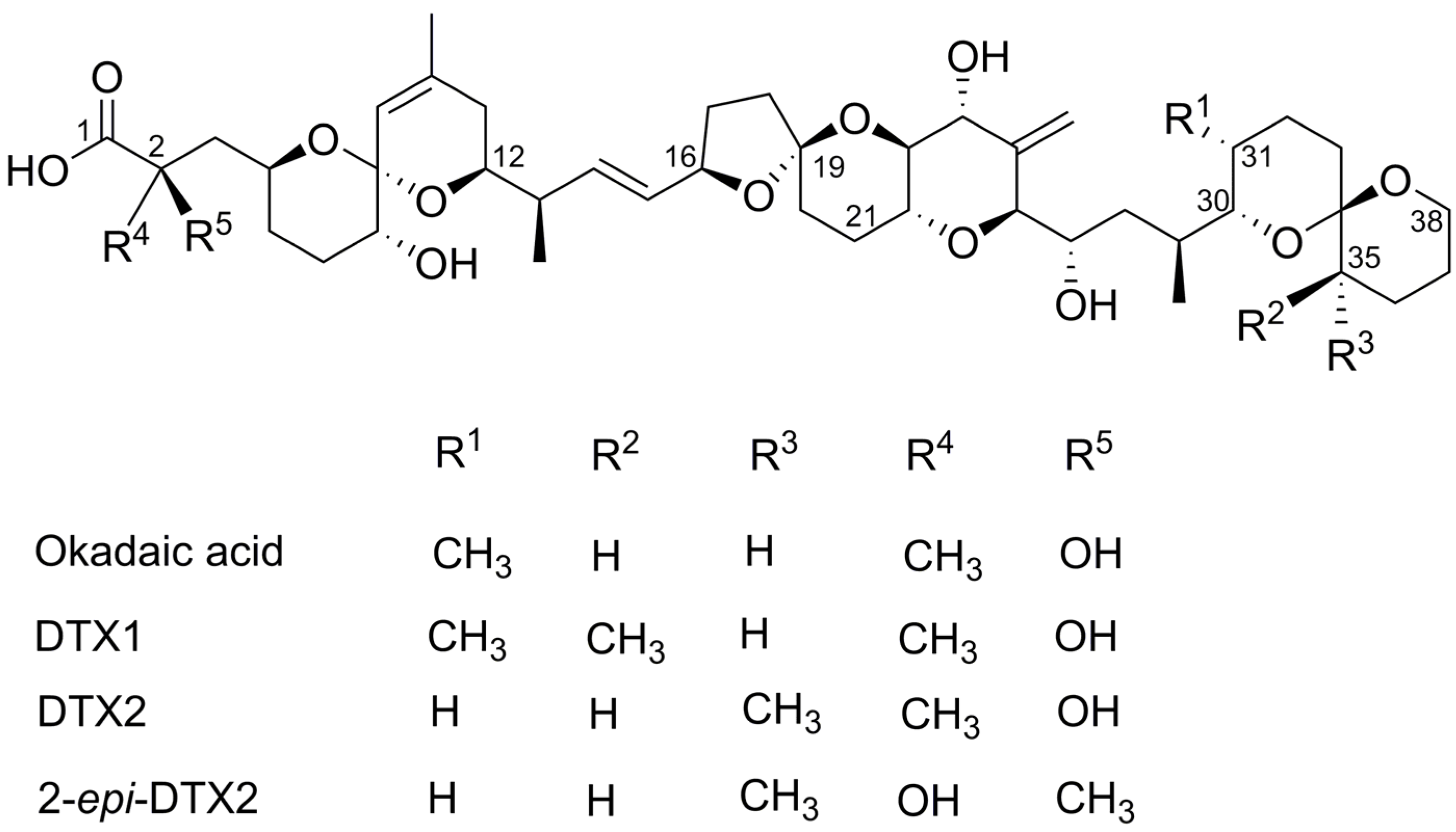
Structure–Activity Relationship Studies Using Natural and Synthetic Okadaic Acid/Dinophysistoxin Toxins

 ,
,
Abstract
:1. Introduction
2. Results
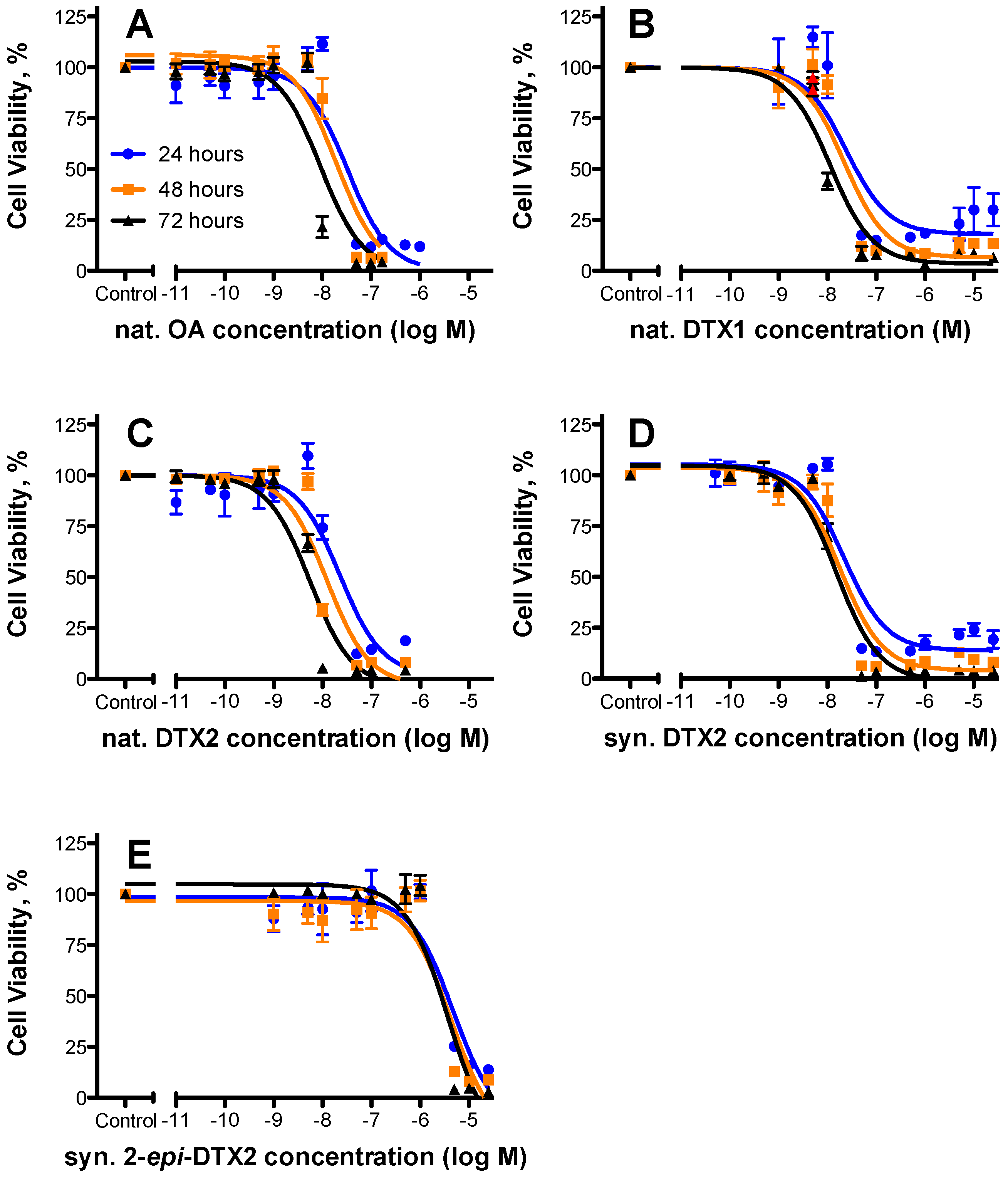
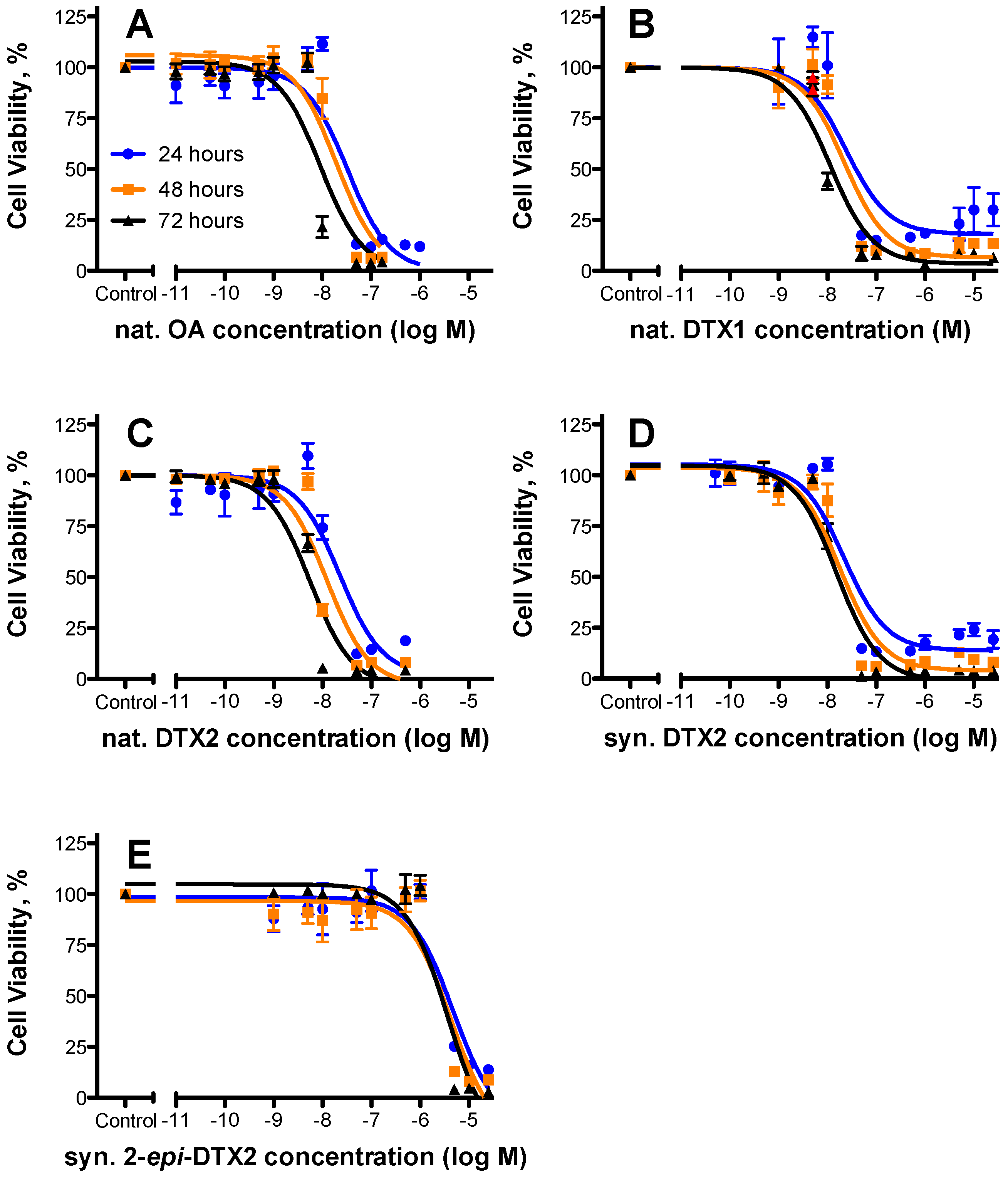
2.1. Cytotoxicty
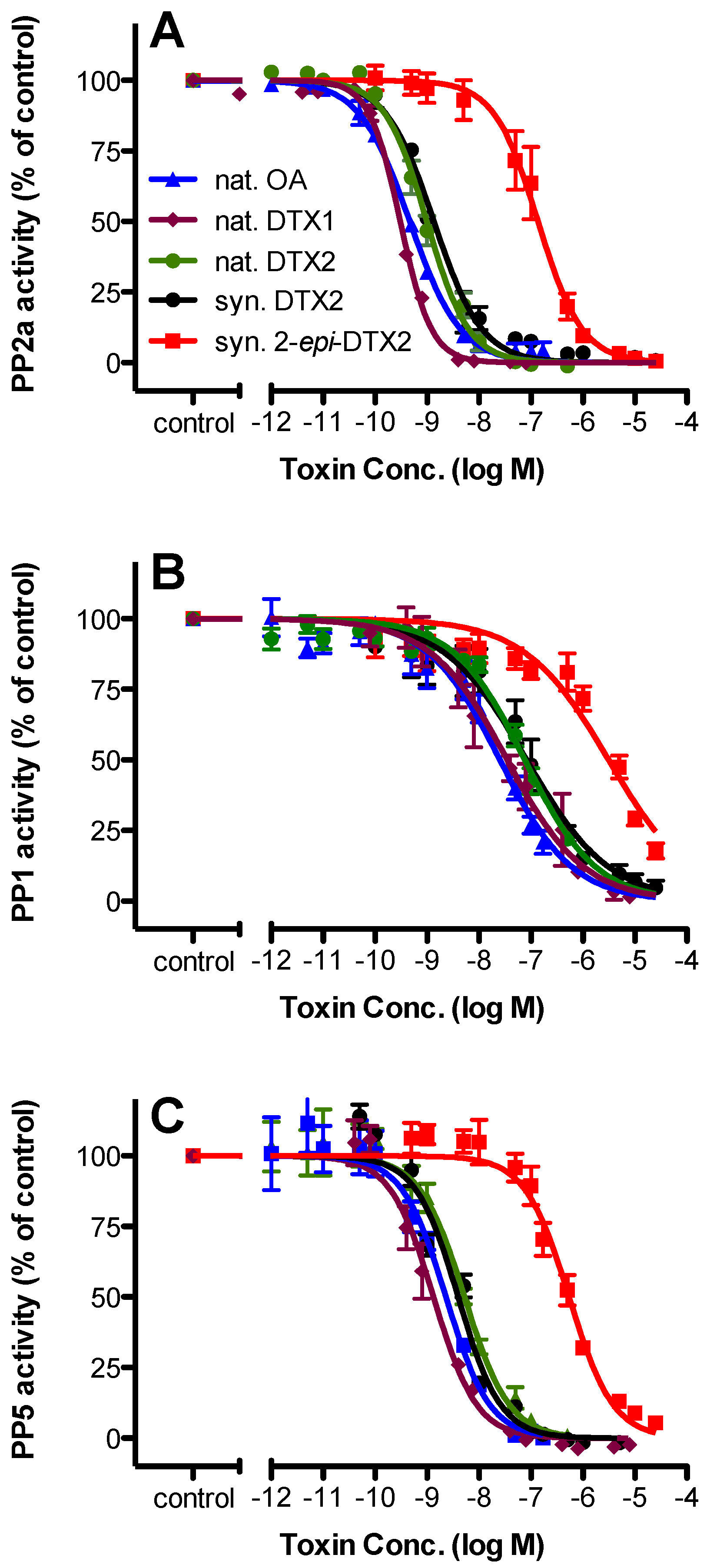
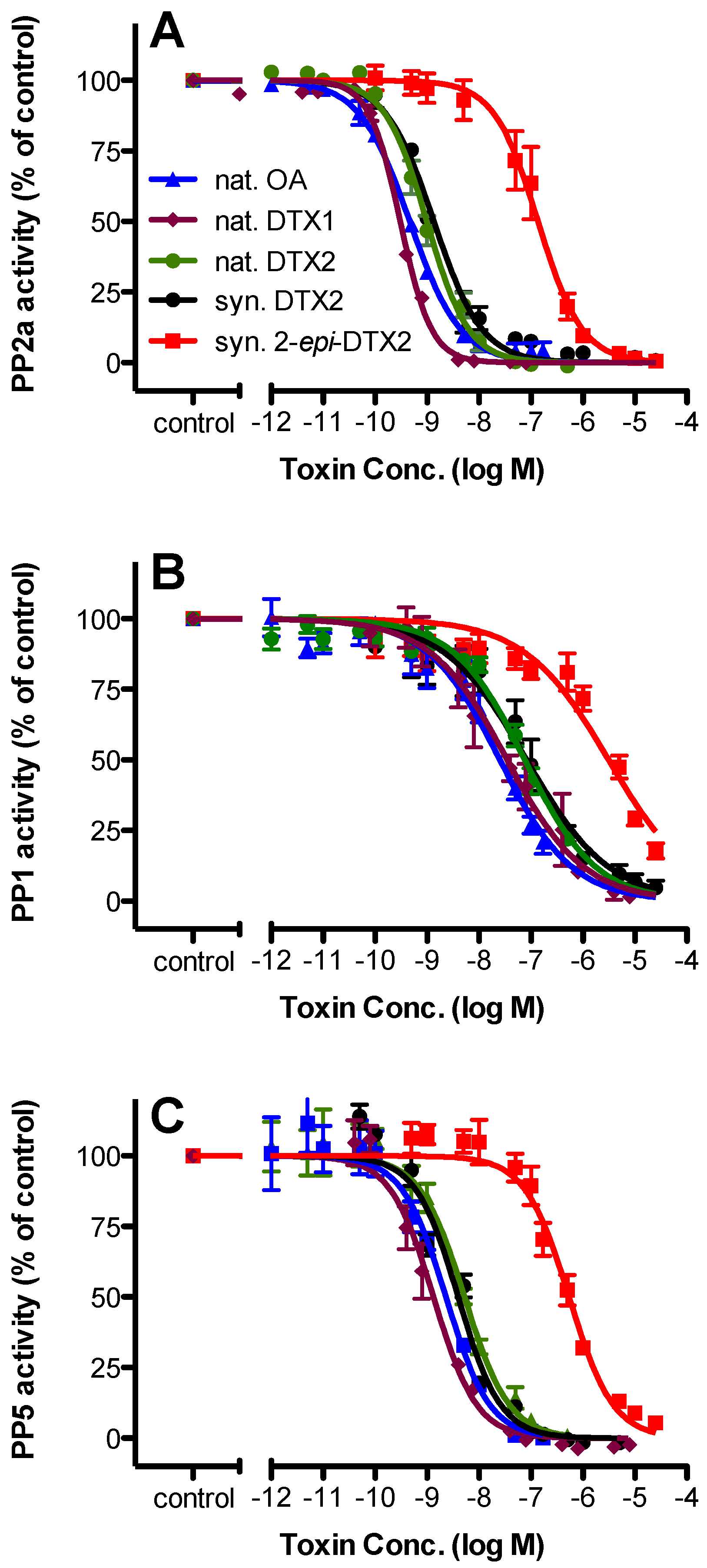
2.2. Protein Phosphatase Inhibition
3. Discussion
4. Materials and Methods
4.1. Toxins
4.2. Cytotoxicty Assays
4.3. Protein Phosphatase Inhibition Assays
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DTX1 | Dinophysistoxin-1 |
| DTX2 | Dinophysistoxin-2 |
| EC50 | 50% Maximal Effective Concentration |
| IC50 | 50% Maximal Inhibitory Concentration |
| MTS | 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium |
| OA | Okadaic Acid |
| PBS | Phosphate Buffered Saline |
| SAR | Structure–Activity Relationship |
| TEF | Toxic Equivalence Factor |
References
- Yasumoto, T.; Murata, M.; Oshima, Y.; Sano, M.; Matsumoto, G.; Clardy, J. Diarrhetic shellfish toxins. Tetrahedron 1985, 41, 1019–1025. [Google Scholar] [CrossRef]
- Subba Rao, D.; Pan, Y.; Zitko, V.; Mackeigan, K. Diarrhetic shellfish poisoning (DSP) associated with subsurface bloom of Dinophysis norvegica in Bedford Basin, eastern Canada. Mar. Ecol. Prog. Ser. 1993, 97, 117–126. [Google Scholar] [CrossRef]
- Park, M.G.; Kim, S.; Kim, H.S.; Myung, G.; Kang, Y.G.; Yih, W. First successful culture of the marine dinoflagellate Dinophysis acuminata. Aquat. Microb. Ecol. 2006, 45, 101–106. [Google Scholar] [CrossRef]
