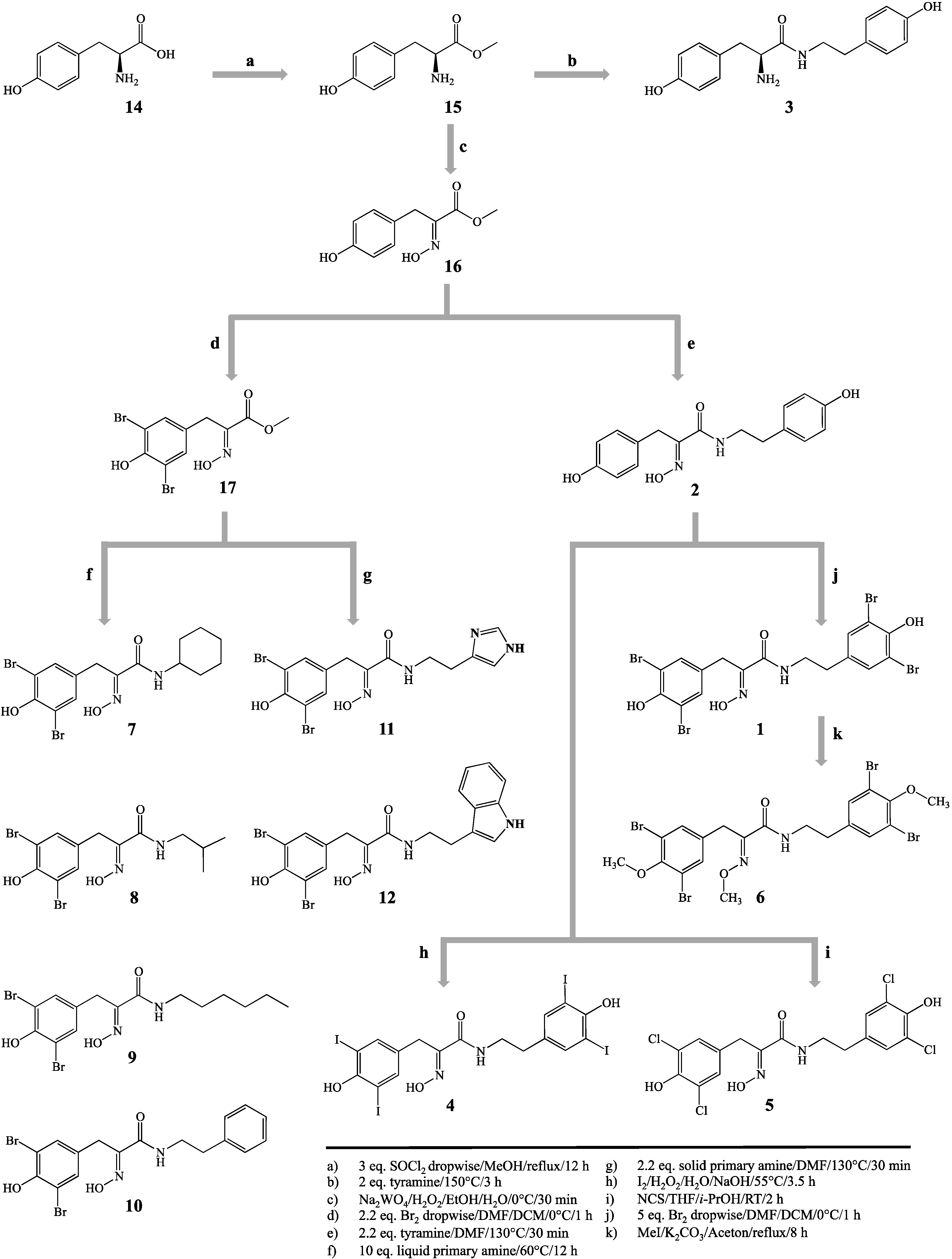
3.3. Synthetic Procedures
Hemibastadin analogues were synthesized by an optimized and extended method, as described earlier [
7,
8] (
Figure 1). Structures were confirmed via LC-ESI-MS and
1H-NMR (for spectra of new hemibastadin congeners see
supporting information).
