
Xyloketal B Suppresses Glioblastoma Cell Proliferation and Migration in Vitro through Inhibiting TRPM7-Regulated PI3K/Akt and MEK/ERK Signaling Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results and Discussion
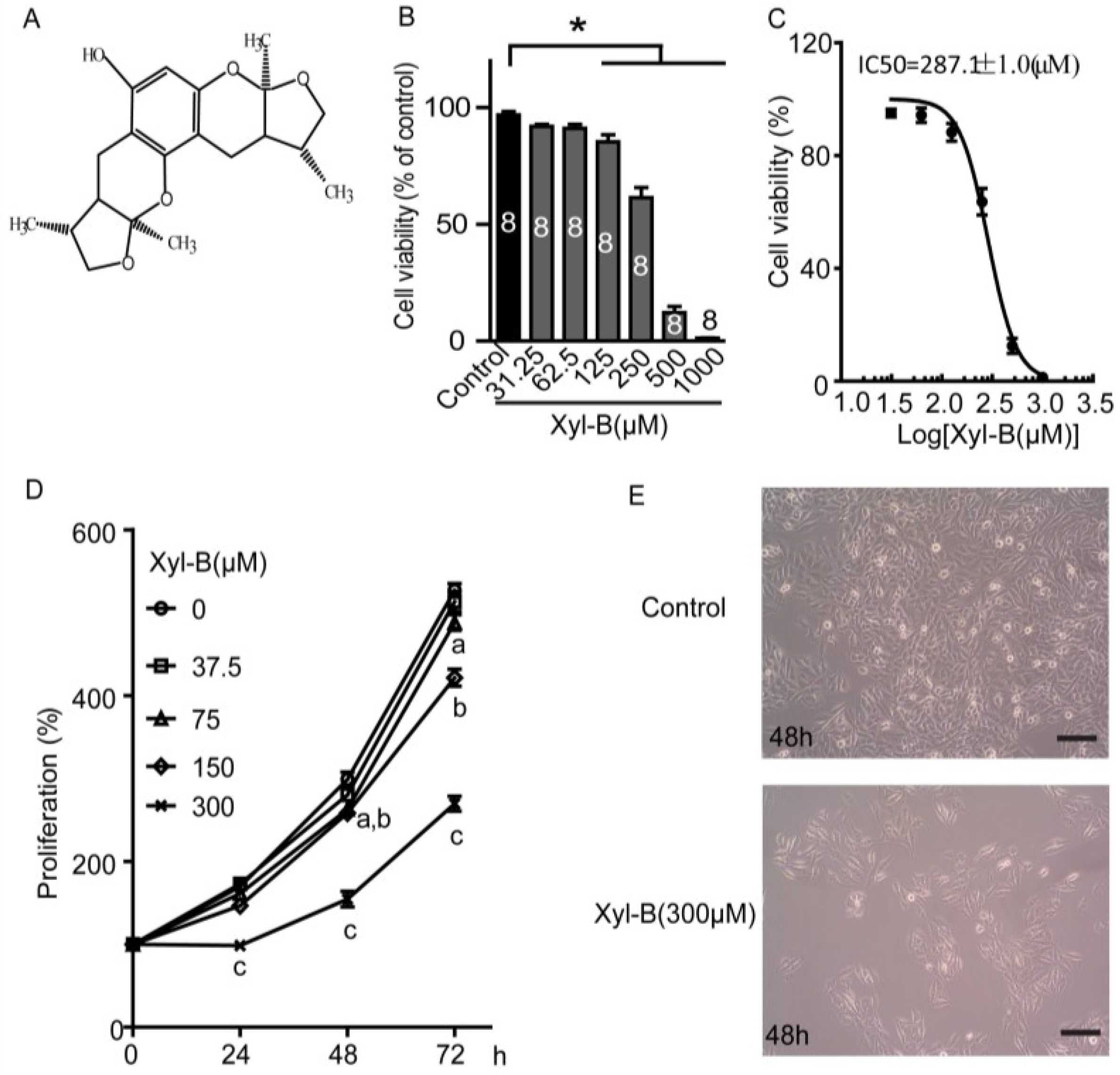
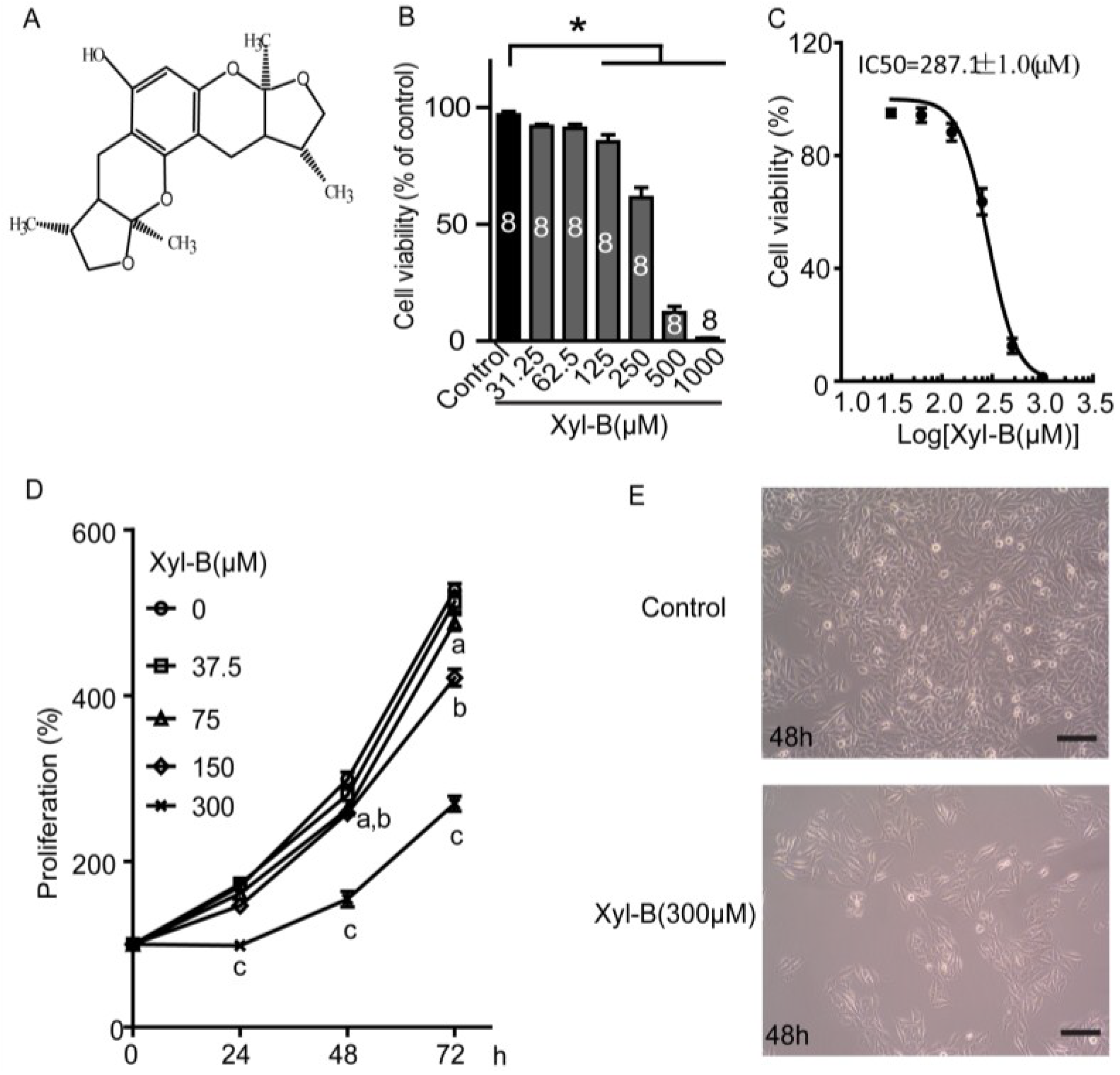
2.1. Xyloketal B Reduces U251 Cell Viability
2.2. Xyloketal B Inhibits U251 Cell Proliferation
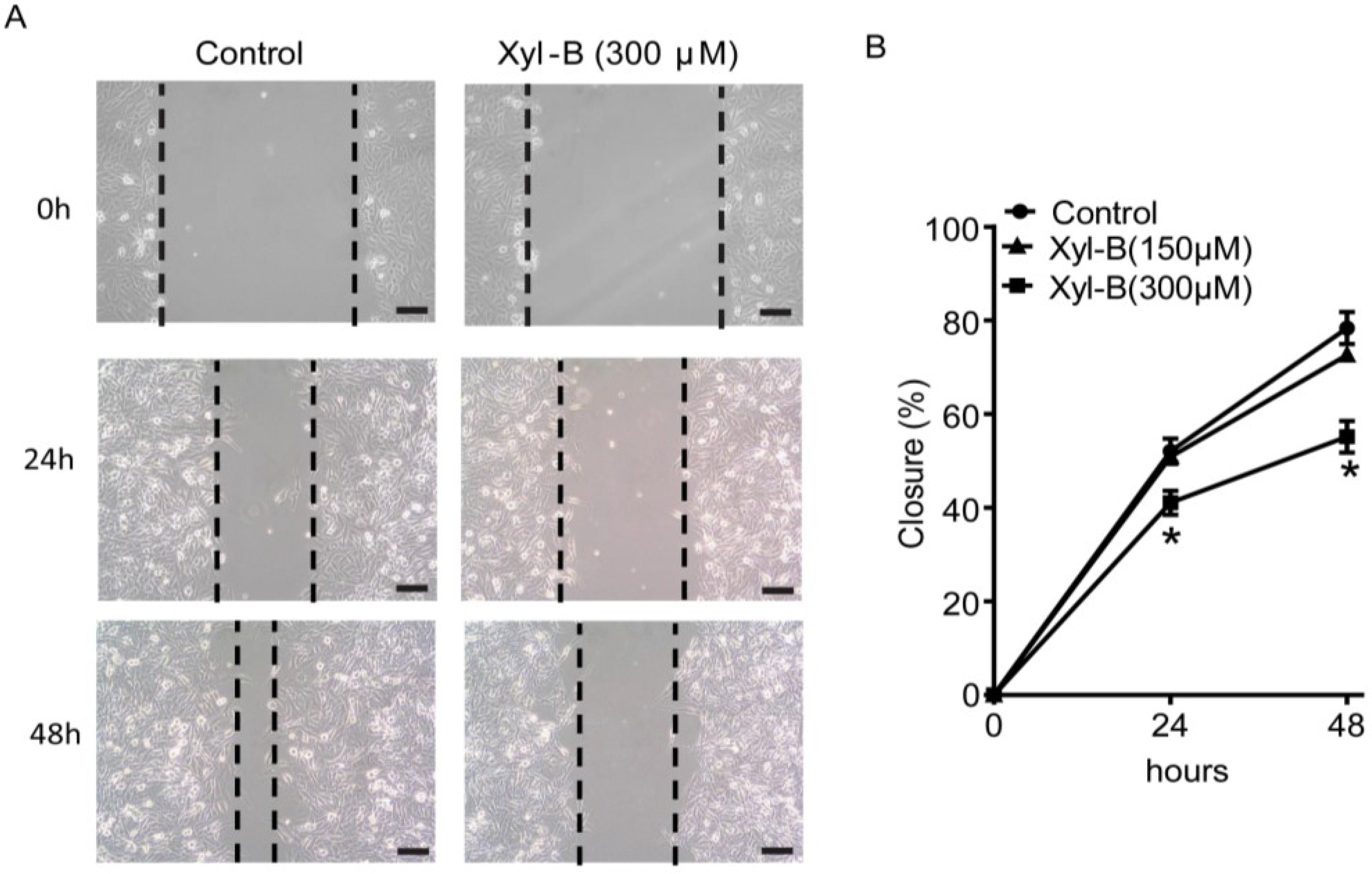
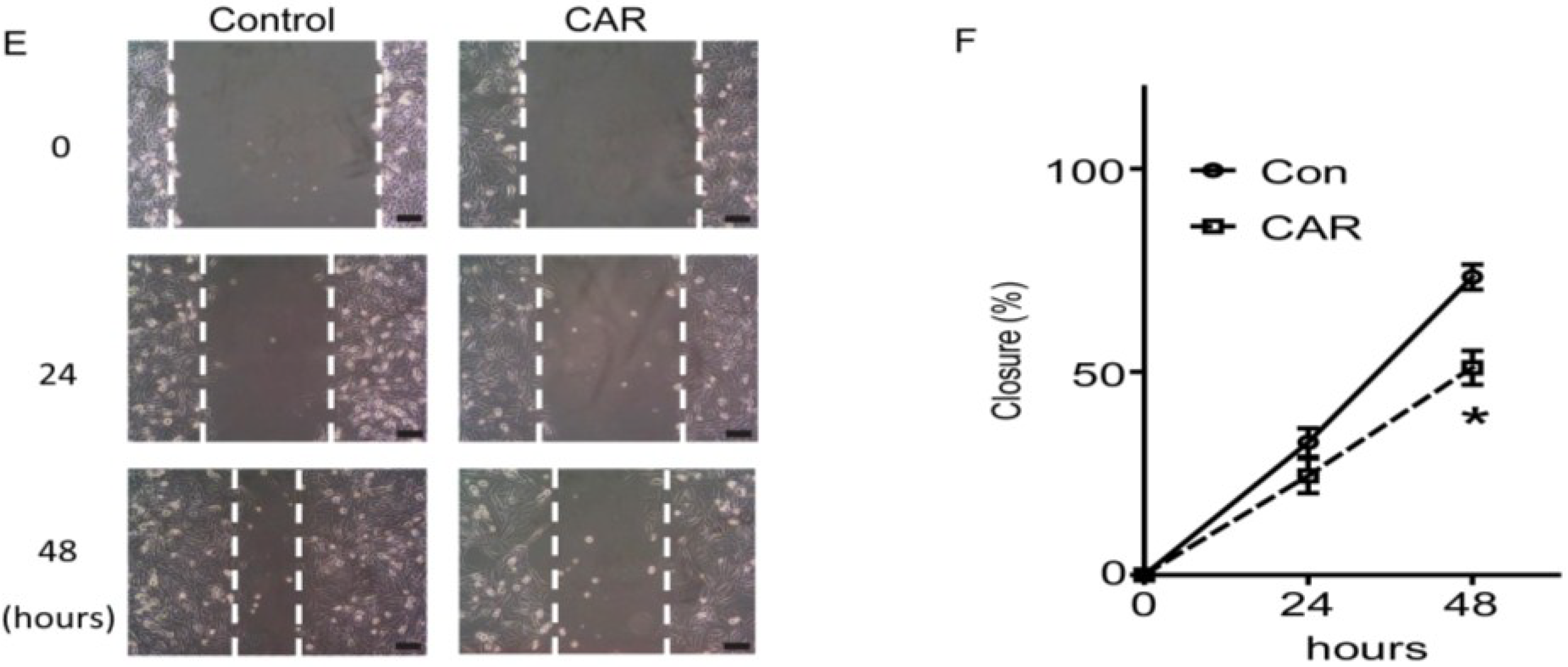
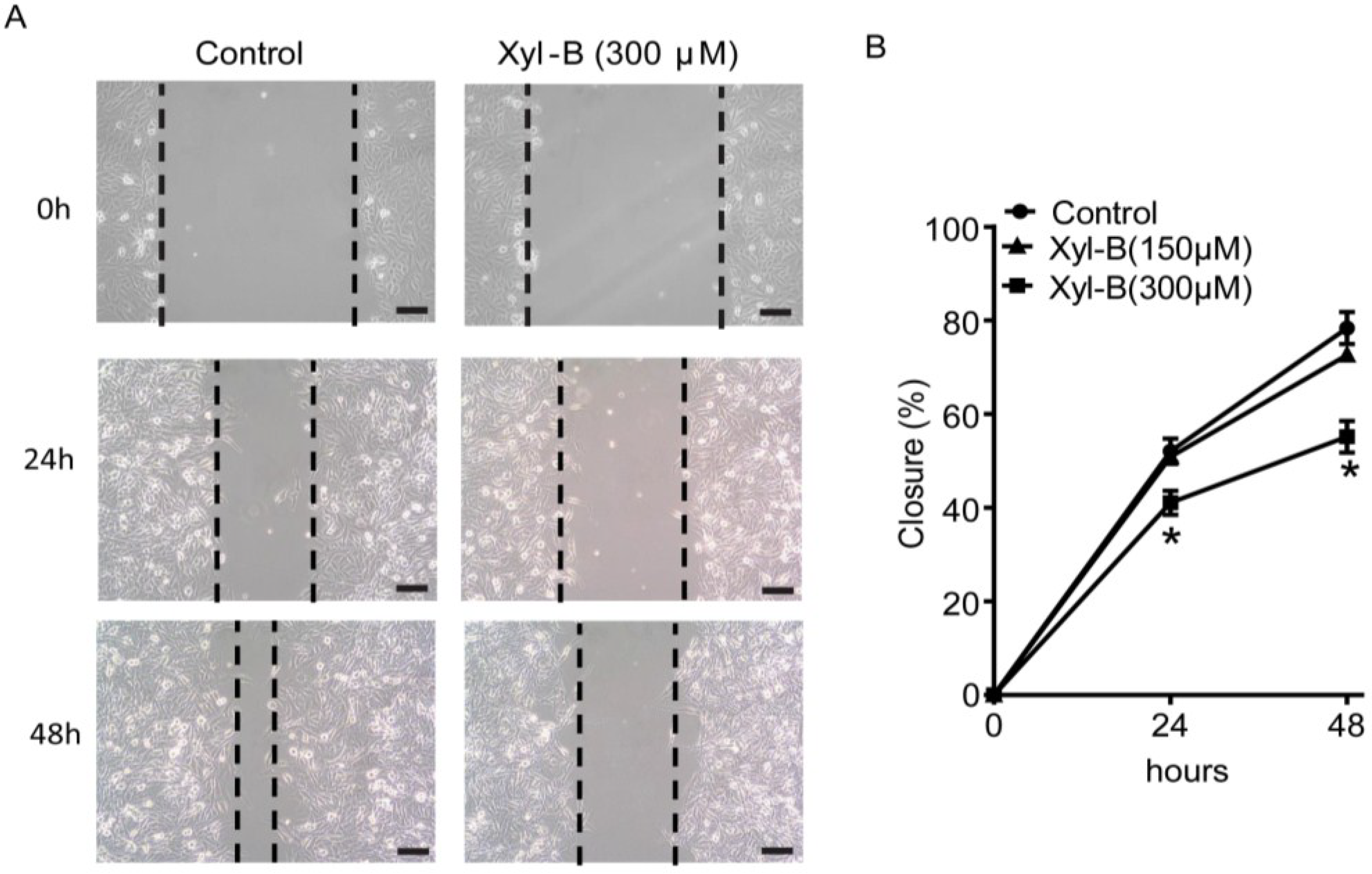
2.3. Xyloketal B Inhibits U251 Cell Migration
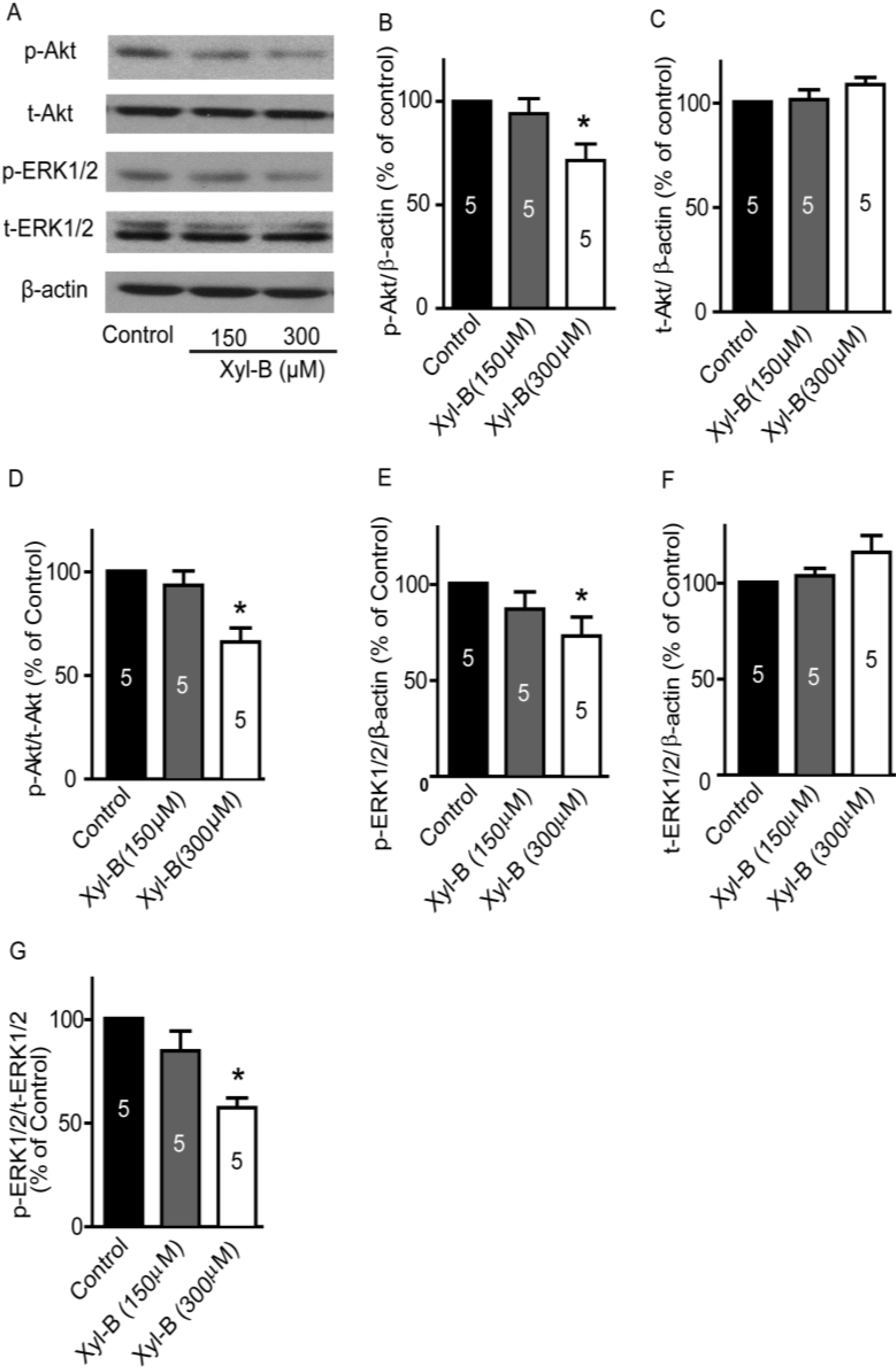
2.4. Xyloketal B Suppresses the PI3K/Akt and MEK/ERK Signaling Pathways

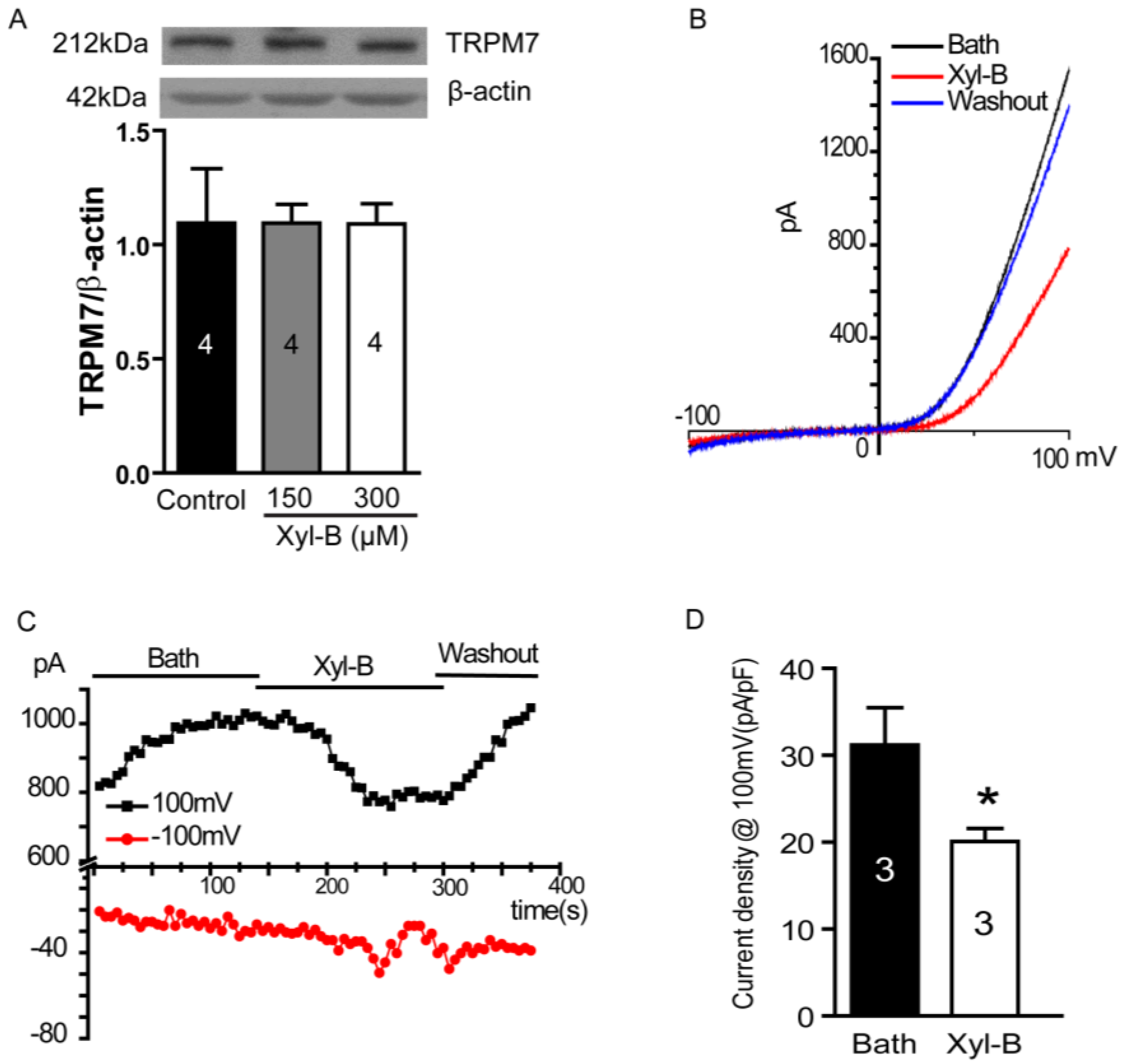
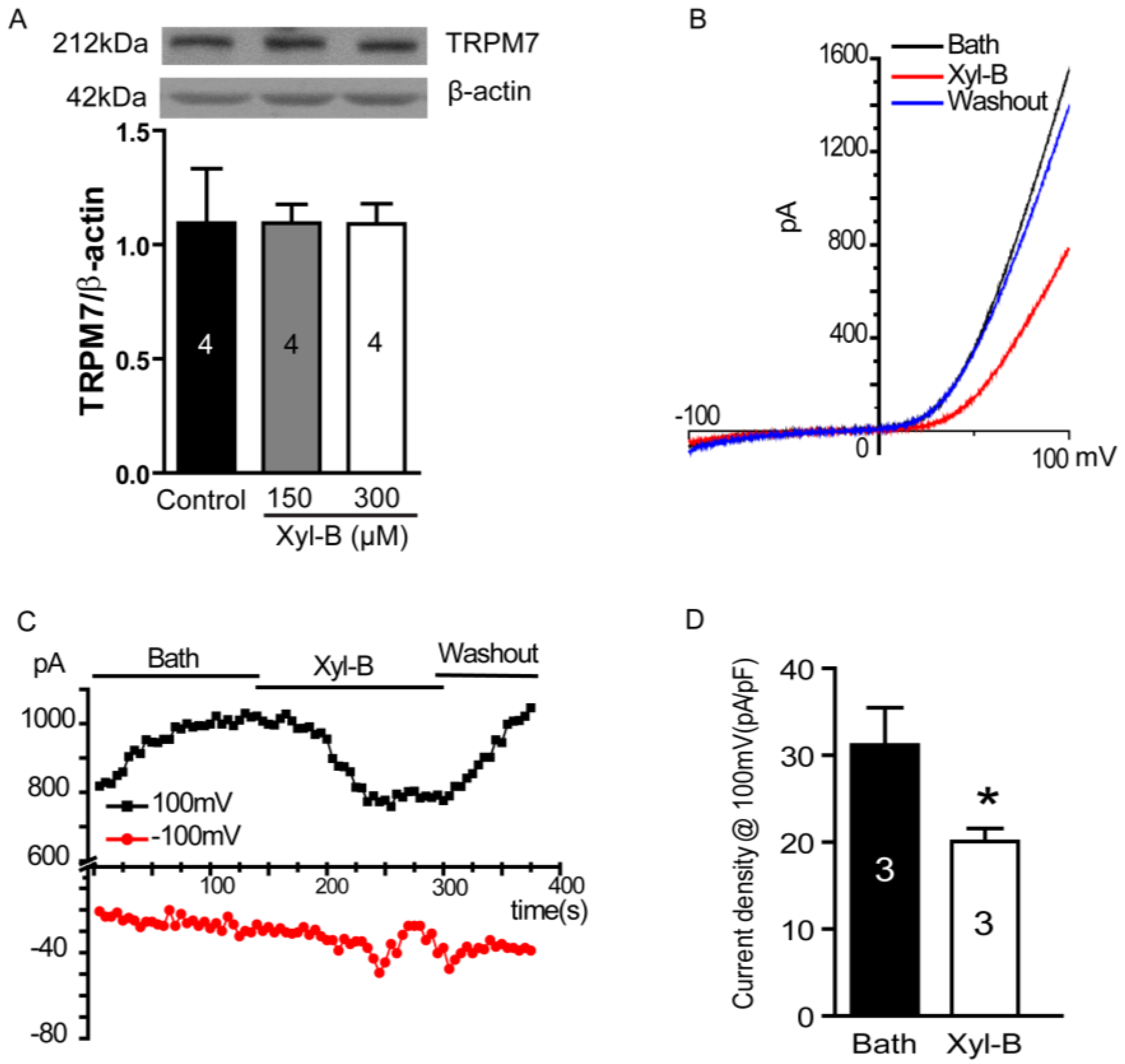
2.5. Xyloketal B Blocks the TRPM7 Current
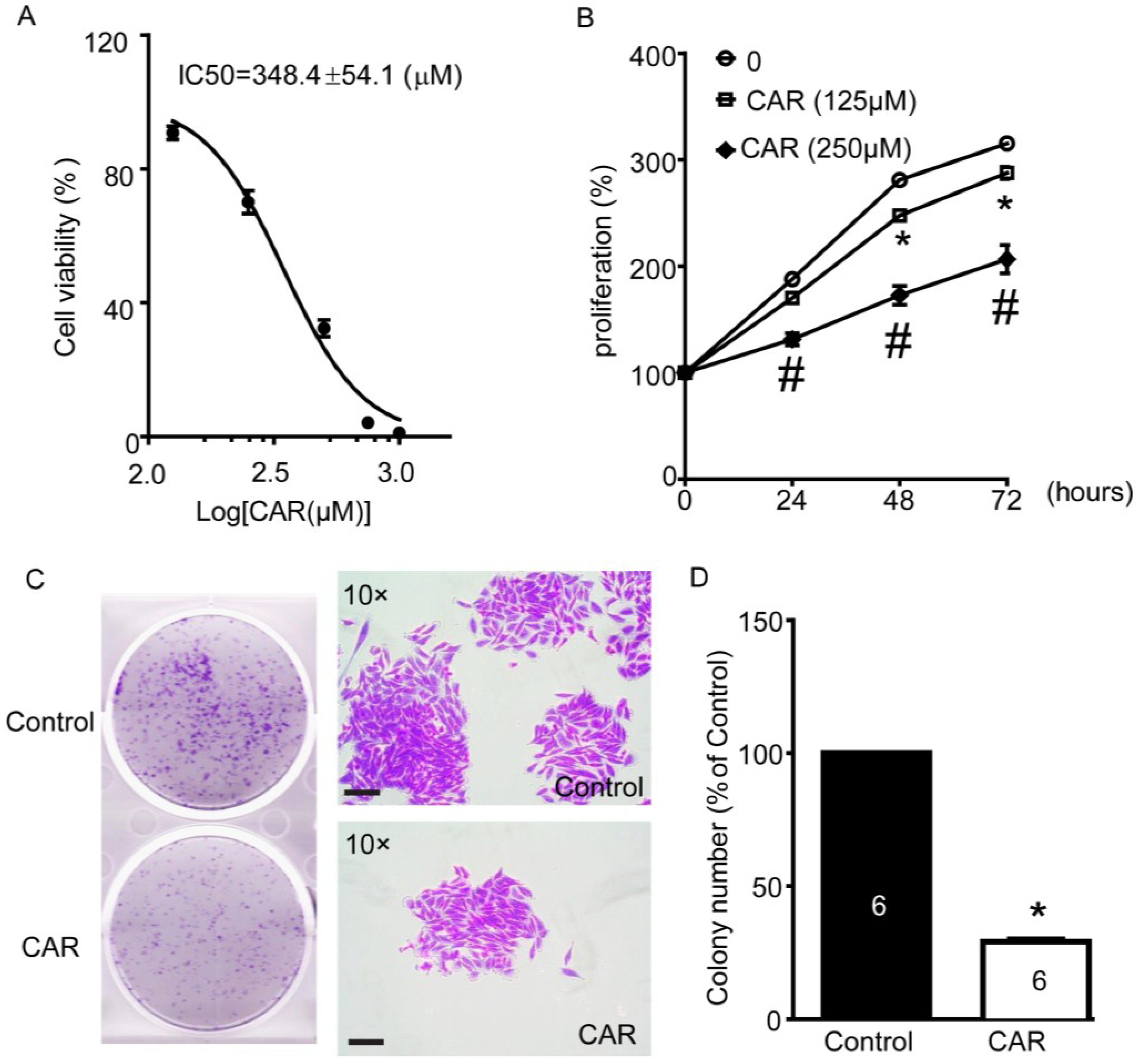
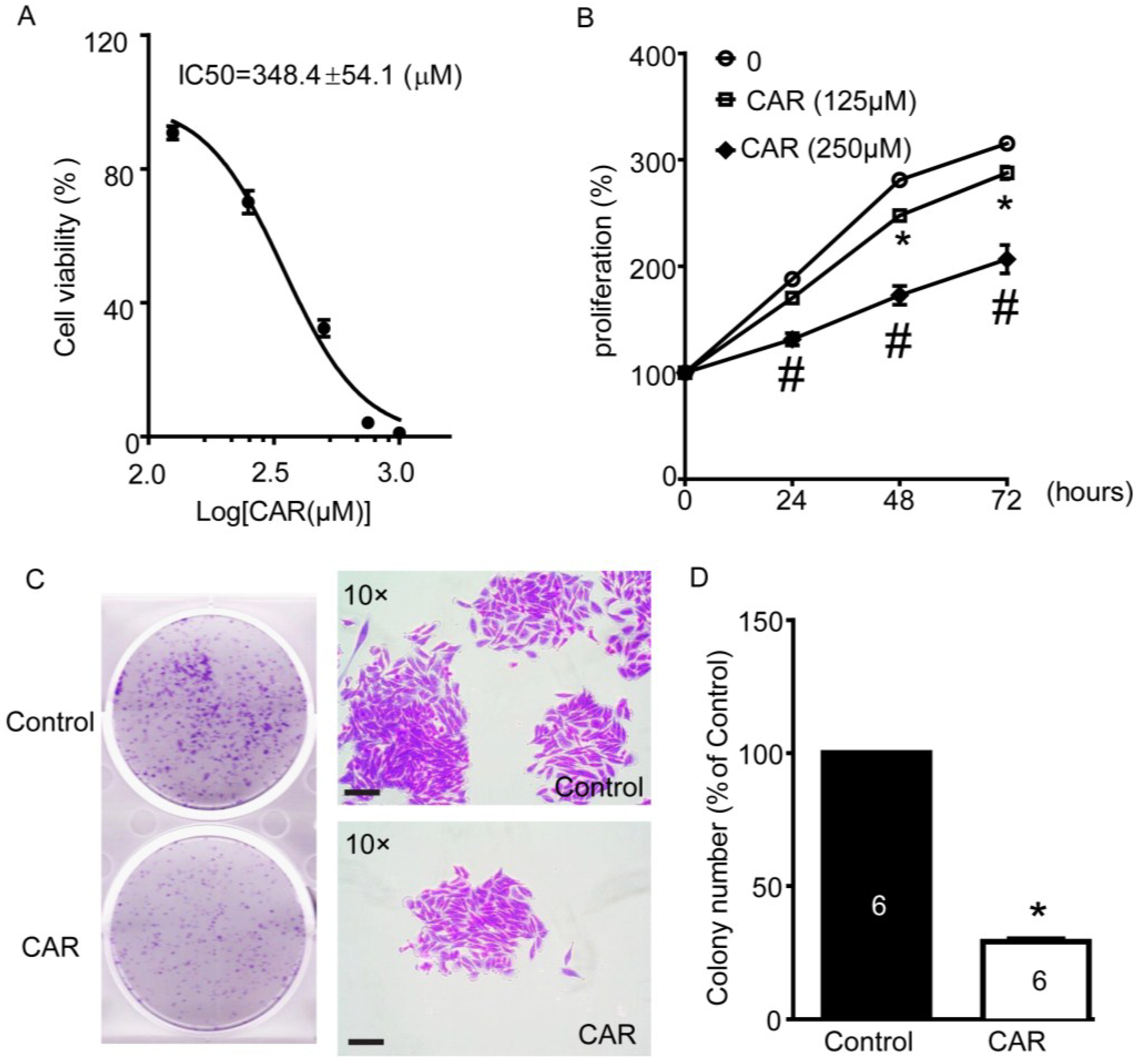
2.6. TRPM7 Inhibitor Carvacrol Reduces U251 Cell Viability, Proliferation, and Migration

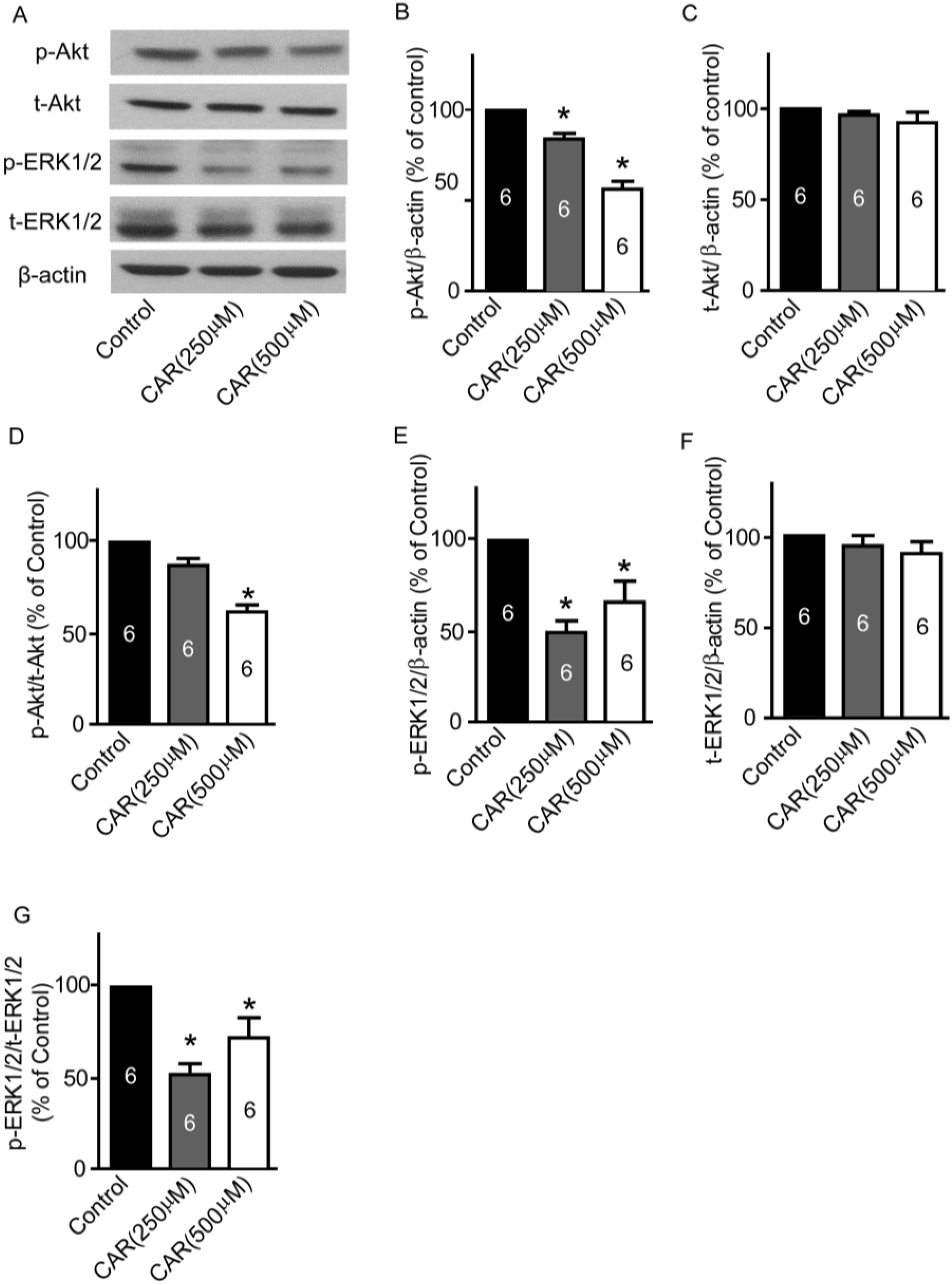
2.7. TRPM7 Inhibitor Carvacrol Suppresses the PI3K/Akt and MEK/ERK Signaling Pathways

2.8. Discussion
3. Experimental Section
3.1. Reagents
3.2. Cell Culture
3.3. Cell Viability and Proliferation
3.4. Colony Formation
3.5. Cell Migration
3.6. Western Blotting
3.7. Patch-Clamp Recording
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase iii study: 5-Year analysis of the eortc-ncic trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Soni, D.; King, J.A.; Kaye, A.H.; Hovens, C.M. Genetics of glioblastoma multiforme: Mitogenic signaling and cell cycle pathways converge. J. Clin. Neurosci. 2005, 12, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.P.; Zhu, J.; Black, P.M. Genetic aberrations in human brain tumors. Neurosurgery 1994, 34, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.P.; Saxena, A.; Clark, W.C.; Robertson, J.T.; Oldfield, E.H.; Aaronson, S.A.; Ali, I.U. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992, 52, 4550–4553. [Google Scholar] [PubMed]