- Trainer, V.L.; Moore, L.; Bill, B.D.; Adams, N.G.; Harrington, N.; Borchert, J.; da Silva, D.A.; Eberhart, B.T. Diarrhetic shellfish toxins and other lipophilic toxins of human health concern in Washington State. Mar. Drugs 2013, 11, 1815–1835. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, J.K.; Duchin, J.S.; Borchert, J.; Quintana, H.F.; Robertson, A. Diarrhetic shellfish poisoning, Washington, USA, 2011. Emerg. Infect. Dis. 2013, 19, 1314–1316. [Google Scholar] [CrossRef] [PubMed]
- Scoging, A.; Bahl, M. Diarrhetic shellfish poisoning in the UK. Lancet 1998, 352, 117. [Google Scholar] [CrossRef]
- Fujiki, H.; Suganuma, M.; Yoshizawa, S.; Nishiwaki, S.; Winyar, B.; Sugimura, T. Mechanisms of action of okadaic acid class tumor promoters on mouse skin. Environ. Health Perspect. 1991, 93, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Bialojan, C.; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 1988, 256, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.T.W. The structure and regulation of protein phosphatases. Annu. Rev. Biochem. 1989, 58, 453–508. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Scheuer, P.J.; Tsukitani, Y.; Kikuchi, H.; Van Engen, D.; Clardy, J.; Gopichand, Y.; Schmitz, F.J. Okadaic acid, a cytotoxic polyether from two marine sponges of the genus Halichondria. J. Am. Chem. Soc. 1981, 103, 2469–2471. [Google Scholar] [CrossRef]