3.3.1. l-Tyrosine-methyl Ester (15)
l-Tyrosine (14, 10.06 g, 55.53 mmol) was converted into the methyl ester (15) by dropwise addition of three equivalents thionyl chloride (SOCl2, 12.08 mL, 166.59 mmol) in methanol (100 mL) on ice. After complete addition of SOCl2, the suspension was heated under reflux for 12 h. The resulting solution was concentrated in vacuo and the pH was adjusted to 8 with NaHCO3, followed by exhaustive extraction with ethyl acetate. The organic phase was separated and dried over anhydrous magnesium sulfate. After solvent evaporation, 9.86 g of l-tyrosine-methyl ester (15) were obtained as a white, amorphous powder (50.53 mmol, 91.0%). Positive mode ESI-MS analysis revealed the pseudomolecular ion [M + H]+ at m/z 196.
3.3.2. (E)-Methyl 2-(hydroxyimino)-3-(4-hydroxyphenyl)propanoate (16)
Oxidation of
15 to
16 was carried out according to a modified procedure of Boehlow
et al. [
10]. The ester
15 (8.59 g, 44 mmol) was dissolved in ethanol (100 mL) and stirred on ice. After the addition of H
2O (76 mL), equimolar amounts of sodium tungstate dihydrate (Na
2WO
4·2H
2O, 44 mmol) and H
2O
2 (30% aqueous solution, 44 mL), the solution was stirred until the color changed into pale yellow, and TLC analysis (SiO
2, dichloromethane, ethyl acetate 3:1) revealed a complete turnover of the educt. After exhaustive extraction with ethyl acetate, the combined organic phases were washed with aqueous sodium hydrogen sulfite (2 × 100 mL) and H
2O (2 × 100 mL) and dried over anhydrous magnesium sulfate. The solvent was removed
in vacuo to yield a pale yellow powder. The crude product was further purified by column chromatography (SiO
2, dichloromethane, ethyl acetate 3:1), and 7.09 g of pure
16 (33.90 mmol, 77.0%) were obtained as a nearly white amorphous powder. All spectral data were in accordance with previously reported values [
10].
3.3.3. (E)-Methyl 3-(3,5-dibromo-4-hydroxyphenyl)-2-(hydroxyimino)propanoate (17)
The ester
16 (1 g, 4.78 mmol) was dissolved in dimethylformamide (10 mL) and diluted with dichloromethane (60 mL). After cooling on crushed ice, a solution of bromine (11 mL 1M-Br
2 in dichloromethane) was added dropwise during 20 min of stirring and cooling in the absence of light. TLC analysis was used to control complete bromination. Excessive bromine was reduced by the addition of aqueous 10% sodium hydrogen sulfite solution until the brown color was converted to pale yellow. The water phase was separated and extracted exhaustively with ethyl acetate. Both organic phases were washed with water, then combined and dried over anhydrous magnesium sulfate. After solvent evaporation, the resulting crude product was purified by column chromatography (SiO
2, dichloromethane, ethyl acetate 5:1) to yield 1.53 g of pure
17 (4.17 mmol, 87.3%) as an amorphous white powder. All spectral data were in accordance with previously reported values [
10].
