
Marine Bromophenol Derivative 3,4-Dibromo-5-(2-bromo-3,4-dihydroxy-6-isopropoxymethyl benzyl)benzene-1,2-diol Protects Hepatocytes from Lipid-Induced Cell Damage and Insulin Resistance via PTP1B Inhibition


 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
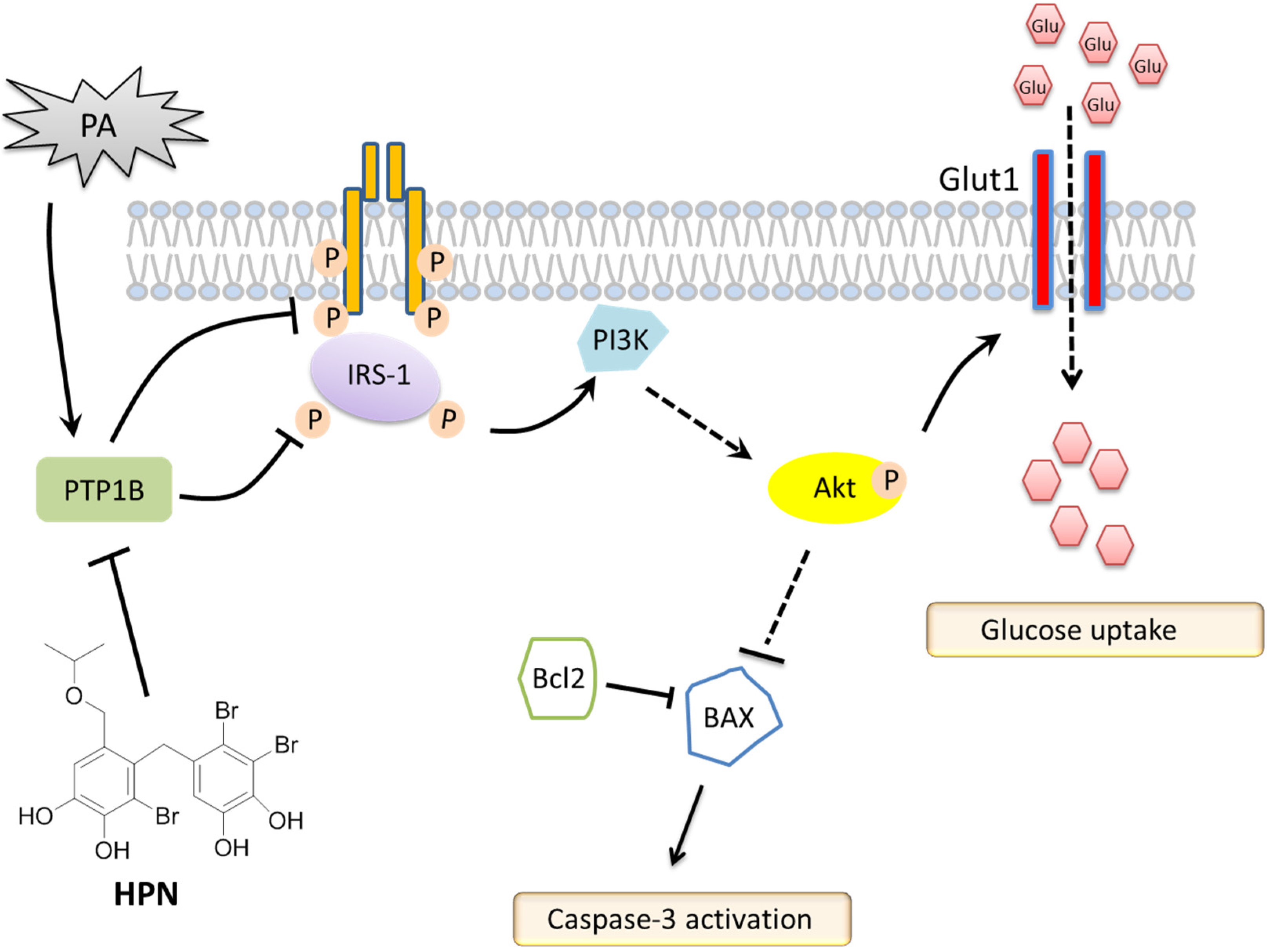{kind=link}
1. Introduction
2. Results
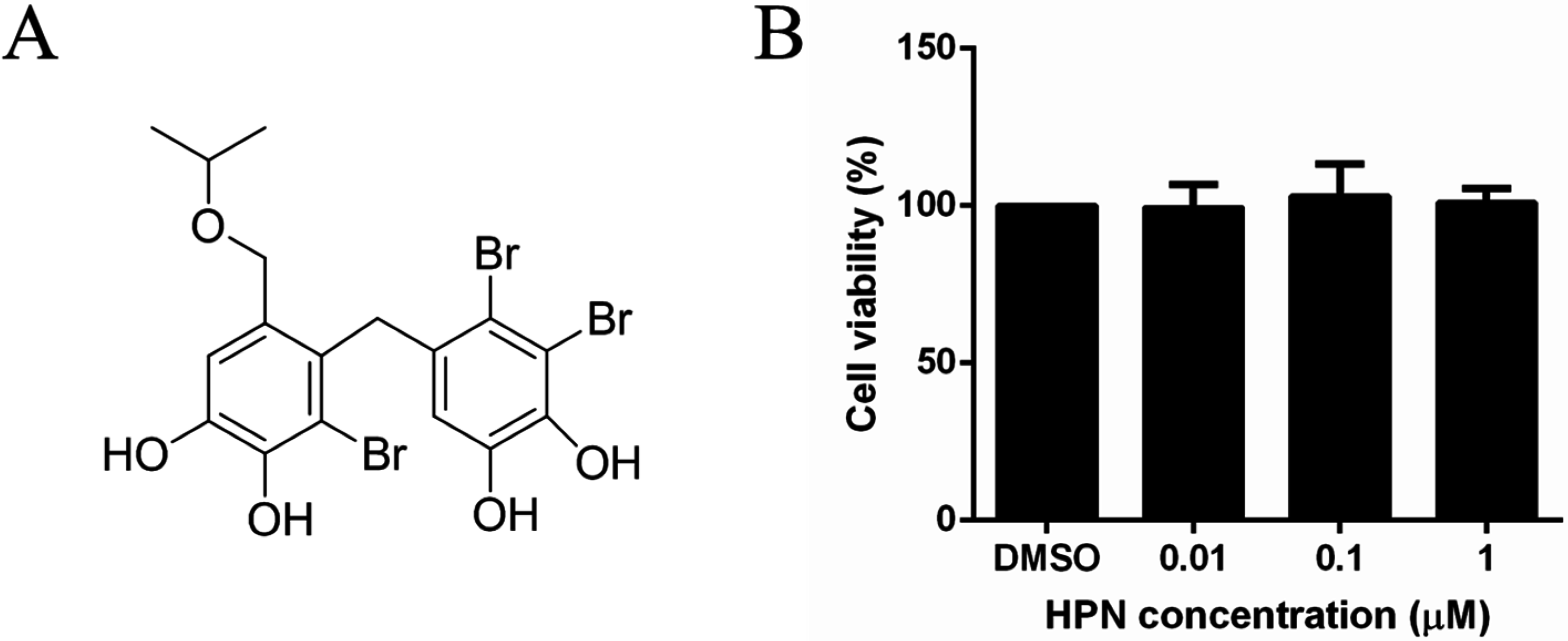
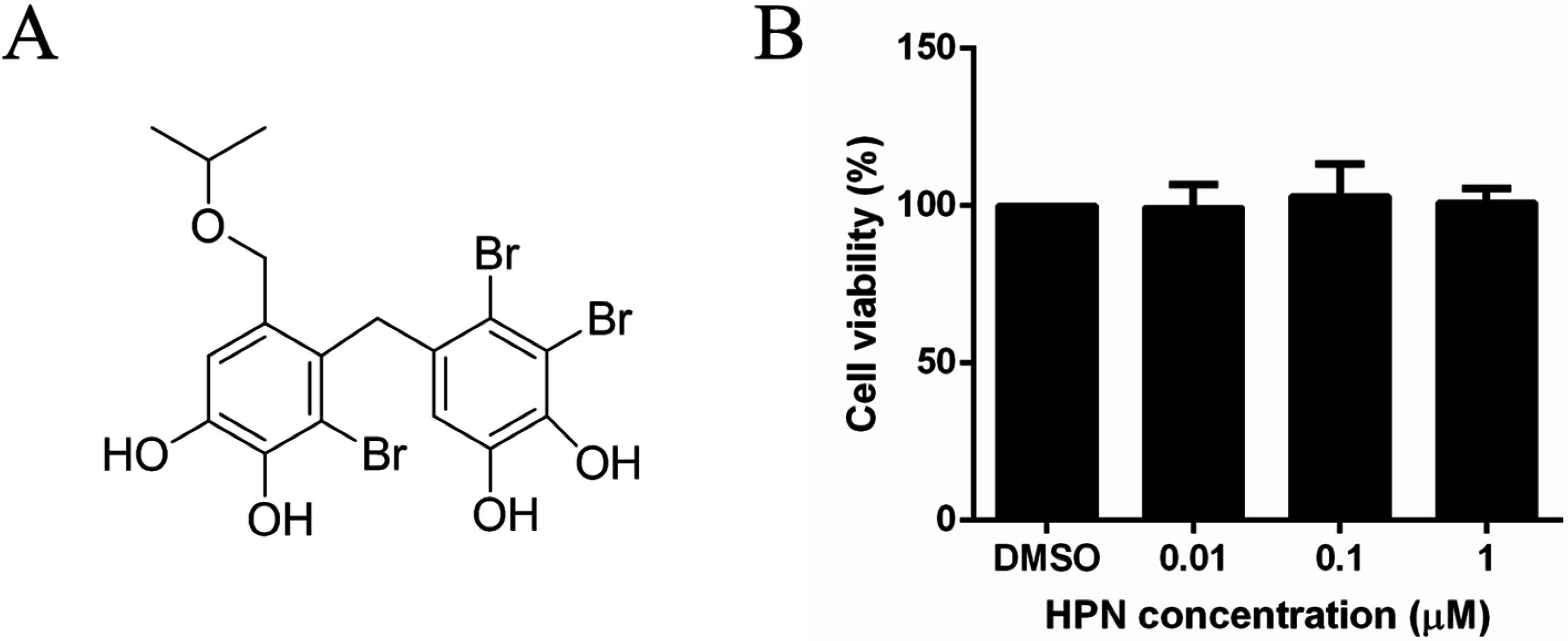
2.1. HPN Shows No Effect on HepG2 Cell Proliferation
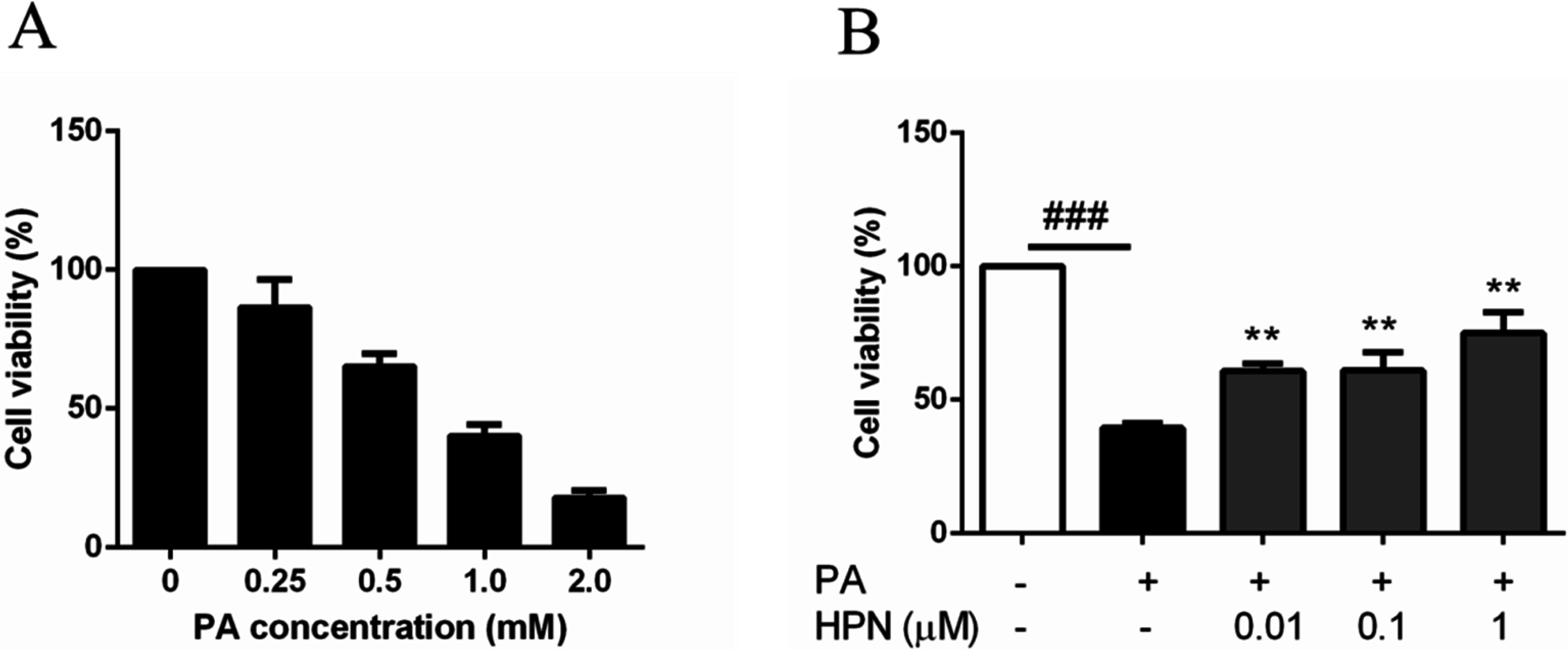
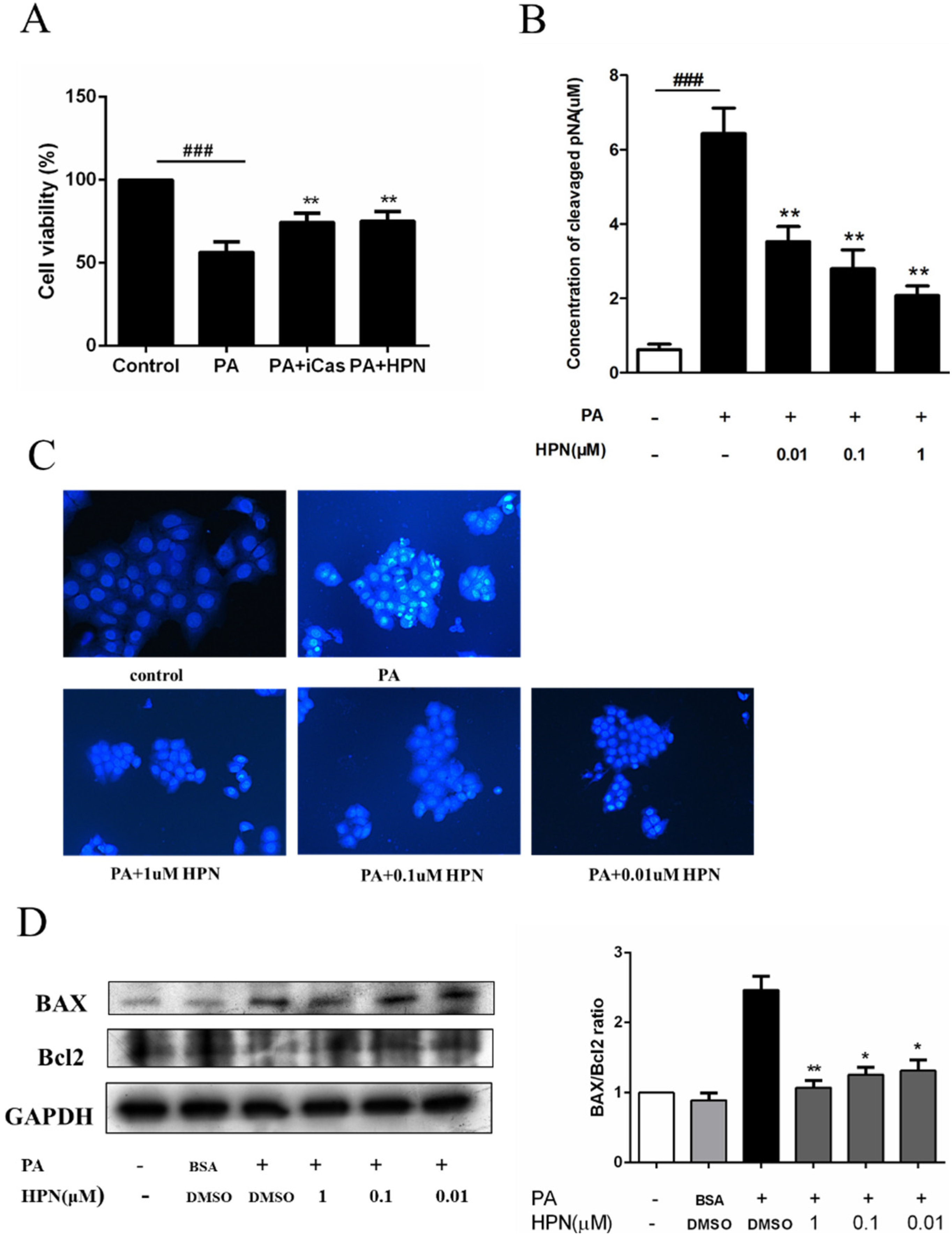
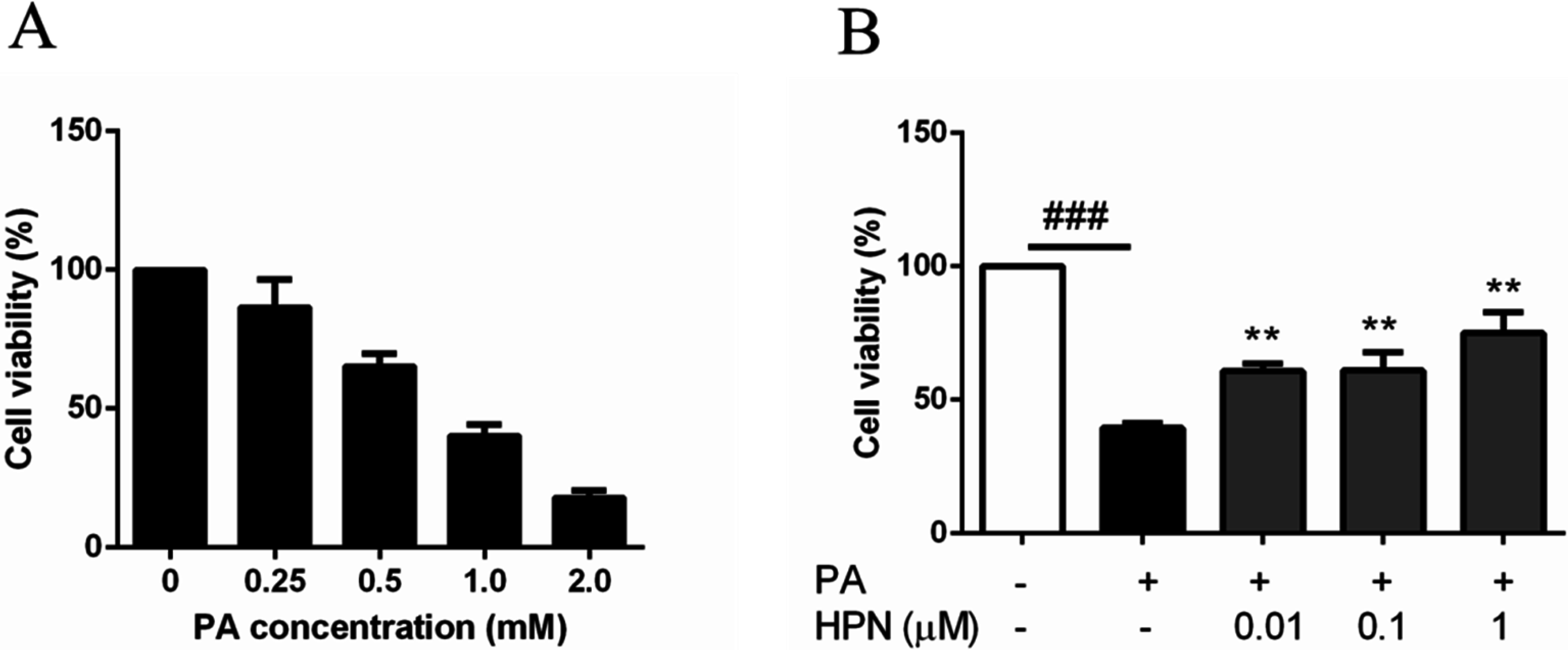
2.2. HPN Inhibits PA-Induced Cell Death in HepG2 Cells
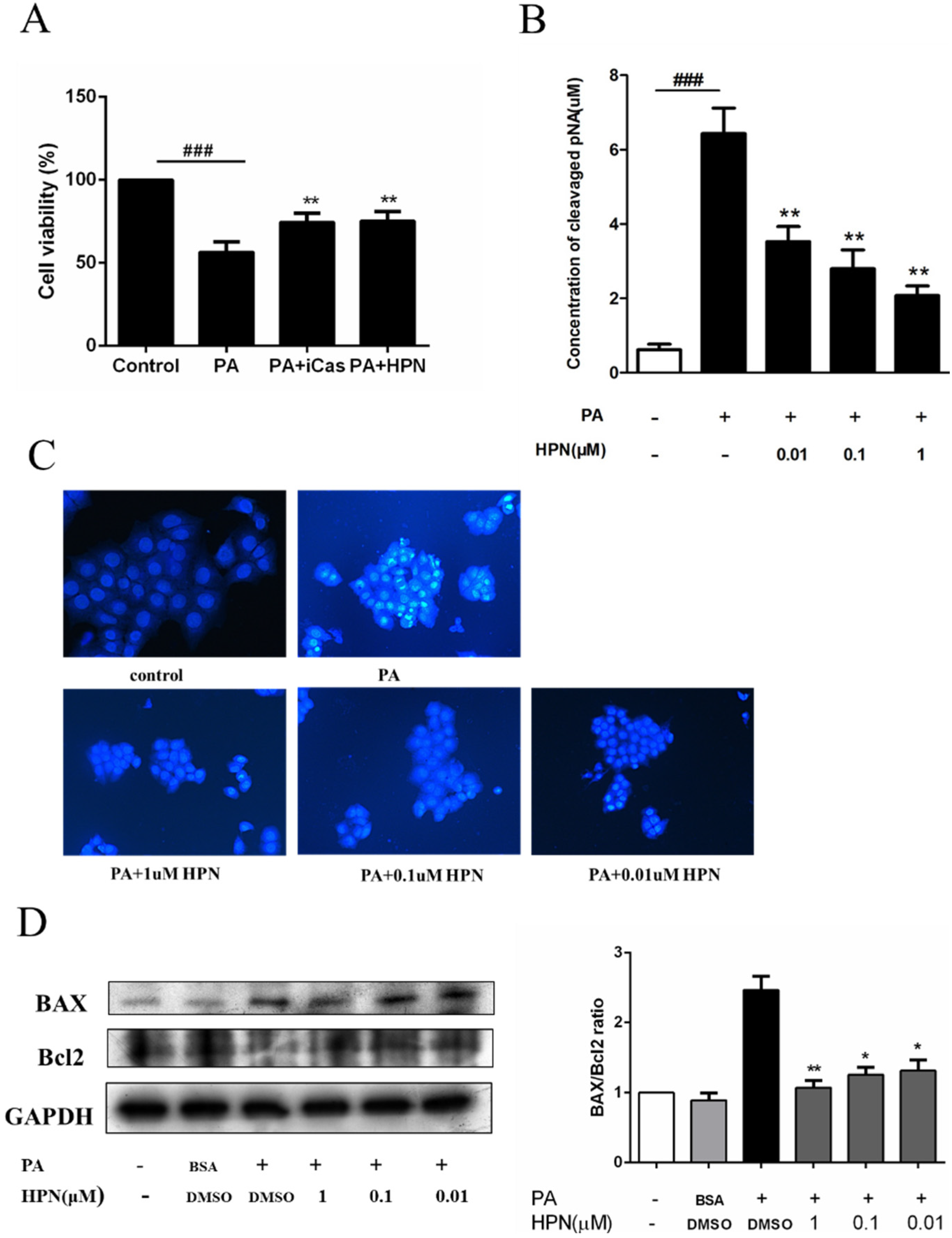
2.3. HPN Prevents PA-Induced Caspase-3 Activation
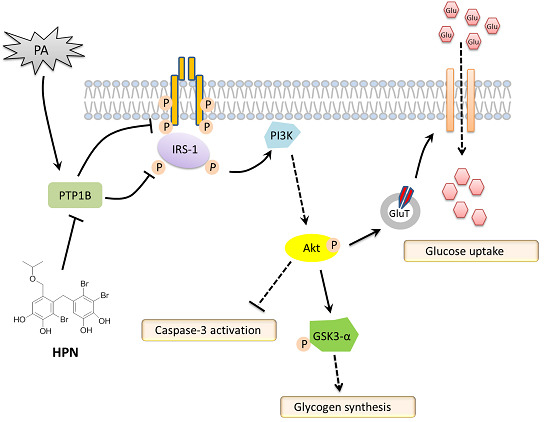
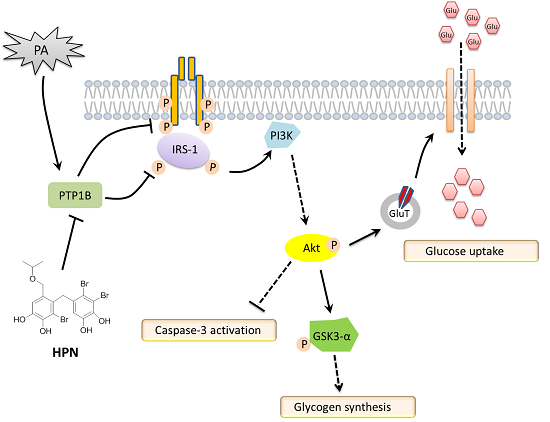
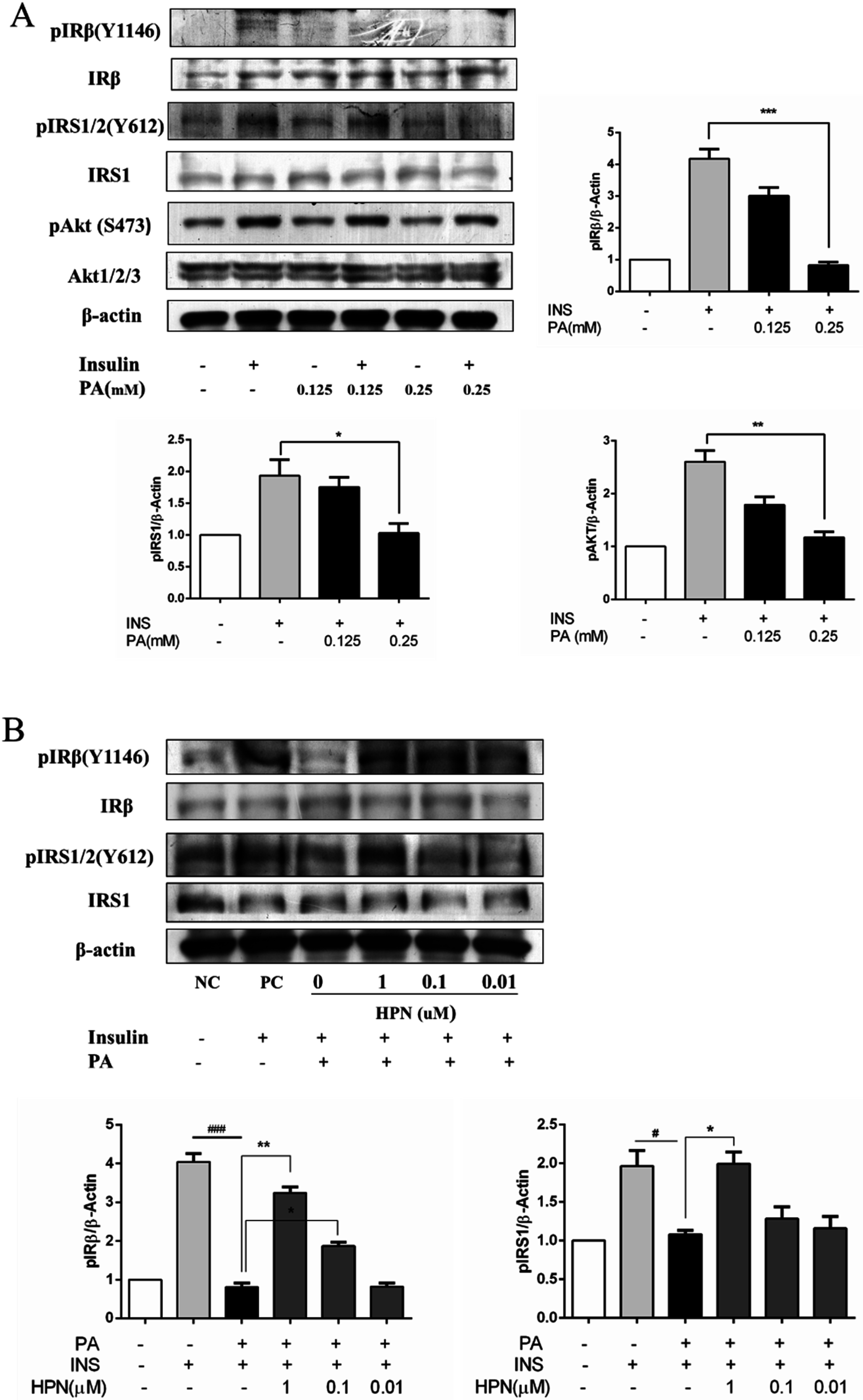
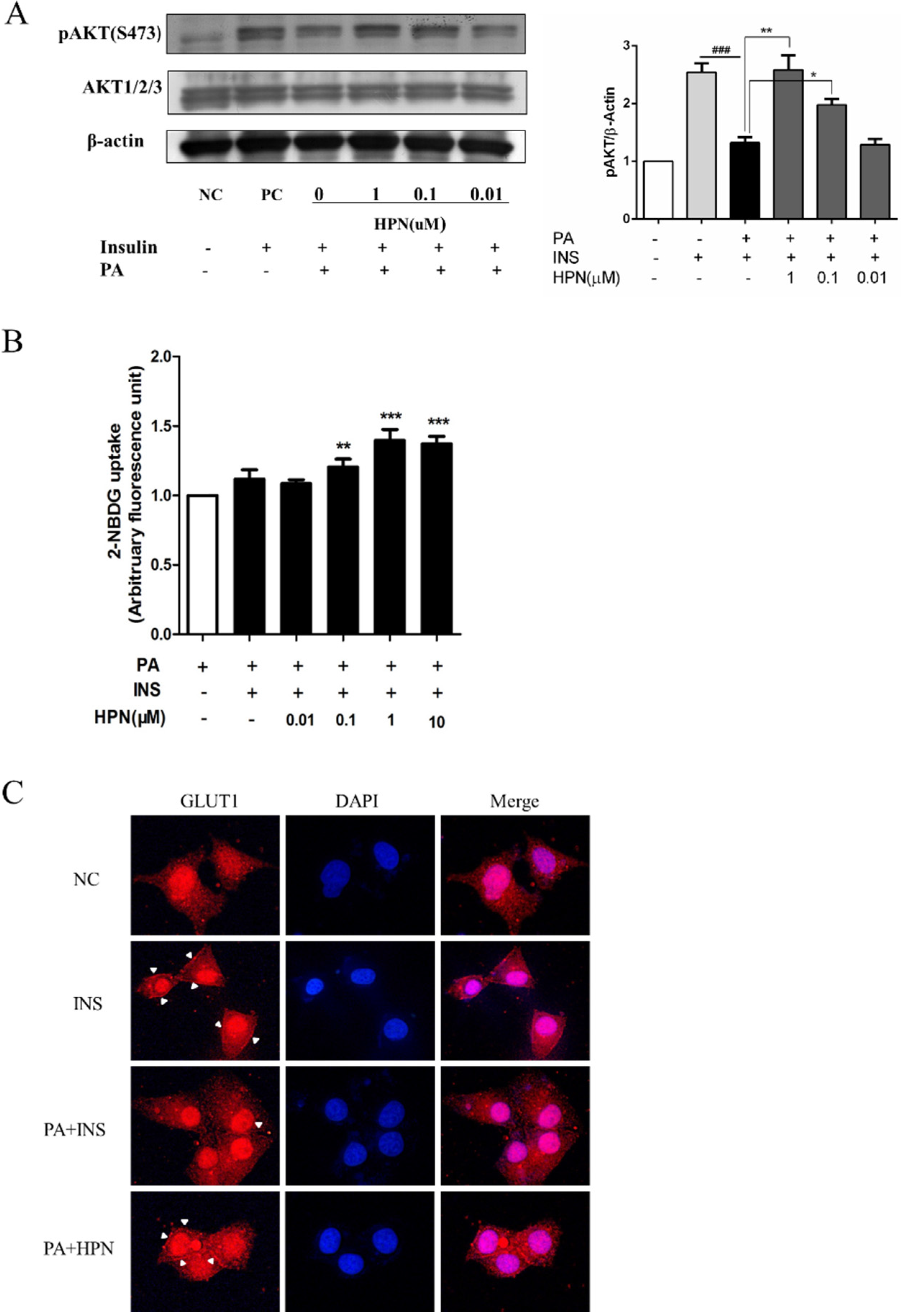
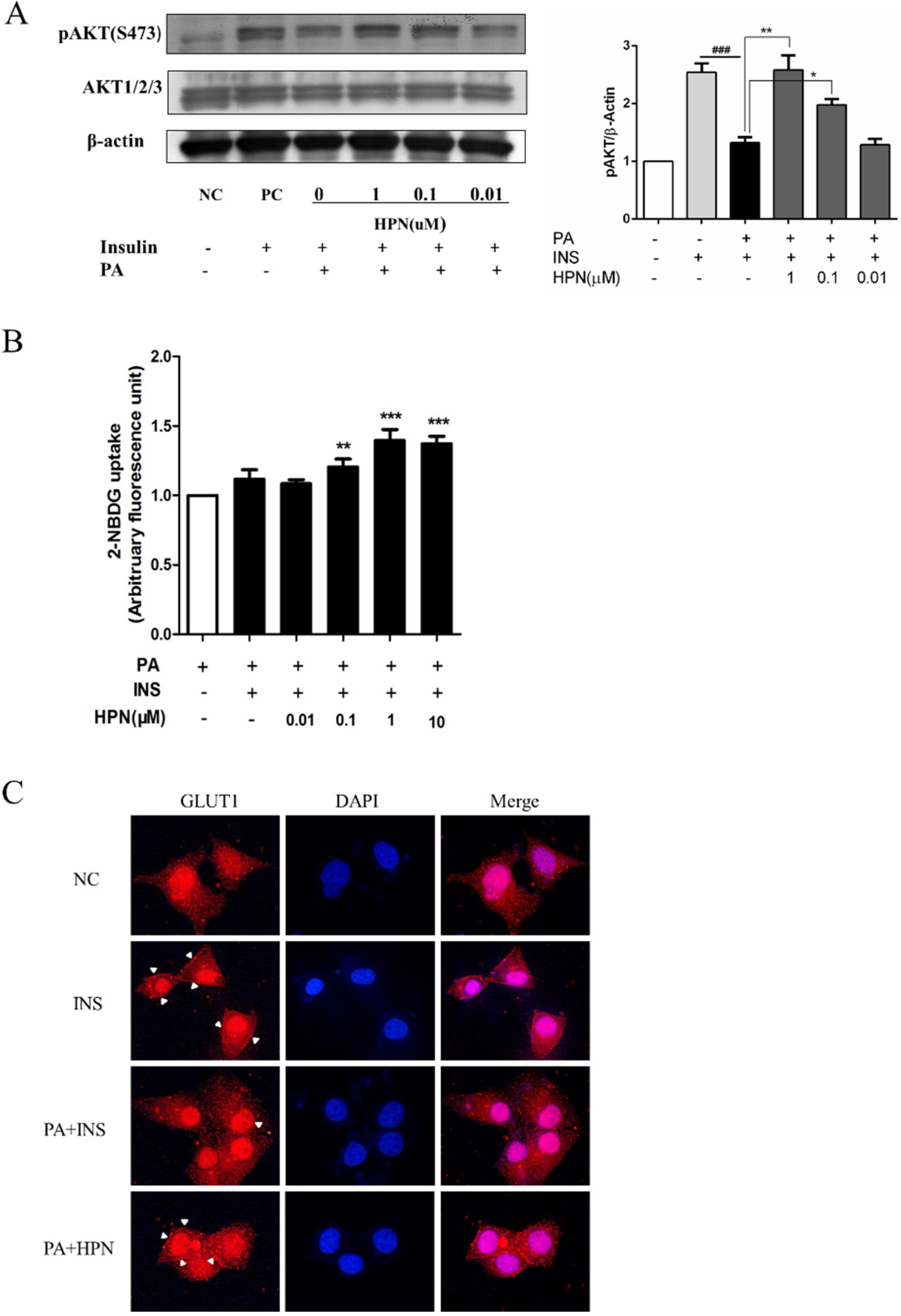
2.4. HPN Ameliorates PA-Induced Insulin Resistance with Up-Regulated Insulin Signaling

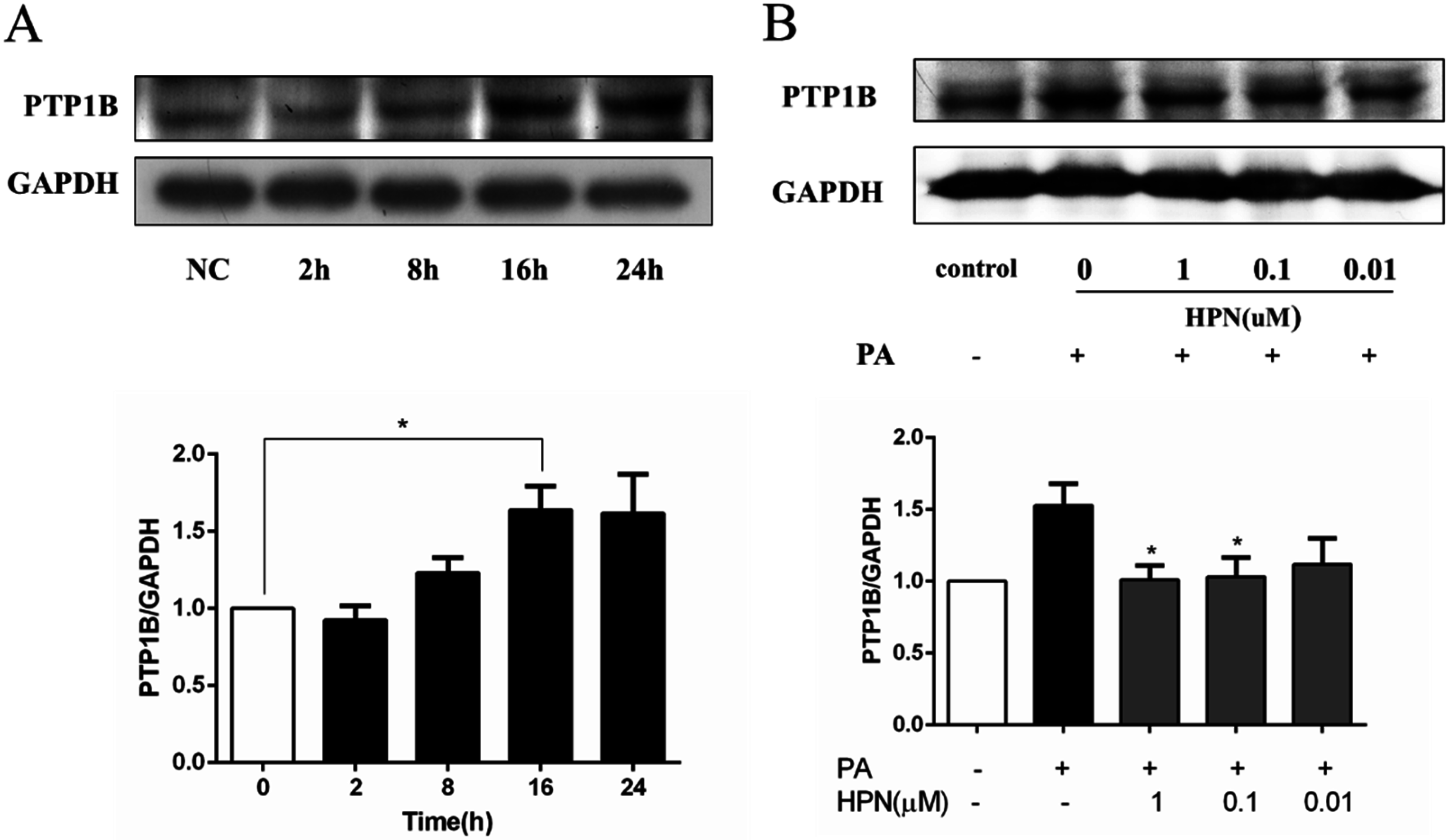
2.5. HPN Inhibits PA-Stimulated PTP1B Expression
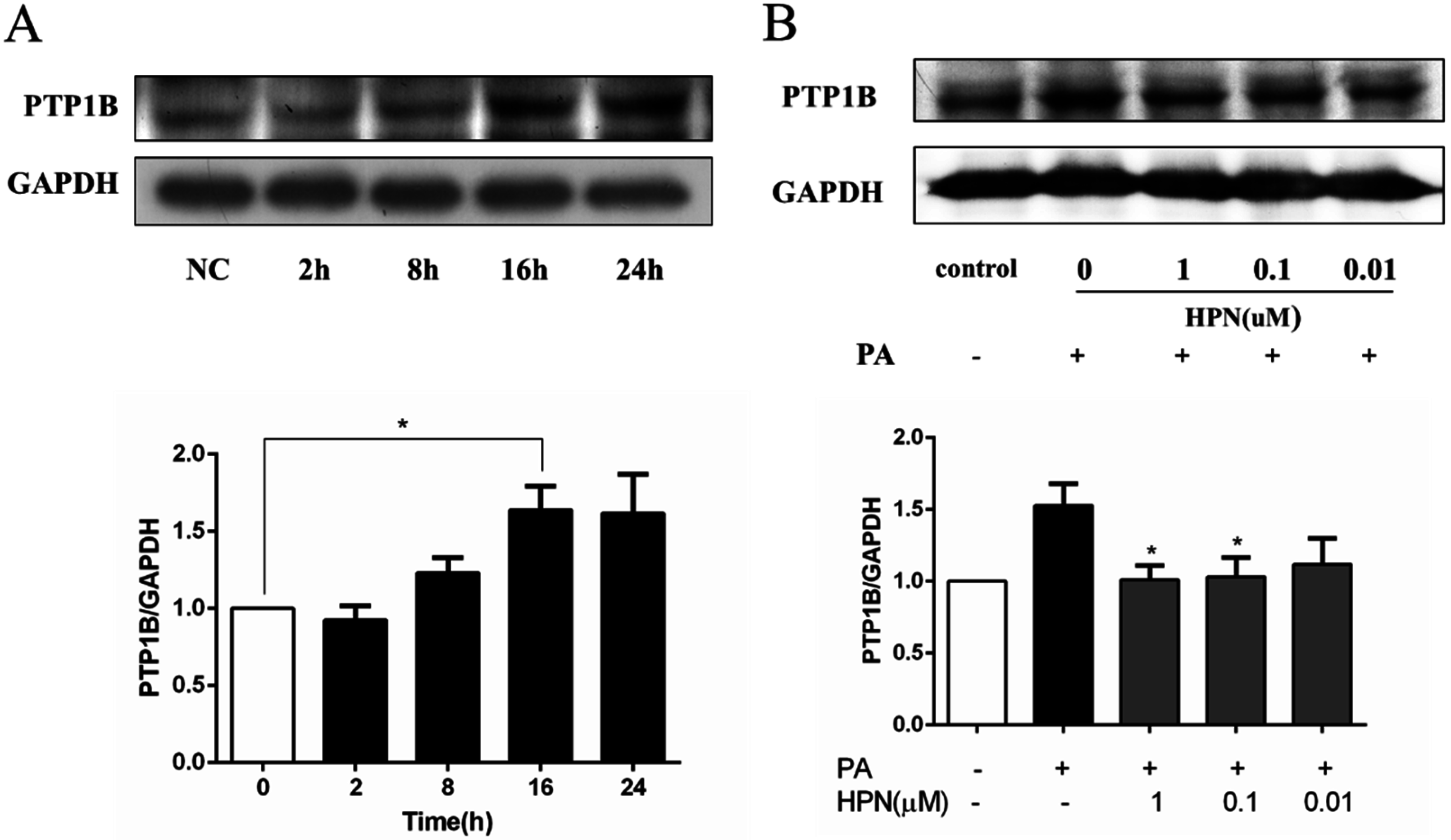
2.6. HPN Improves Glucose Uptake in HepG2 Cells
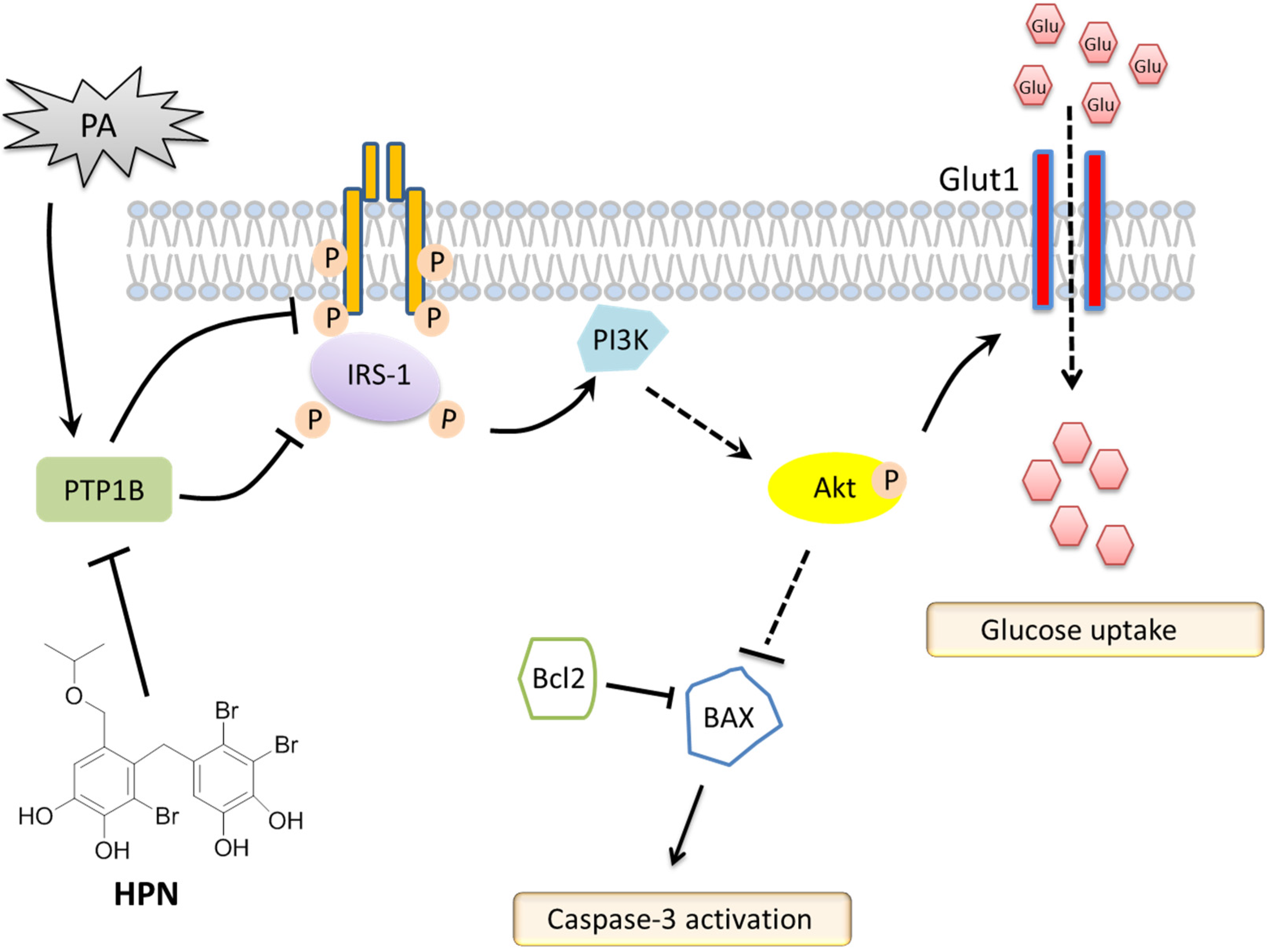
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Preparation of BSA-Conjugated Palmitate Complexes
4.3. MTT Assay
4.4. Caspase-3 Activity Assay
4.5. Hoechst 33258 Staining Assay
4.6. Western Blot Analysis
4.7. Glucose Uptake Assay
4.8. Immunofluorescence
