1. Introduction
The Red Sea serves as an epicenter for marine bio-diversity, with a high endemic biota. Indeed, of the 180 soft coral species identified world-wide, approximately 40% are native to the Red Sea [
1]. Soft coral (Cnidaria: Anthozoa: Octocorallia) are an important structural component of coral reef communities contributing significantly to coral-reef biomass [
2,
3]. Soft coral have also been the subject of biological studies since the 19th century with coral belonging to the genus
Sarcophyton (Alcyoniidae) well recognized as a rich source of terpenoids [
4]. For cembranoid-type diterpenes, a wide range of biological activities have been reported including antitumor, ichthyotoxic, anti-inflammatory, neuroprotective, antibacterial, antiangiogenic, antimetastatic, anti-osteoporotic, and cytotoxic properties [
5]. Diterpenes have been isolated previously from
Sarcophyton ehrenbergi [
6,
7,
8,
9,
10]; however, to the best of our knowledge, this is the first chemical investigation of the Red Sea soft coral
Sarcophyton ehrenbergi (
Figure 1). In the course of our research for bioactive substances from marine sources [
11,
12], chromatographic separation of an ethyl acetate extract of the alcyonacean soft coral,
S. ehrenbergi has led to the isolation of three compounds
1–
3 as well as the previously reported marine cembranoid diterpene sarcophine (
4) (
Figure 2). Structures of these isolated metabolites were elucidated by comprehensive NMR and HRMS techniques. Identified compounds were
in vitro assayed for cytotoxic activity in two human cancer cell lines.
Figure 1.
Soft coral Sarcophyton ehrenbergi.
Figure 1.
Soft coral Sarcophyton ehrenbergi.
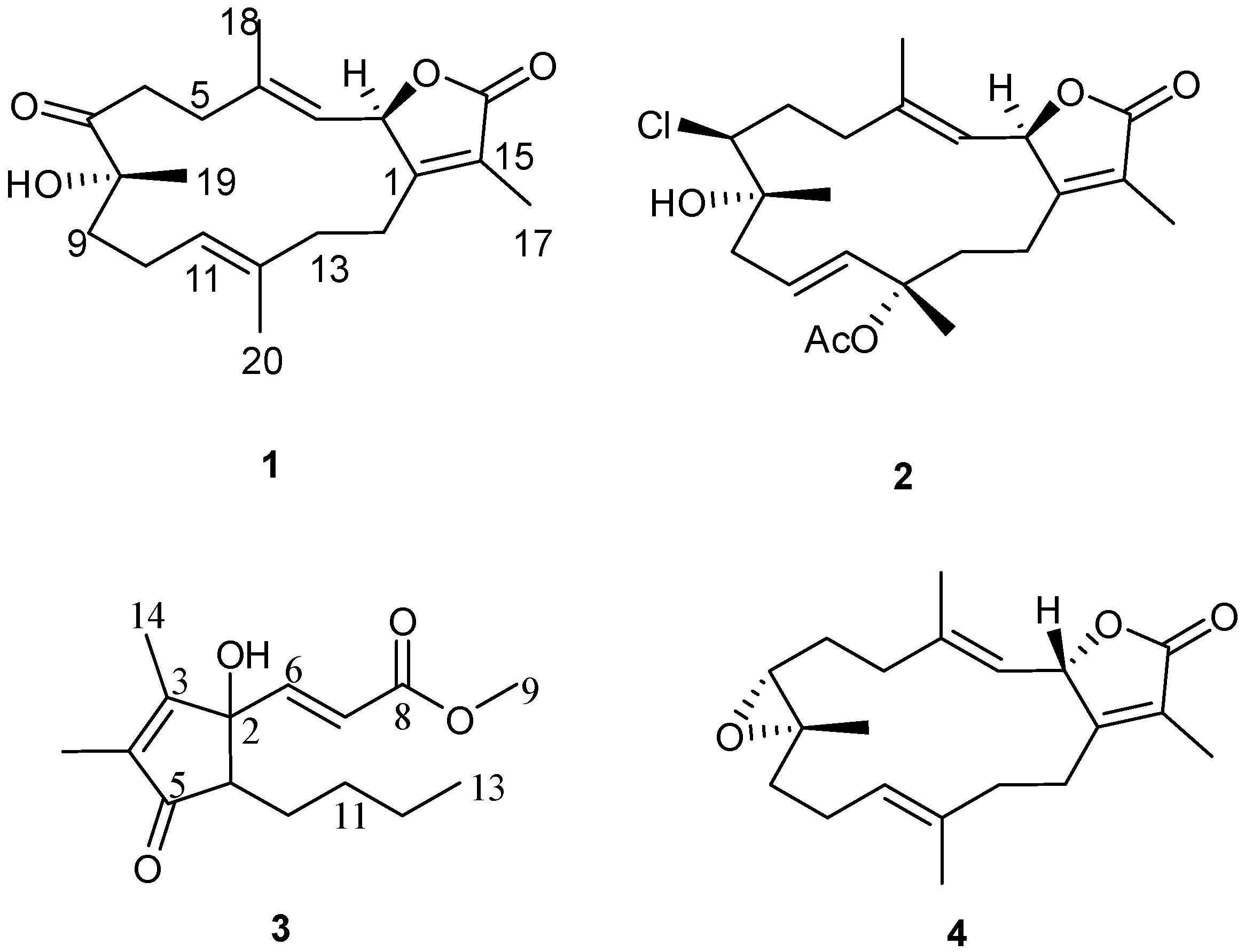
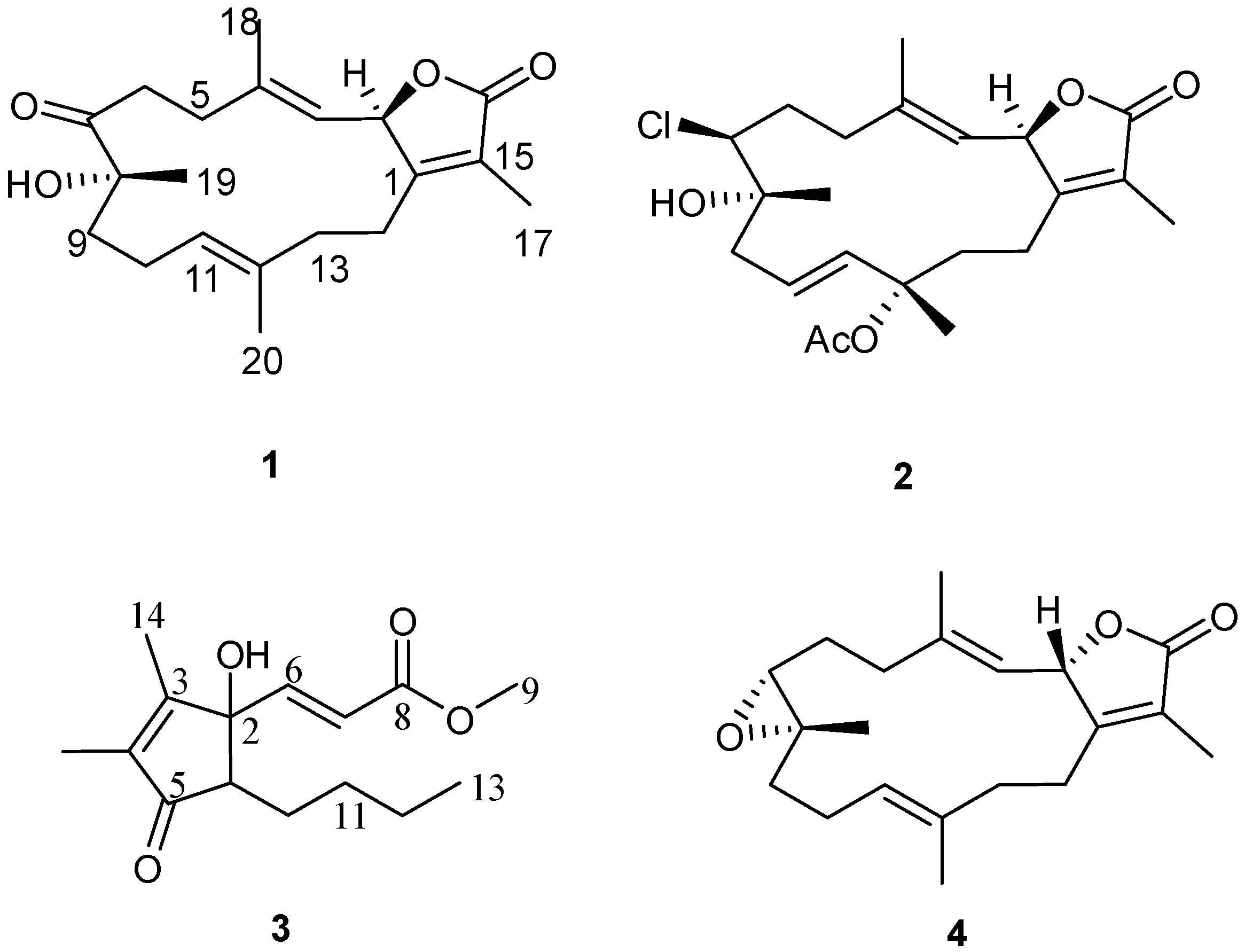
Figure 2.