- Xing, Y.; Xu, Y.; Chen, Y.; Jeffrey, P.D.; Chao, Y.; Lin, Z.; Li, Z.; Strack, S.; Stock, J.B.; Shi, Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 2006, 127, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Swingle, M.R.; Amable, L.; Lawhorn, B.G.; Buck, S.B.; Burke, C.P.; Ratti, P.; Fischer, K.L.; Boger, D.L.; Honkanen, R.E. Structure-activity relationship studies of fostriecin, cytostatin, and key analogs, with PP1, PP2A, PP5, and (β12–β13)-chimeras (PP1/PP2A and PP5/PP2A), provide further insight into the inhibitory actions of fostriecin family inhibitors. J. Pharmacol. Exp. Ther. 2009, 331, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Swingle, M.R.; Honkanen, R.E.; Ciszak, E.M. Structural basis for the catalytic activity of human serine/threonine protein phosphatase-5. J. Biol. Chem. 2004, 279, 33992–33999. [Google Scholar] [CrossRef] [PubMed]
- Honkanan, R.E.; Codispoti, B.A.; Tse, K.; Boynton, A.L. Characterization of natural toxins with inhibitory activity against serine/threonine protein phosphatases. Toxicon 1994, 32, 339–350. [Google Scholar] [CrossRef]
- Maynes, J.T.; Bateman, K.S.; Cherney, M.M.; Das, A.K.; Luu, H.A.; Holmes, C.F.B.; James, M.N.G. Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J. Biol. Chem. 2001, 276, 44078–44082. [Google Scholar] [CrossRef] [PubMed]
- Dounay, A.B.; Forsyth, C.J. Okadaic acid: The archetypal serine/threonine protein phosphatase inhibitor. Curr. Med. Chem. 2002, 9, 1939–1980. [Google Scholar] [CrossRef] [PubMed]