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tahrani, A.A.; Bailey, C.J.; Del Prato, S.; Barnett, A.H. Management of type 2 diabetes: New and future developments in treatment. Lancet 2011, 378, 182–197. [Google Scholar] [CrossRef]
- Rochester, C.D.; Akiyode, O. Novel and emerging diabetes mellitus drug therapies for the type 2 diabetes patient. World J. Diabetes 2014, 5, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Moller, D.E. New drug targets for type 2 diabetes and the metabolic syndrome. Nature 2001, 414, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 2001, 104, 517–529. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Hajduch, E.; Litherland, G.J.; Hundal, H.S. Protein kinase B (PKB/Akt)—A key regulator of glucose transport? FEBS Lett. 2001, 492, 199–203. [Google Scholar] [CrossRef]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, Z.Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002, 1, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. PTP1B: From the sidelines to the front lines! FEBS Lett. 2003, 546, 140–148. [Google Scholar] [CrossRef]
- Byon, J.C.; Kusari, A.B.; Kusari, J. Protein-tyrosine phosphatase-1B acts as a negative regulator of insulin signal transduction. Mol. Cell. Biochem. 1998, 182, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kenner, K.A.; Anyanwu, E.; Olefsky, J.M.; Kusari, J. Protein-tyrosine phosphatase 1B is a negative regulator of Insulin- and Insulin-like growth factor-I-stimulated signaling. J. Biol. Chem. 1996, 271, 19810–19816. [Google Scholar] [CrossRef] [PubMed]