- Pelloski, C.E.; Lin, E.; Zhang, L.; Yung, W.K.; Colman, H.; Liu, J.L.; Woo, S.Y.; Heimberger, A.B.; Suki, D.; Prados, M.; et al. Prognostic associations of activated mitogen-activated protein kinase and akt pathways in glioblastoma. Clin. Cancer Res. 2006, 12, 3935–3941. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Sathornsumetee, S.; Rich, J.N. Designer therapies for glioblastoma multiforme. Ann. N. Y. Acad. Sci. 2008, 1142, 108–132. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Sontheimer, H. Molecular interaction and functional regulation of clc-3 by Ca2+/calmodulin-dependent protein kinase ii (camkii) in human malignant glioma. J. Biol. Chem. 2010, 285, 11188–11196. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, B.; Li, W.; Guo, H.; Zou, F. Atp-sensitive potassium channels control glioma cells proliferation by regulating erk activity. Carcinogenesis 2009, 30, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Leng, T.D.; Li, M.H.; Shen, J.F.; Liu, M.L.; Li, X.B.; Sun, H.W.; Branigan, D.; Zeng, Z.; Si, H.F.; Li, J.; et al. Suppression of trpm7 inhibits proliferation, migration, and invasion of malignant human glioma cells. CNS Neurosci. Ther. 2015, 21, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Inoue, K.; Leng, T.; Guo, S.; Xiong, Z.G. Trpm7 channels regulate glioma stem cell through stat3 and notch signaling pathways. Cell. Signal. 2014, 26, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.S.; Jackson, M.F.; Martin, L.J.; Jansen, K.; Teves, L.; Cui, H.; Kiyonaka, S.; Mori, Y.; Jones, M.; Forder, J.P.; et al. Suppression of hippocampal trpm7 protein prevents delayed neuronal death in brain ischemia. Nat. Neurosci. 