Structures of metabolites 1–4.
Figure 2.
Structures of metabolites 1–4.
2. Results and Discussion
The EtOAc extract of freshly collected S. ehrenbergi was subjected to normal and reverse phase chromatography to afford the isolated metabolites as reported.
Compound
1 was obtained as a colorless oil. The molecular formula C
20H
28O
4, determined by HR-EI-MS (
m/z 332.1935, [M]
+, C
20H
28O
4; calcd. 332.1988) points to seven degrees of unsaturation. NMR data were consistent with a sarchophine skeleton previously isolated from other
Sarcophyton species [
11,
12] and reported here (
4). Select
13C NMR signals were consistent with an α,β-unsaturated-γ-lactone functionality: δ 123.1 (C-15), δ 162.9 (C-1) δ 174.9 (C-16). Carbon NMR chemical shifts (Supplementary Figure S2) established olefinic carbons at δ
C 120.2/143.9 (C-3/C-4) and δ
C 123.9/135.2 (C-11/C-12), a ketone functionality at δ
C 213.5 (C-7) and oxygen functionalities at δ
C 79.4 (C-2) and δ
C 78.6 (C-8); all signals were consistent with a sarchophine skeleton. The olefin carbons at C-3 and C-11 were assumed to be in the
E configuration based on other sarchophine derivatives [
11,
12,
13]; this orientation was confirmed by NOESY correlations. The
1H,
1H COSY spectrum revealed the coupling of four hydrocarbon regions common to sarchophine skeletons: δ
H 5.47 (d,
J = 10.3 Hz, H-2) and 4.96 (br. d,
J = 10.3 Hz, H-3); δ
H 2.29 (m, H
2-5) and 2.46 (m, H
2-6); δ
H 1.87/1.98 (m, H
2-9), 2.04/2.18 (m, H
2-10) and 4.72 (br. t,
J = 7.6 Hz, H-11) and δ
H 2.04 (m, H