- Zabolotny, J.M.; Haj, F.G.; Kim, Y.B.; Kim, H.J.; Shulman, G.I.; Kim, J.K.; Neel, B.G.; Kahn, B.B. Transgenic overexpression of protein-tyrosine phosphatase 1B in muscle causes insulin resistance, but overexpression with leukocyte antigen-related phosphatase does not additively impair insulin action. J. Biol. Chem. 2004, 279, 24844–24851. [Google Scholar] [CrossRef] [PubMed]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C.; et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.B.; Sharpe, A.H.; et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Hoffstedt, J.; Deeb, W.; Singh, R.; Sedkova, N.; Zilbering, A.; Zhu, L.; Park, P.K.; Arner, P.; Goldstein, B.J. Depot-specific variation in protein-tyrosine phosphatase activities in human omental and subcutaneous adipose tissue: A potential contribution to differential insulin sensitivity. J. Clin. Endocrinol. Metab. 2001, 86, 5973–5980. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Azevedo, J.L.; Cortright, R.; Dohm, G.L.; Goldstein, B.J. Alterations in skeletal muscle protein-tyrosine phosphatase activity and expression in insulin-resistant human obesity and diabetes. J. Clin. Invest. 1997, 100, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Escribano, O.; Alba, J.; Rondinone, C.M.; Benito, M.; Valverde, A.M. Levels of protein tyrosine phosphatase 1B determine susceptibility to apoptosis in serum-deprived hepatocytes. J. Cell. Physiol. 