2009, 12, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Desai, B.N.; Navarro, B.; Donovan, A.; Andrews, N.C.; Clapham, D.E. Deletion of trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 2008, 322, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Lehen’kyi, V.; Shapovalov, G.; Skryma, R.; Prevarskaya, N. Ion channnels and transporters in cancer. 5. Ion channels in control of cancer and cell apoptosis. Am. J. Physiol. Cell Physiol. 2011, 301, C1281–C1289. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, L.; Luo, C.H.; Zhou, H.; Hu, J.; Tang, Y.X.; Fang, K.N.; Zhang, Y. Overexpression of trpm7 is associated with poor prognosis in human ovarian carcinoma. Asian Pac. J. Cancer Prev. APJCP 2014, 15, 3955–3958. [Google Scholar] [CrossRef]
- Rybarczyk, P.; Gautier, M.; Hague, F.; Dhennin-Duthille, I.; Chatelain, D.; Kerr-Conte, J.; Pattou, F.; Regimbeau, J.M.; Sevestre, H.; Ouadid-Ahidouch, H.; et al. Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int. J. Cancer 2012, 131, E851–E861. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.S.; Kazi, A.A.; Yee, R.K. Cellular and developmental biology of trpm7 channel-kinase: Implicated roles in cancer. Cells 2014, 3, 751–777. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Guerrero, J.A.; Romero, I.; Poveda, A. Trabectedin therapy as an emerging treatment strategy for recurrent platinum-sensitive ovarian cancer. Chin. J. Cancer 2015, 34, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Alicino, I.; Giglio, M.; Manca, F.; Bruno, F.; Puntillo, F. Intrathecal combination of ziconotide and morphine for refractory cancer pain: A rapidly acting and effective choice. Pain 2012, 153, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, X.; Feng, S.; Jiang, G.; Luo, J.; Zhou, S.; Vrijmoed, L.L.; Jones, E.B.; Krohn, K.; Steingrover, K.; et al. Five unique compounds: Xyloketals from mangrove fungus Xylaria sp. From the south China sea coast. J. Org. Chem. 2001, 66, 6252–6256. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Qian, Y.; Meng, W.F.; Pang, J.Y.; Lin, Y.C.; Guan, Y.Y.; Chen, S.P.; Liu, J.; Pei, Z.; Wang, G.L.; et al. A novel marine compound xyloketal b protects against oxidized ldl-induced cell injury in vitro. Biochem. Pharmacol. 2009, 78, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, L.; Ling, C.; Li, J.; Pang, J.Y.; Lin, Y.C.; Liu, J.; Huang, R.; Wang, G.L.; Pei, Z.; et al. Marine compound xyloketal B protects pc12 cells against ogd-induced cell damage. Brain Res. 2009, 1302, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.L.; Yao, X.L.; Liu, Z.; Zhang, H.; Li, W.; Li, Z.; Wang, G.L.; Pang, J.; Lin, Y.; Xu, Z.; et al. Protective effects of xyloketal B against mpp+-induced neurotoxicity in caenorhabditis elegans and pc12 cells. Brain Res. 2010, 1332, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Chen, J.W.; Yuan, F.; Huang, Y.Y.; Zhao, L.Y.; Li, J.; Su, H.X.; Liu, J.; Pang, J.Y.; Lin, Y.C.; et al. Xyloketal B exhibits its antioxidant activity through induction of ho-1 in vascular endothelial cells and zebrafish. Mar. Drugs 2013, 11, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.J.; Chen, W.; Xu, B.; Liu, R.; Turlova, E.; Barszczyk, A.; Sun, C.L.; Liu, L.; Deurloo, M.; Wang, G.L.; et al. Marine compound xyloketal b reduces neonatal hypoxic-ischemic brain injury. Mar. Drugs 2014, 13, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Yajima, I.; Kumasaka, M.Y.; Thang, N.D.; Goto, Y.; Takeda, K.; Yamanoshita, O.; Iida, M.; Ohgami, N.; Tamura, H.; Kawamoto, Y.; et al. Ras/raf/mek/erk and pi3k/pten/akt signaling in malignant melanoma progression and therapy. Dermatol. Res. Pract. 2012, 2012, 354191. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Bigner, D.D. Development of novel targeted therapies in the treatment of malignant glioma. Nat. Rev. Drug Discov. 