- Huhn, J.; Jeffrey, P.D.; Larsen, K.; Rundberget, T.; Rise, F.; Cox, N.R.; Arcus, V.; Shi, Y.; Miles, C.O. A structural basis for the reduced toxicity of dinophysistoxin-2. Chem. Res. Toxicol. 2009, 22, 1782–1786. [Google Scholar] [CrossRef] [PubMed]
- Takai, A.; Murata, M.; Torigoe, K.; Isobe, M.; Mieskes, G.; Yasumoto, T. Inhibitory effect of okadaic acid derivatives on protein phosphatases. A study on structure-affinity relationship. Biochem. J. 1992, 284, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Aune, T.; Larsen, S.; Aasen, J.A.B.; Rehmann, N.; Satake, M.; Hess, P. Relative toxicity of dinophysistoxin-2 (DTX-2) compared with okadaic acid, based on acute intraperitoneal toxicity in mice. Toxicon 2007, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, R.E.; Golden, T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr. Med. Chem. 2002, 9, 2055–2075. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Fang, C.; Twiner, M.J.; Miles, C.O.; Forsyth, C.J. Total synthesis and PPase inhibition of dinophysistoxin-2 and 2-epi-dinophysistoxin-2. Angew. Chem. Int. Ed. Eng. 2011, 50, 7631–7635. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Marine biotoxins in shellfish—Okadaic acid and analogues—Scientific Opinion of the Panel on Contaminants in the Food chain. EFSA J. 2008, 589, 1–62. [Google Scholar]
- Chen, M.X.; McPartlin, A.E.; Brown, L.; Chen, Y.H.; Barker, H.M.; Cohen, P.T. A novel human protein serine/threonine phosphatase, which possesses four tetratricopeptide repeat motifs and localizes to the nucleus. EMBO J. 1994, 13, 4278–4290. [Google Scholar] [PubMed]