2007, 212, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhang, Z.; Wang, L. Small interference RNA against PTP-1B reduces hypoxia/reoxygenation induced apoptosis of rat cardiomyocytes. Apoptosis 2008, 13, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, V.; Paliouras, G.N.; Cheng, A.; Dube, N.; Tremblay, M.L.; Park, M. Protein-tyrosine phosphatase 1B deficiency protects against Fas-induced hepatic failure. J. Biol. Chem. 2006, 281, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [PubMed]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic β-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, D.B.; Sparagna, G.C.; Amoscato, A.A.; McMillin, J.B.; Dowhan, W. Decreased cardiolipin synthesis corresponds with cytochrome c release in Palmitate-induced cardiomyocyte apoptosis. J. Biol. Chem. 2001, 276, 38061–38067. [Google Scholar] [PubMed]
- Yuzefovych, L.; Wilson, G.; Rachek, L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: Role of oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E1096–E1105. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, Y.W.; Lee, I.K.; Kim, J.Y.; Kang, Y.J.; Park, S.Y. Amp-activated protein kinase activation by 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (aicar) inhibits Palmitate-induced endothelial cell apoptosis through reactive oxygen species suppression. J. Pharmacol. Sci. 2008, 106, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Role of fatty acids in the pathogenesis of insulin resistance and niddm. Diabetes 1997, 46, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Pardo, V.; Gonzalez-Rodriguez, A.; Muntane, J.; Kozma, S.C.; Valverde, A.M. Role of hepatocyte S6K1 in palmitic acid-induced endoplasmic reticulum stress, lipotoxicity, insulin resistance and in oleic acid-induced protection. Food Chem. Toxicol. 