2004, 3, 430–446. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhan, S.; Huang, C.; Cheng, X.; Lv, X.; Si, H.; Li, J. Trpm7 channel regulates pdgf-bb-induced proliferation of hepatic stellate cells via pi3k and erk pathways. Toxicol. Appl. Pharmacol. 2013, 272, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Parnas, M.; Peters, M.; Dadon, D.; Lev, S.; Vertkin, I.; Slutsky, I.; Minke, B. Carvacrol is a novel inhibitor of drosophila trpl and mammalian trpm7 channels. Cell Calcium 2009, 45, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, J.D.; Wilson, P.D. Synthesis of xyloketal a, b, c, d, and g analogues. J. Org. Chem. 2006, 71, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Chang, C.; Xiang, Q.; Zhou, Z.W.; Luo, R.; Yang, L.; He, Z.X.; Yang, H.; Li, J.; Bei, Y.; et al. Xyloketal B, a marine compound, acts on a network of molecular proteins and regulates the activity and expression of rat cytochrome p450 3a: A bioinformatic and animal study. Drug Des. Dev. Ther. 2014, 8, 2555–2602. [Google Scholar]
- Li, S.; Shen, C.; Guo, W.; Zhang, X.; Liu, S.; Liang, F.; Xu, Z.; Pei, Z.; Song, H.; Qiu, L.; et al. Synthesis and neuroprotective action of xyloketal derivatives in parkinson’s disease models. Mar. Drugs 2013, 11, 5159–5189. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, Y.; Xiang, Q.; Pei, Z.; Liu, X.; Lu, B.; Chen, L.; Wang, G.; Pang, J.; Lin, Y.; et al. Design and synthesis of novel xyloketal derivatives and their vasorelaxing activities in rat thoracic aorta and angiogenic activities in zebrafish angiogenesis screen. J. Med. Chem. 2010, 53, 4642–4653. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhao, J.; Qiao, W.; Chen, K. Recent advances in diagnosis and treatment of gliomas using chlorotoxin-based bioconjugates. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 385–405. [Google Scholar] [PubMed]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular determinants of the response of glioblastomas to egfr kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Klingler-Hoffmann, M.; Bukczynska, P.; Tiganis, T. Inhibition of phosphatidylinositol 3-kinase signaling negates the growth advantage imparted by a mutant epidermal growth factor receptor on human glioblastoma cells. Int. J. Cancer 2003, 105, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Lee, E.Q.; Reardon, D.A.; Ligon, K.L.; Alfred Yung, W.K. Current clinical development of pi3k pathway inhibitors in glioblastoma. Neuro-Oncology 2012, 14, 819–829. [Google Scholar] [CrossRef] [PubMed]
- McDowell, K.A.; Riggins, G.J.; Gallia, G.L. Targeting the akt pathway in glioblastoma. Curr. Pharm. Des. 2011, 17, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The ras/raf/mek/erk and the pi3k/akt signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S17–S27. [Google Scholar]
- Sathornsumetee, S.; Reardon, D.A.; Desjardins, A.; Quinn, J.A.; Vredenburgh, J.J.; Rich, J.N. Molecularly targeted therapy for malignant glioma. Cancer 2007, 110, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Sunayama, J.; Matsuda, K.; Sato, A.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Crosstalk between the pi3k/mtor and mek/erk pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 2010, 28, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Hottinger, A.F.; Stupp, R.; Homicsko, K. Standards of care and novel approaches in the management of glioblastoma multiforme. Chin. J. Cancer 2014, 33, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liao, Q.J.; Zhang, Y.; Zhou, H.; Luo, C.H.; Tang, J.; Wang, Y.; Tang, Y.; Zhao, M.; Zhao, X.H.; et al. Trpm7 is required for ovarian cancer cell growth, migration and invasion. Biochem. Biophys. Res. Commun. 