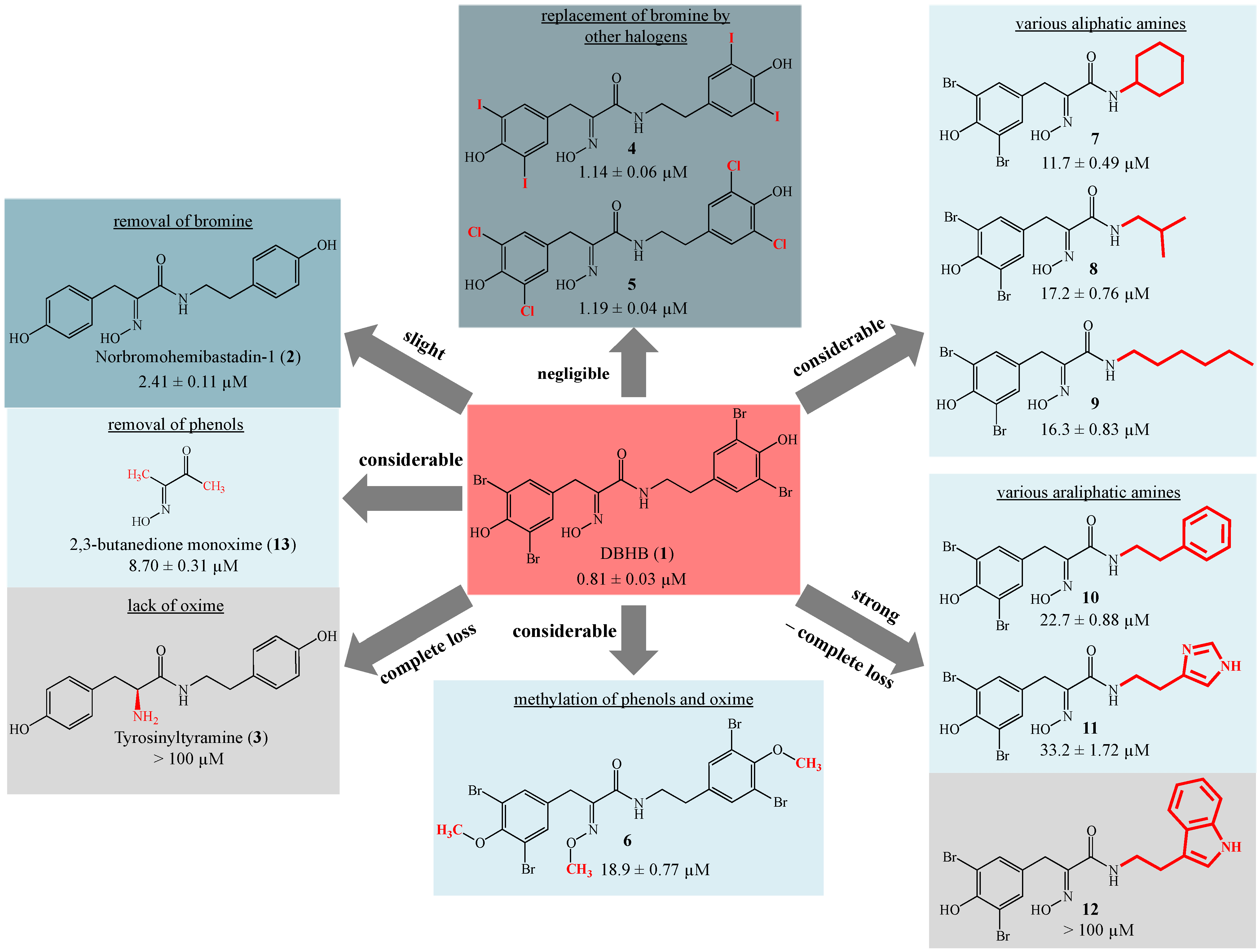
3.3.4. 5,5′-Dibromohemibastadin-1 (1) and Norbromohemibastadin-1 (2)
DBHB (
1) and nobromohemibastadin-1 (
2) were synthesized by replication of the protocol published earlier [
8]. All spectral data were in accordance with previously reported values [
8].
3.3.5. Tyrosinyltyramine (3)
Tyrosinyltyramine (
3) was synthesized by replication of the protocol published earlier, and all spectral data were in accordance with [
7].
3.3.6. Tetraiodo-norbromohemibastadin-1 (4)
Tetraiodo-norbromohemibastadin-1 (
4) was synthesized by iodination of
2, utilizing a modified method of Wada
et al. [
11]. Briefly,
2 (157.0 mg, 0.5 mmol) was dissolved in diluted sodium hydroxide solution (5 mL, pH 10), and H
2O
2 (30% aqueous solution, 0.12 mL) and iodine (I
2, 152.0 mg, 0.6 mmol) were added. The solution was stirred at 55 °C for 3 h. Excessive iodine was reduced by the addition of aqueous 10% sodium hydrogen sulfite solution. After solvent evaporation
in vacuo, the resulting crude product was purified by column chromatography (SiO
2, hexan, ethyl acetate, 4:1), and pure
4 (3.7 mg, 4.5 µmol, 0.9%) was obtained.