2015, 80, 298–309. [Google Scholar] [CrossRef] [PubMed]
- MohammadTaghvaei, N.; Taheripak, G.; Taghikhani, M.; Meshkani, R. Palmitate-induced PTP1B expression is mediated by ceramide-JNK and nuclear factor κB (NF-κB) activation. Cell. Signal. 2012, 24, 1964–1970. [Google Scholar] [CrossRef] [PubMed]
- Parvaneh, L.; Meshkani, R.; Bakhtiyari, S.; Mohammadtaghvaie, N.; Gorganifiruzjaee, S.; Taheripak, G.; Golestani, A.; Foruzandeh, M.; Larijani, B.; Taghikhani, M. Palmitate and inflammatory state additively induce the expression of PTP1B in muscle cells. Biochem. Biophys. Res. Commun. 2010, 396, 467–471. [Google Scholar] [CrossRef] [PubMed]
- MohammadTaghvaei, N.; Meshkani, R.; Taghikhani, M.; Larijani, B.; Adeli, K. Palmitate enhances protein tyrosine phosphatase 1B (PTP1B) gene expression at transcriptional level in C2C12 skeletal muscle cells. Inflammation 2011, 34, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xu, N.; Guo, J.; Yan, X. Antitumor effects and the mechanism of two kinds of bromophenols from marine algae. Chin. Med. J. 2010, 35, 1173–1176. [Google Scholar]
- Li, K.; Li, X.M.; Gloer, J.B.; Wang, B.G. Isolation, characterization, and antioxidant activity of bromophenols of the marine red alga Rhodomela confervoides. J. Agric. Food Chem. 2011, 59, 9916–9921. [Google Scholar] [CrossRef] [PubMed]