2014, 454, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Huang, C.; Huang, Y.; Wu, X.; Li, X.; Li, J. Inhibition of trpm7 channels prevents proliferation and differentiation of human lung fibroblasts. Inflamm. Res. 2013, 62, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Cai, C.; Wu, J.; Cai, S.; Ye, C.; Chen, H.; Yang, Z.; Zeng, H.; Shen, Q.; Zou, F.; et al. Trpm7 mediates breast cancer cell migration and invasion through the mapk pathway. Cancer Lett. 2013, 333, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Xiong, Z.G. Silencing trpm7 promotes growth/proliferation and nitric oxide production of vascular endothelial cells via the erk pathway. Cardiovasc. Res. 2009, 83, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Inoue, K.; Sun, H.; Leng, T.; Feng, X.; Zhu, L.; Xiong, Z.G. Trpm7 regulates vascular endothelial cell adhesion and tube formation. Am. J. Physiol. Cell Physiol. 2015, 308, C308–C318. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.R.; Follo, M.Y.; Cocco, L.; Suh, P.G. The physiological roles of primary phospholipase c. Adv. Biol. Regul. 2013, 53, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Middelbeek, J.; Lasonder, E.; Dulyaninova, N.G.; Morrice, N.A.; Ryazanov, A.G.; Bresnick, A.R.; Figdor, C.G.; van Leeuwen, F.N. Trpm7 regulates myosin iia filament stability and protein localization by heavy chain phosphorylation. J. Mol. Biol. 2008, 378, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Deason-Towne, F.; Perraud, A.L.; Schmitz, C. Identification of ser/thr phosphorylation sites in the c2-domain of phospholipase c gamma2 (plcgamma2) using trpm7-kinase. Cell. Signal. 2012, 24, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Langeslag, M.; Clark, K.; Moolenaar, W.H.; van Leeuwen, F.N.; Jalink, K. Activation of trpm7 channels by phospholipase c-coupled receptor agonists. J. Biol. Chem. 2007, 282, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, L.; Kloog, Y. A ras inhibitor tilts the balance between rac and rho and blocks phosphatidylinositol 3-kinase-dependent glioblastoma cell migration. Cancer Res. 2006, 66, 11709–11717. [Google Scholar] [PubMed]