- Cohen, P.T.W. Novel protein serine/threonine phosphatases: Variety is the spice of life. Trends Biochem. Sci. 1997, 22, 245–251. [Google Scholar] [CrossRef]
- Larsen, K.; Petersen, D.; Wilkins, A.L.; Samdal, I.A.; Sandvik, M.; Rundberget, T.; Goldstone, D.; Arcus, V.; Hovgaard, P.; Rise, F.; et al. Clarification of the C-35 stereochemistries of dinophysistoxin-1 and dinophysistoxin-2 and its consequences for binding to protein phosphatase. Chem. Res. Toxicol. 2007, 20, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Silverstein, A.M.; Pratt, W.B.; Chinkers, M. The tetratricopeptide repeat domain of protein phosphatase 5 mediates binding to glucocorticoid receptor heterocomplexes and acts as a dominant negative mutant. J. Biol. Chem. 1996, 271, 32315–32320. [Google Scholar] [CrossRef] [PubMed]
- Chinkers, M. Protein phosphatase 5 in signal transduction. Trends Endocrinol. Metabol. 2001, 12, 28–32. [Google Scholar] [CrossRef]
- Bertini, I.; Calderone, V.; Fragai, M.; Luchinat, C.; Talluri, E. Structural basis of serine/threonine phosphatase inhibition by the archetypal small molecules cantharidin and norcantharidin. J. Med. Chem. 2009, 52, 4838–4843. [Google Scholar] [CrossRef] [PubMed]
- Valdiglesias, V.; Prego-Faraldo, M.V.; Pasaro, E.; Mendez, J.; Laffon, B. Okadaic acid: More than a diarrheic toxin. Mar. Drugs 2013, 11, 4328–4349. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.; Wang, C. Synthesis and stereochemistry of the terminal spiroketal domain of the phosphatase inhibitor dinophysistoxin-2. Bioorg. Med. Chem. Lett. 2008, 18, 3043–3046. [Google Scholar] [CrossRef] [PubMed]