2-13) and 2.29/2.46 (m, H
2-14). HMBC correlations (Supplementary Figure S4) between H-2 and δ
C 162.9 (C-1), H-3 and δ
C 31.5 (C-5), H
2-5 and δ
C 143.9 (C-4) and H
2-6/H
2-5 and δ
C 213.5 (C-7) established the carbon linkages from C-1 to C-7. Additionally, correlations between H
2-9 and δ
C 213.5 (C-7)/78.6 (C-8)/123.9 (C-11), H-11 and δ
C 35.9 (C-13), H
2-13 and δ
C 135.2 (C-12)/162.9 (C-1) and H
2-14 and δ
C 162.9 (C-1)/79.4 (C-2) completed connectivities of the 14-membered ring. The methyl groups’ positions were established by HMBC correlations between C-3, C-4 and C-5 and δ
H 1.94 (H
3-18, br. s); C-7, C-8 and C-9 and δ
H 1.31 (H
3-19, s); C-11, C-12 and C-13 and δ
H 1.56 (H
3-20, br. s); and C-1, C-15 and C-16 and δ
H 1.81 (H
3-17, s). The relative configuration of
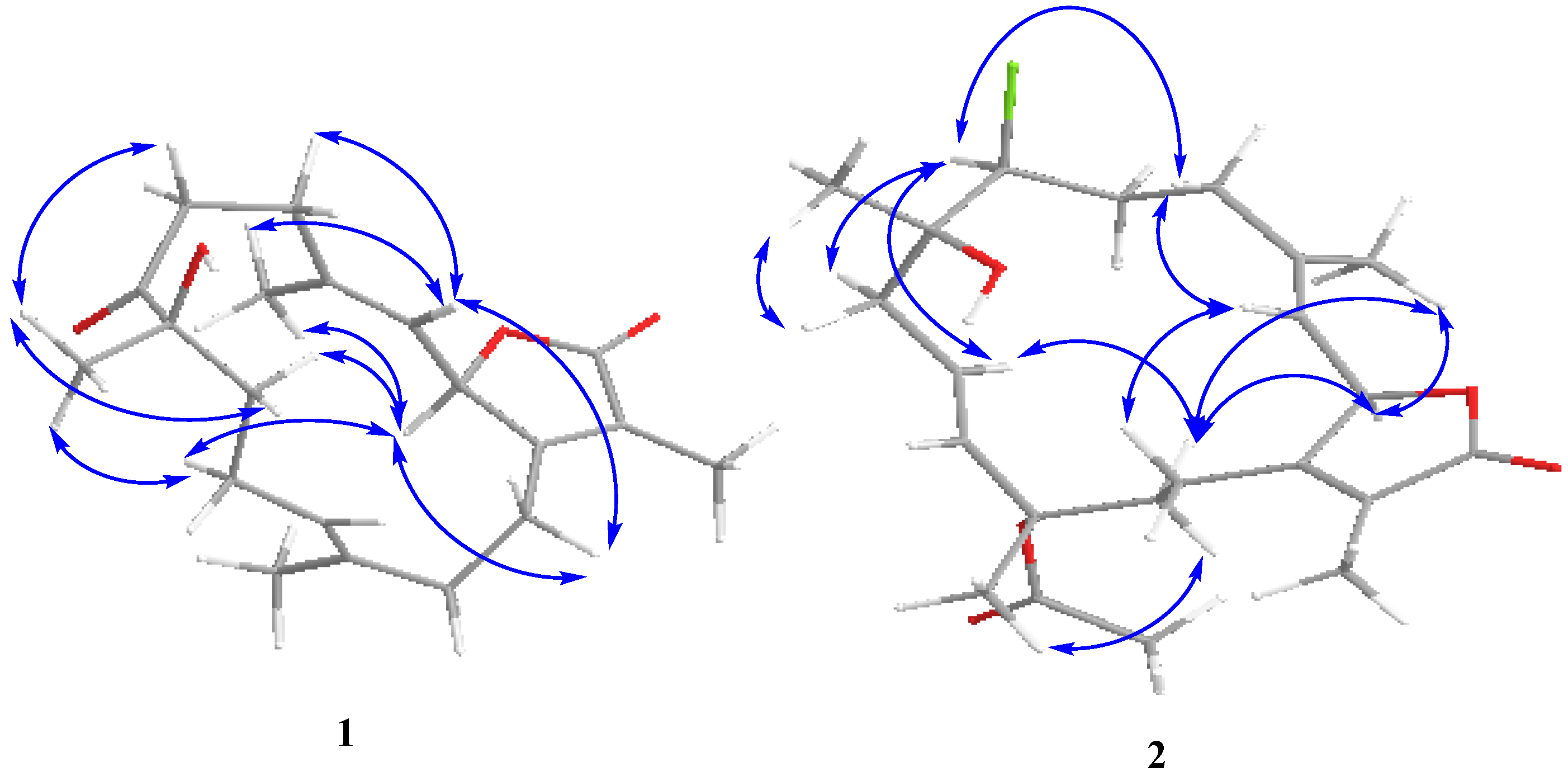
1 was determined on the basis of coupling constants and NOESY experiments. The vicinal coupling constant of 10.3 Hz between H-2 and H-3 suggested a
cis configuration between the γ-lactone proton (H-2) and the olefinic proton (H-3) which was confirmed by observed NOESY correlations (
Figure 3). Correlations were observed between Me-19 and H-6 (δ
H 2.91) as well as between H-9 (δ
H 1.98) and H-10 (δ
H 2.04); additionally, H-2 (δ
H 5.47) showed a correlation with H-18 (δ
H 1.94) and H-10 (δ
H 2.18) which indicated that Me-19 and an OH group at C-8 (δ
C 78.6) were positioned in β and α configurations, respectively [
11,
12,
13].
Figure 3.
NOESY correlations for 1–2.
Figure 3.
NOESY correlations for 1–2.