- Pearce, A.N.; Chia, E.W.; Berridge, M.V.; Maas, E.W.; Page, M.J.; Webb, V.L.; Harper, J.L.; Copp, B.R. E/Z-Rubrolide O, an anti-inflammatory halogenated furanone from the new zealand ascidian Synoicum n. sp. J. Nat. Prod. 2007, 70, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, G.; Xiao, L.; Xu, X.; Liu, X.; Xu, P.; Lin, X. Bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, a marine algae derived bromophenol, inhibits the growth of botrytis cinerea and interacts with DNA molecules. Mar. Drugs 2014, 12, 3838–3851. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Nguyen, T.H.; Kurihara, H.; Kim, S.M. α-Glucosidase inhibitory activity of bromophenol purified from the red alga polyopes lancifolia. J. Food Sci. 2010, 75, H145–H150. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, W.; Wei, J.; Lin, X. Synthesis and α-glucosidase inhibitory mechanisms of Bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, a potential marine bromophenol α-glucosidase inhibitor. Mar. Drugs 2011, 9, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Xu, F.; He, J.; Li, J.; Fan, X.; Han, L. Inhibition of bromophenols against PTP1B and anti-hyperglycemic effect of Rhodomela confervoides extract in diabetic rats. Chin. Sci. Bull. 2008, 53, 2476–2479. [Google Scholar] [CrossRef]
- Shi, D.; Li, J.; Jiang, B.; Guo, S.; Su, H.; Wang, T. Bromophenols as inhibitors of protein tyrosine phosphatase 1B with antidiabetic properties. Bioorg. Med. Chem. Lett. 2012, 22, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Shi, D.; Cui, Y.; Guo, S. Design, synthesis, and biological evaluation of bromophenol derivatives as protein tyrosine phosphatase 1B inhibitors. Arch. Pharm. 