- Trygstad, T.; Pang, Y.; Forsyth, C.J. Versatile synthesis of the C3–C14 domain of 7-deoxy okadaic acid. J. Org. Chem. 2009, 74, 910–913. [Google Scholar] [CrossRef] [PubMed]
- Dounay, A.; Urbanek, R.; Frydrychowski, V.; Forsyth, C. Expedient access to the okadaic acid architecture: A novel synthesis of the C1–C27 domain. J. Org. Chem. 2001, 66, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.; Sabes, S.; Urbanek, R. An efficient total synthesis of okadaic acid. J. Am. Chem. Soc. 1997, 119, 8381–8382. [Google Scholar] [CrossRef]
- Dounay, A.; Urbanek, R.; Sabes, S.; Forsyth, C. Total synthesis of the natural product 7-deoxy-okadaic acid: A potent inhibitor of protein serine/threonine phosphatases. Angew. Chem. 1999, 38, 2258–2262. [Google Scholar] [CrossRef]
- Cruz, P.; Daranas, A.; Fernández, J.; Norte, M. 19-epi-Okadaic acid, a novel protein phosphatase inhibitor with enhanced selectivity. Org. Lett. 2007, 9, 3045–3048. [Google Scholar] [CrossRef] [PubMed]
- Twiner, M.J.; El-Ladki, R.; Kilcoyne, J.; Doucette, G.J. Comparative effects of the marine algal toxins azaspiracid-1, -2, and -3 on Jurkat T lymphocyte cells. Chem. Res. Toxicol. 2012, 25, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Vieytes, M.R.; Fontal, O.I.; Leira, F.; Baptista de Sousa, J.M.V.; Botana, L.M. A fluorescent microplate assay for diarrheic shellfish toxins. Anal. Biochem. 