- Chen, W.L.; Huang, X.Q.; Zhao, L.Y.; Li, J.; Chen, J.W.; Xiao, Y.; Huang, Y.Y.; Liu, J.; Wang, G.L.; Guan, Y.Y.; et al. Involvement of kv1.5 protein in oxidative vascular endothelial cell injury. PLoS ONE 2012, 7, e49758. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Iihara, K.; Wei, W.L.; Xiong, Z.G.; Arundine, M.; Cerwinski, W.; MacDonald, J.F.; Tymianski, M. A key role for trpm7 channels in anoxic neuronal death. Cell 2003, 115, 863–877. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-L.; Turlova, E.; Sun, C.L.F.; Kim, J.-S.; Huang, S.; Zhong, X.; Guan, Y.-Y.; Wang, G.-L.; Rutka, J.T.; Feng, Z.-P.; et al. Xyloketal B Suppresses Glioblastoma Cell Proliferation and Migration in Vitro through Inhibiting TRPM7-Regulated PI3K/Akt and MEK/ERK Signaling Pathways. Mar. Drugs 2015, 13, 2505-2525. https://doi.org/10.3390/md13042505
Chen W-L, Turlova E, Sun CLF, Kim J-S, Huang S, Zhong X, Guan Y-Y, Wang G-L, Rutka JT, Feng Z-P, et al. Xyloketal B Suppresses Glioblastoma Cell Proliferation and Migration in Vitro through Inhibiting TRPM7-Regulated PI3K/Akt and MEK/ERK Signaling Pathways. Marine Drugs. 2015; 13(4):2505-2525. https://doi.org/10.3390/md13042505
Chicago/Turabian StyleChen, Wen-Liang, Ekaterina Turlova, Christopher L. F. Sun, Ji-Sun Kim, Sammen Huang, Xiao Zhong, Yong-Yuan Guan, Guan-Lei Wang, James T. Rutka, Zhong-Ping Feng, and et al. 2015. "Xyloketal B Suppresses Glioblastoma Cell Proliferation and Migration in Vitro through Inhibiting TRPM7-Regulated PI3K/Akt and MEK/ERK Signaling Pathways" Marine Drugs 13, no. 4: 2505-2525. https://doi.org/10.3390/md13042505