2012, 345, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Guo, S.; Jiang, B.; Guo, C.; Wang, T.; Zhang, L.; Li, J. HPN, a synthetic analogue of bromophenol from red alga Rhodomela confervoides: Synthesis and anti-diabetic effects in C57BL/KSJ-db/db mice. Mar. Drugs 2013, 11, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M.; Hollenbeck, C.; Jeng, C.Y.; Wu, M.S.; Chen, Y.D. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 1988, 37, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- De Vries, J.E.; Vork, M.M.; Roemen, T.H.; de Jong, Y.F.; Cleutjens, J.P.; van der Vusse, G.J.; van Bilsen, M. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J. Lipid Res. 1997, 38, 1384–1394. [Google Scholar]
- Shao, J.; Gao, Y.; Yuan, Z. Free fatty acids promoting PTP1B expression in rat skeletal muscle and hepatic cells. Chin. Med. J. 1998, 78, 753–755. [Google Scholar]
- Wu, N.; Luo, J.; Jiang, B.; Wang, L.; Wang, S.; Wang, C.; Fu, C.; Li, J.; Shi, D. Marine bromophenol Bis(2,3-dibromo-4,5-dihydroxy-phenyl)-methane inhibits the proliferation, migration, and invasion of hepatocellular carcinoma cells via modulating β1-Integrin/FAK signaling. Mar. Drugs 2015, 13, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, R.G.; Wu, L.; Mullani, N. Uptake of a fluorescent deoxyglucose analog (2-NBDG) in tumor cells. Mol. Imaging Biol. 2005, 7, 388–392. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Wu, N.; Jiang, B.; Wang, L.; Wang, S.; Li, X.; Wang, B.; Wang, C.; Shi, D. Marine Bromophenol Derivative 3,4-Dibromo-5-(2-bromo-3,4-dihydroxy-6-isopropoxymethyl benzyl)benzene-1,2-diol Protects Hepatocytes from Lipid-Induced Cell Damage and Insulin Resistance via PTP1B Inhibition. Mar. Drugs 2015, 13, 4452-4469. https://doi.org/10.3390/md13074452
Luo J, Wu N, Jiang B, Wang L, Wang S, Li X, Wang B, Wang C, Shi D. Marine Bromophenol Derivative 3,4-Dibromo-5-(2-bromo-3,4-dihydroxy-6-isopropoxymethyl benzyl)benzene-1,2-diol Protects Hepatocytes from Lipid-Induced Cell Damage and Insulin Resistance via PTP1B Inhibition. Marine Drugs. 2015; 13(7):4452-4469. https://doi.org/10.3390/md13074452
Chicago/Turabian StyleLuo, Jiao, Ning Wu, Bo Jiang, Lijun Wang, Shuaiyu Wang, Xiangqian Li, Baocheng Wang, Changhui Wang, and Dayong Shi. 2015. "Marine Bromophenol Derivative 3,4-Dibromo-5-(2-bromo-3,4-dihydroxy-6-isopropoxymethyl benzyl)benzene-1,2-diol Protects Hepatocytes from Lipid-Induced Cell Damage and Insulin Resistance via PTP1B Inhibition" Marine Drugs 13, no. 7: 4452-4469. https://doi.org/10.3390/md13074452
APA StyleLuo, J., Wu, N., Jiang, B., Wang, L., Wang, S., Li, X., Wang, B., Wang, C., & Shi, D. (2015). Marine Bromophenol Derivative 3,4-Dibromo-5-(2-bromo-3,4-dihydroxy-6-isopropoxymethyl benzyl)benzene-1,2-diol Protects Hepatocytes from Lipid-Induced Cell Damage and Insulin Resistance via PTP1B Inhibition. Marine Drugs, 13(7), 4452-4469. https://doi.org/10.3390/md13074452