1997, 248, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Twiner, M.J.; Hess, P.; Bottein Dechraoui, M.-Y.; McMahon, T.; Samons, M.S.; Satake, M.; Yasumoto, T.; Ramsdell, J.S.; Doucette, G.J. Cytotoxic and cytoskeletal effects of azaspiracid-1 on mammalian cell lines. Toxicon 2005, 45, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Heresztyn, T.; Nicholson, B.C. A colorimetric protein phosphatase inhibition assay for the determination of cyanobacterial peptide hepatotoxins based on the dephosphorylation of phosvitin by recombinant protein phosphatase 1. Environ. Toxicol. 2001, 16, 242–252. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Toxin | 24 h | 48 h | 72 h | Rel. Potency (as per 48 h Data) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (nM) | 95% Confidence Intervals | n | EC50 (nM) | 95% Confidence Intervals | n | EC50 (nM) | 95% Confidence Intervals | n | ||
| nat. OA | 32.8 | 19.4–55.3 | 3 | 19.5 | 11.7–32.4 | 3 | 9.16 | 5.43–15.5 | 3 | 1.000 |
| nat. DTX1 | 24.6 | 8.60–70.2 | 2 | 22.2 | 11.6–42.5 | 2 | 11.4 | 7.39–17.6 | 2 | 0.87 |
| nat. DTX2 | 22.8 | 11.1–46.6 | 3 | 12.3 | 6.71–22.5 | 3 | 5.52 | 3.55–8.61 | 3 | 1.58 |
| syn. DTX2 | 21.8 | 11.7–40.7 | 3 | 18.2 | 10.8–30.6 | 3 | 15.4 | 10.2–23.0 | 3 | 1.07 |
| syn. 2-epi-DTX2 | 4750 | 2010–11,200 | 3 | 4260 | 1710–10,600 | 3 | 3740 | 1880–7470 | 3 | 0.004 |