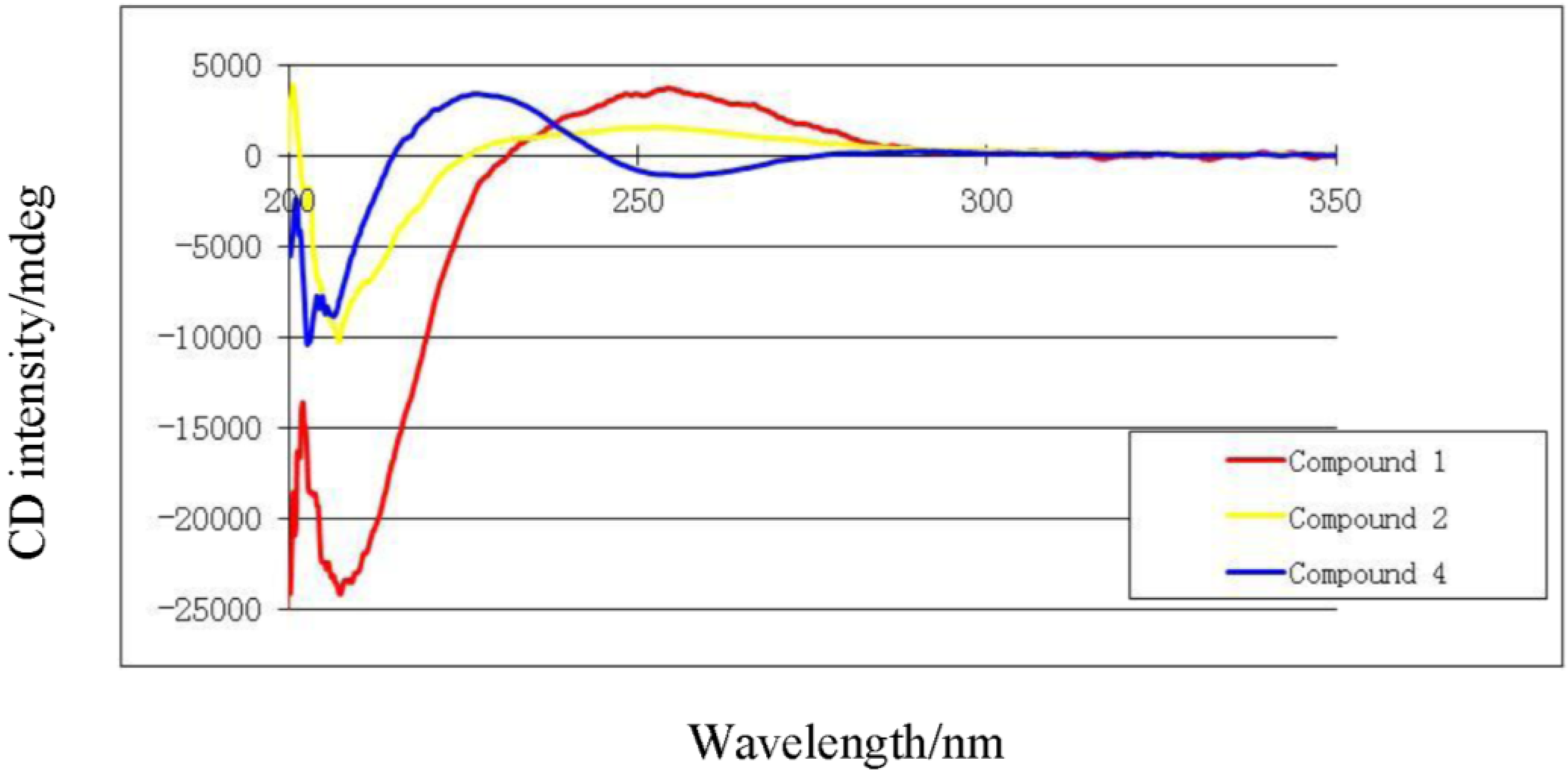
The absolute stereochemistry of
1 at C-2 was determined via circular dichroism (CD) analysis (
Figure 3). The observed positive Cotton effect ([θ]
255 +4) followed by a negative value ([θ]
225 −24.8) observed in the CD spectrum (
Figure 4) for the electronic transitions of the 2(5
H)-furanone moiety, indicated a left-handed (
M) helix configuration for the five-membered α,β-unsaturated-γ-lactone ring [
14]. CD spectral comparisons between
1 and sarcophine (
4) show a shifted spectrum establishing an inverted stereochemistry at C-2 for
1 and
4 (
Figure 4); with
4 having a literature assigned
S absolute configuration, the absolute configuration for
1 at C-2 was confirmed to be
R [
12,
14,
15]. From the above evidence, the structure of
1 was confirmed to be 7-keto-8α-hydroxy-deepoxysarcophine (
Figure 2).