4:
1H (500 MHz, DMSO-
d6) δ 11.90 (s, 1H), 9.40 (s, 1H), 9.35 (s, 1H), 8.02 (t,
J = 5.9 Hz, 1H), 7.56 (s, 2H), 7.54 (s, 2H), 3.65 (s, 2H), 3.30–3.26 (m, 2H), 2.61 (t,
J = 7.2 Hz, 2H); ESI-MS [M + H]
+ m/
z 818.7.
3.3.7. Tetrachloro-norbromohemibastadin-1 (5)
To a solution of 2 (157.0 mg 0.5 mmol) in a mixture of tetrahydrofuran (3 mL) and isopropanol (1 mL), N-chlorosuccinimide (333.8 mg, 2.5 mmol) was added in portions, and the suspension was stirred at room temperature for 2 h. After solvent evaporation in vacuo, the crude product was purified via column chromatography (SiO2, dichloromethane, ethyl acetate 1:1), and semipreparative HPLC (gradient system of 0.1% trifluoro acetic acid and methanol) was conducted to obtain pure 5 as a yellow solid (3.7 mg, 8.2 µmol, 1.6%). 5: 1H (500 MHz, DMSO-d6) δ 11.90 (s, 1H), 9.93 (s, 1H), 9.85 (s, 1H), 7.99 (t, J = 5.8 Hz, 1H), 7.15 (s, 2H), 7.14 (s, 2H) 3.69 (s, 2H), 3.33 (m, 2H), 2.67 (t, J = 7.0 Hz, 2H); ESI-MS [M + H]+ 453.3, [M − H]− m/z 451.3.
3.3.8. Tri-O-methyl-5,5′-dibromohemibastadin-1 (6)
Methylation of both phenolic hydroxyls and of the oxime function of 1 (189 mg, 0.3 mmol) was achieved in acetone (6 mL) by the addition of potassium carbonate (207.0 mg, 1.5 mmol) and iodomethane (63.9 mg, 0.45 mmol), heating under reflux for 8 h and additional stirring at RT for 12 h. After solvent evaporation and subsequent purification of the crude product via semipreparative HPLC (gradient system of 0.1% trifluoro acetic acid and methanol), pure 6 (46.0 mg, 0.07 mmol, 23.0%) was obtained as a white powder. 6: 1H (500 MHz, DMSO-d6) δ 8.27 (t, J = 5.8 Hz, 1H), 7.47 (s, 2H), 7.42 (s, 2H), 3.97 (s, 3H), 3.76–3.75 (m, 8H), 3.35 (t, J = 6.9 Hz, 2H), 2.73 (t, J = 6.9 Hz, 1H); ESI-MS [M + H]+ m/z 672.9.
3.3.9. Amides Resulting from Liquid Primary Amines (7–10)
For the preparation of Compounds 7–10, four aliquots of 17 (200 mg, 0.96 mmol) were dissolved in ten equivalents (9.6 mmol) of a liquid primary amine (for 7: 1.11 mL cyclohexylamine; for 8: 0.82 mL isobutylamine; for 9: 1.26 mL n-hexylamine; for 10: 1.21 mL phenethylamine), respectively, and stirred at 60 °C for 12 h in an open flask. For the general work-up, the crude products (7–10) were diluted with ethyl acetate (50 mL) and aqueous 10% HCl (10 mL). After separation, the aqueous phase was extracted exhaustively with ethyl acetate. The organic phases were combined and dried over anhydrous magnesium sulfate. After solvent evaporation, the resulting solids were further purified by column chromatography (SiO2, dichloromethane, ethyl acetate 3:1) to obtain the pure compounds:
N-cyclohexyl-3-(3,5-dibromo-4-hydroxyphenyl)-2-(2-hydroxyimino)-propanamide (7): 371.8 mg (89.2%), 1H-NMR (500 MHz, DMSO-d6) δ 11.83 (s, 1H), 9.77 (s, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.35 (s, 2H), 3.69 (s, 2H), 3.66–3.53 (m, 1H), 1.72–1.62 (m, 3H), 1.55 (d, J = 11.0 Hz, 1H), 1.32–1.18 (m, 5H), 1.13–1.02 (m, 2H); ESI-MS [M + H]+ m/z 435.1.