| Toxin | PP2a | PP1 | PP5 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | 95% Confidence Intervals | n | Rel. Potency | IC50 (nM) | 95% Confidence Intervals | n | Rel. Potency | IC50 (nM) | 95% Confidence Intervals | n | Rel. Potency | |
| nat. OA | 0.466 | 0.402–0.539 | 3 | 1.000 | 25.2 | 18.2–34.8 | 3 | 1.000 | 2.30 | 1.66–3.17 | 3 | 1.000 |
| nat. DTX1 | 0.306 | 0.283–0.330 | 4 | 1.523 | 34.8 | 21.9–55.2 | 4 | 0.724 | 1.30 | 1.06–1.66 | 4 | 1.769 |
| nat. DTX2 | 0.987 | 0.844–1.16 | 3 | 0.472 | 76.4 | 57.9–101 | 3 | 0.330 | 5.25 | 3.96–6.98 | 4 | 0.438 |
| syn. DTX2 | 1.35 | 1.12–1.63 | 3 | 0.345 | 82.6 | 55.4–123 | 3 | 0.305 | 3.95 | 3.18–5.00 | 4 | 0.582 |
| syn. 2-epi-DTX2 | 137 | 106–178 | 3 | 0.003 | 3110 | 2080–4660 | 3 | 0.008 | 541 | 433–675 | 4 | 0.004 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Twiner, M.J.; Doucette, G.J.; Pang, Y.; Fang, C.; Forsyth, C.J.; Miles, C.O. Structure–Activity Relationship Studies Using Natural and Synthetic Okadaic Acid/Dinophysistoxin Toxins. Mar. Drugs 2016, 14, 207. https://doi.org/10.3390/md14110207
Twiner MJ, Doucette GJ, Pang Y, Fang C, Forsyth CJ, Miles CO. Structure–Activity Relationship Studies Using Natural and Synthetic Okadaic Acid/Dinophysistoxin Toxins. Marine Drugs. 2016; 14(11):207. https://doi.org/10.3390/md14110207
Chicago/Turabian StyleTwiner, Michael J., Gregory J. Doucette, Yucheng Pang, Chao Fang, Craig J. Forsyth, and Christopher O. Miles. 2016. "Structure–Activity Relationship Studies Using Natural and Synthetic Okadaic Acid/Dinophysistoxin Toxins" Marine Drugs 14, no. 11: 207. https://doi.org/10.3390/md14110207