Figure 4.
Circular dichroism spectra of compounds 1, 2 and 4.
Figure 4.
Circular dichroism spectra of compounds 1, 2 and 4.
Compound
2 was obtained as a colorless oil. The molecular formula C
22H
32ClO
5 was determined by HR-FAB-MS (
m/z 411.1943, [M + H]
+, C
22H
32ClO
5+; calcd. 411.1938) and was consistent with seven degrees of unsaturation. The NMR spectrum of
2 was quite similar to
1 except for several noted differences. First, there was a disappearance of the C-7 ketone signal accompanied by the appearance of an up-field signal at δ
C 62.9 (C-7). This C-7 carbon signal was coupled by HMQC (Supplementary Figure S7) with a downfield doublet signal at δ
H 3.45 (
J = 11.0 Hz) consistent with a chlorine atom at C-7 [
13,
14,
15]. HMBC correlations (Supplementary Figure S8) between δ
H 3.45 and δ
C 36.0 (C-5), 26.3 (C-6) and 74.8 (C-8) confirmed the proton signal assignment to H-7. Second, there was a downfield shift of C-20 by 9 ppm to δ
C 24.4 accompanied by the appearance of carbonyl and methyl groups at δ
C 170.0 and 22.2, respectively. These data were consistent with an acetoxy group at C-12 and the displacement of an olefinic bond between C-11 and C-12 (compound
1) to a C-10-C-11 double bond position. This double bond shift was confirmed by COSY correlations starting from H
2-9 (δ
H 2.20/2.62, m) correlating with δ
H 5.53 (H-10, ddd,
J = 15.8, 10.3, 4.8 Hz) and δ
H 5.82 (H-11, d,
J = 15.8 Hz). HMBC correlations were also observed between H
2-9 and δ
C 138.2 (C-11) and H-20 and δ
C 138.2 (C-11), 82.0 (C-12) and 41.7 (C-13). HMBC correlations were also observed between the acetoxy methyl signal (δ
C 2.05) and the carbonyl ester (δ
C 170) as shown in
Figure 5 (Supplementary Figure S8). The relative configuration of
2 was determined on the basis of coupling constants, and NOESY experiments. The vicinal coupling constant of 10.3 Hz between H-2 and H-3 suggested a
cis configuration between the γ-lactone proton (H-2) and the olefinic proton (H-3) that was confirmed by observed NOESY correlations (
Figure 3). The NOE correlations between H-7 and H-10 (δH 5.53), H-9 (δH 2.20) and Me-18 (δH 1.76), Me-18 and H-2 (δH 5.42) and Me-19 and H-9 (δH 2.62), indicate a β chlorine conformation at C-7 (δC 62.9). In addition, the NOE interactions between H-2 and H-14 (δH 2.20) and H-14 (δH 2.62) and Me-20 (δH 1.63) reveal an α orientation for the acetoxy group at C-12. With nearly equivalent CD spectra observed for
2 compared with
1 (
Figure 4) assignment of an (
R) configuration at C-2 as well as absolute stereochemistry at the other stereo centers was possible [
12,
14,
15]. From the above spectral data,
2 was identified as 7β-chloro-8α-hydroxy-12-acetoxy-deepoxysarcophine (
Figure 2).