N-(2-methyl-propyl)-3-(3,5-dibromo-4-hydroxyphenyl)-2-(2-hydroxyimino)-propanamide (8): 385.5 mg (98.4%), 1H (500 MHz, DMSO-d6) δ 11.87 (s, 1H), 9.77 (s, 1H), 8.00 (t, J = 6.6 Hz, 1H), 7.34 (s, 2H), 3.71 (s, 2H), 2.95 (t, J = 6.6 Hz, 2H), 1.82–1.69 (1H, m), 0.80 (d, J = 6.6 Hz, 6H); ESI-MS [M + H]+ m/z 409.0.
N-hexyl-3-(3,5-dibromo-4-hydroxyphenyl)-2-(2-hydroxyimino)-propanamide (9): 383.9 mg (91.7%), 1H (500 MHz, DMSO-d6) δ 11.86 (s, 1H), 9.76 (s, 1H), 7.98 (t, J = 6.6 Hz, 1H), 7.34 (s, 2H), 3.70 (s, 2H), 3.11 (dd, 2H, J = 6.6, 6.9 Hz), 1.40 (t, 2H, J = 6.9 Hz), 0.83 (t, J = 6.5 Hz, 3H); ESI-MS [M + H]+ m/z 436.9.
N-2-phenylethyl-3-(3,5-dibromo-4-hydroxyphenyl)-2-(2-hydroxyimino)-propanamide (10): 409.0 mg (93.4%), 1H (500 MHz, DMSO-d6) δ 11.89 (s, 1H), 9.74 (s, 1H), 7.98 (t, J = 5.9 Hz, 1H), 7.34 (s, 2H), 7.25 (t, J = 7.4 Hz, 2H), 7.20–7.14 (m, 3H), 3.70 (s, 2H), 3.37 (dt, J = 5.9, 7.4 Hz, 2H), 2.75 (t, J = 7.4 Hz, 2H); ESI-MS [M + H]+ m/z 457.2.
3.3.10. Amides Resulting from Solid Primary Amines (11, 12)
Compounds 11 and 12 were obtained by triturating two aliquots of 17 (200 mg, 0.96 mmol) with the corresponding solid primary amine (each 2.11 mmol; for 11: 234.5 mg histamine; for 12: 338.1 mg tryptamine), adding dimethylformamide (3 mL) and melting at 130 °C for 30 min in an open flask. The resulting crude products were purified utilizing preparative HPLC (gradient system of 0.1% trifluoro acetic acid and methanol) to obtain the pure compounds:
N-[2-(4-imidazolyl)-ethyl]-3-(3,5-dibromo-4-hydroxyphenyl)-2-(2-hydroxyimino)-propanamide (11): 274.5 mg (64.1%), 1H (500 MHz, DMSO-d6) δ 14.27 (s, 1H), 11.98 (s, 1H), 9.81 (s, 1H), 8.96 (s, 1H), 8.21 (t, J = 6.0 Hz, 1H), 7.41 (s, 1H), 7.32 (s, 1H), 3.68 (s, 2H), 3.44 (dt, J = 6.0, 6.9 Hz, 2H), 2.83 (t, J = 6.9 Hz, 2H); ESI-MS [M + H]+ m/z 447.2.
N-[2-(3-indolyl)-ethyl]-3-(3,5-dibromo-4-hydroxyphenyl)-2-(2-hydroxyimino)-propanamide (12): 274.8 mg (57.8%), 1H (500 MHz, DMSO-d6) δ 11.92 (s, 1H), 10.79 (s, 1H), 9.78 (s, 1H), 8.06 (t, J = 5.9 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.37 (s, 2H), 7.33 (d, J = 8.0 Hz, 1H), 7.14 (s, 1H), 7.06 (dd, J = 7.5, 7.8 Hz, 1H), 6.96 (dd, J = 7.5, 8.0 Hz, 1H), 3.72 (s, 2H), 3.43 (m, 2H, overlapped with solvent signal), 2.85 (t, J = 7.6 Hz, 2H); ESI-MS [M + H]+ m/z 496.2.


 ,
, {kind=link}
{kind=link}