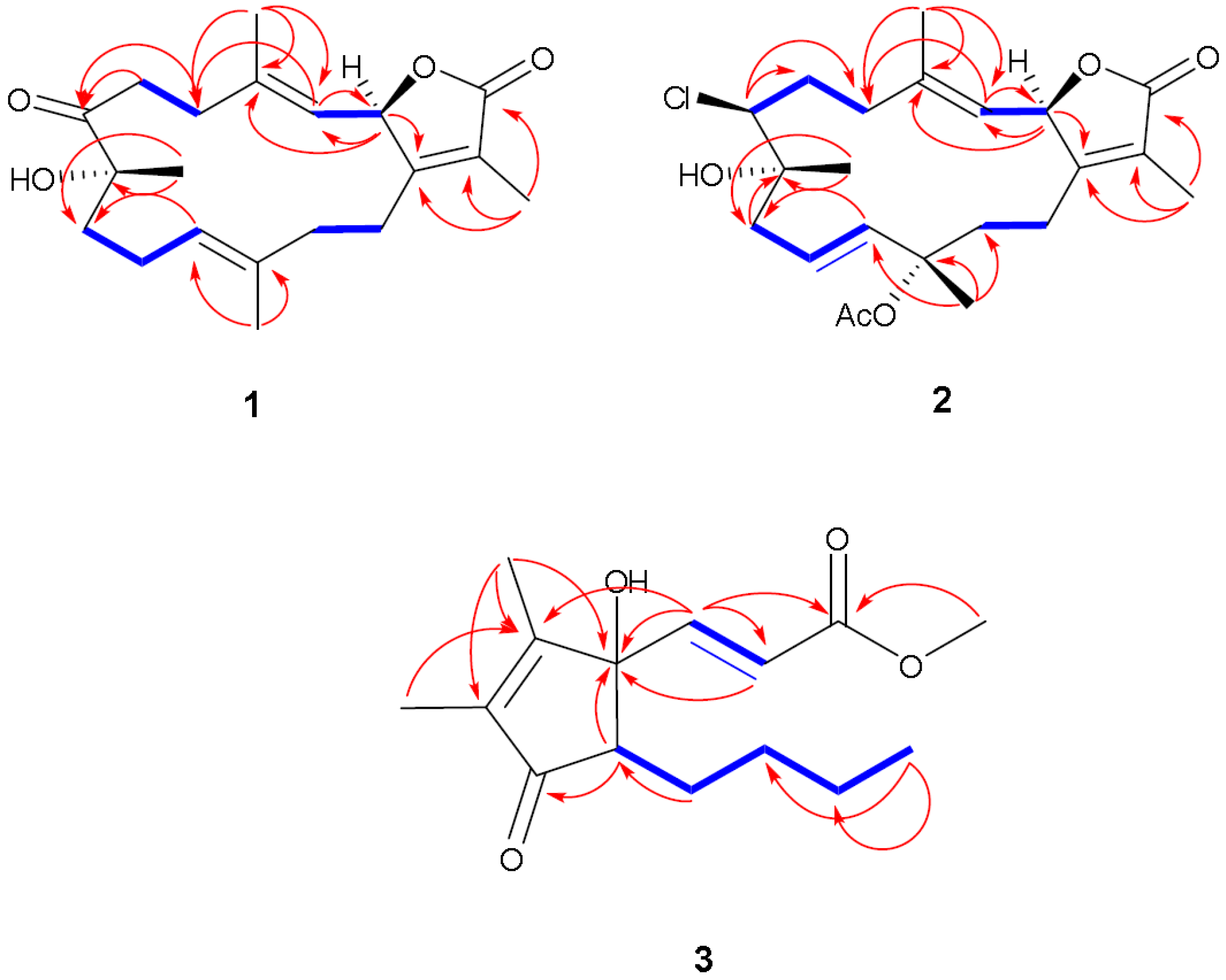
Figure 5.
Selected
1H-
1H COSY (
![Marinedrugs 12 01977 i001]()
) and HMBC (
![Marinedrugs 12 01977 i002]()
) correlations of
1–
3.
Figure 5.
Selected
1H-
1H COSY (
![Marinedrugs 12 01977 i001]()
) and HMBC (
![Marinedrugs 12 01977 i002]()
) correlations of
1–
3.
Compound 3 was obtained as a colorless oil. The molecular formula C15H22O4 was determined by HR-EI-MS (m/z 266.1552, [M]+, C15H22O4; calcd. 266.1518) and consistent with five degrees of unsaturation. Starting with a carbonyl carbon based on a chemical shift of δC 204.7 (C-5) and HMBC data (Supplementary Figure S12), a cyclopentenone ring structure with two methyl groups was deduced. Informative correlations were observed between C-5 and δH 1.73 (H3-15)/2.42 (H-1); H-1 and δC 82.8 (C-2); C-2 and δH 1.87 (H3-14); H3-14 and δC 165.4 (C-3)/137.5 (C-4), C-3 and δH 1.73 (H3-15) and C-4 and H3-15. HMBC data established that the pentone ring was linked to olefinic protons via C-2 with correlations observed between C-2 and δH 6.60 (H-6, d, J = 15.8 Hz) and C-2 and δH 6.21 (H-7, d, J = 15.8 Hz) with the olefin in a Z configuration based on the coupling constant. The methyl ester terminus was linked to an olefinic group through HMBC correlations between H-6 and δC 166.4 (C-8) as well as between C-8 and H-7. From COSY correlations, a four carbon unbranched saturated hydrocarbon originating from C-1 was identified. The H-1 NMR proton signal correlated with δH 1.30 (H-10, m)/1.82 (H-10, m); H-10 correlated with δH 1.32 (H-11, m)/1.45 (H-11, m); H-11 correlated with δH 1.30 (H-12, m); and H-12 correlated with δH 0.87 (H-13, t, J = 7.6 Hz). The relative structure of 3 was elucidated by an analysis of coupling constant. The coupling constant of H-1 (δ 2.42, dd) of 8.9 and 5.5 Hz indicated that H-1 is on the β-face. From the above evidence, the structure of 3 was assigned to be (E)-methyl-3-(5-butyl-1-hydroxy-2,3-dimethyl-4-oxocyclopent-2-enyl) acrylate.
Using a 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide (MTT) spectrophotometric cell-viability assay, the biological activity of the reported coral metabolites to lower human hepatocarcinoma (HepG2) and breast adenocarcinoma (MCF-7) cell-line viability was assayed. The treatment of Hep-G2 cells with metabolites 1–4 (100–12.5 μg/mL) did not exhibit a measureable cytotoxic effect. Metabolites 1–3 exhibited a moderate cytotoxic effect with the MCF-7 cell-line with IC50 values of 192.87, 68.57, and 114.41 μmol/mL, respectively.
3. Experimental Section
3.1. General Experimental Procedures
1H and 13C NMR spectra were recorded in CDCl3 on a JEOL ECA-600 spectrometer (600 MHz for 1H and 150 MHz for 13C, respectively). All chemical shifts (δ) are given in ppm units with reference to TMS as an internal standard and coupling constants (J) are reported in Hz. FAB-MS was performed on a Finnigan LCQ ion trap mass spectrometer and HR-FAB-MS experiments were performed on Fourier transform ion cyclotron mass spectrometer (Ion Spec, Varian, Walnut Creek, CA, USA). EI-MS experiments were performed using a Thermo ISQ Single Quadrupole system (Thermo Scientific, San Jose, CA, USA). High performance liquid chromatography (HPLC) was performed on an Agilent pump equipped with an Agilent-G1314 variable wavelength UV detector at 254 nm and a semi-preparative reverse-phase column (Econosphere™, RP-C18, 5 μm, 250 × 4.6 mm, Alltech, Deerfield, IL, USA). Optical rotation was determined at 589 nm (sodium D line) using a Perkin–Elmer-341 MC digital polarimeter (Wellesley, MA, USA); [α] D-values are given in units of 10 deg−1·cm2 g−1. CD was measured with an OLIS, DSM-10 UV/Vis CD (Olis, Bogart, GA, US). Silica gel 60 (230–400 mesh, Merck, Darmstadt, Germany) and Sephadex LH-20 (Sigma, St. Louis, MO, USA) were used for column chromatography. Pre-coated silica gel plates (Merck, Darmstadt, Germany, Kieselgel 60 F254, 0.25 mm) were used for TLC analyses. Spots were visualized by heating after spraying with 10% H2SO4.
3.2. Animal Material
Soft coral S. ehrenbergi was collected from the Egyptian Red Sea off the coast of Hurghada in March 2012. The soft coral was identified by co-author Alhammady with a voucher specimen (03RS27) deposited in the National Institute of Oceanography and Fisheries, Marine Biological Station, Hurghada, Egypt.
Table 1.
1H and 13C NMR spectral data of 1–3 a.
Table 1.
1H and 13C NMR spectral data of 1–3 a.
| Position | 1 | 2 | 3 |
|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC |
|---|
| 1 | − | 162.9 C | − | 161.9 C | 2.42 (dd, 8.9, 5.5) | 60.0 CH |
| 2 | 5.47 (d, 10.3) | 79.4 CH | 5.42 (d, 10.3) | 78.3 CH | − | 82.8 C |
| 3 | 4.96 (br d, 10.3) | 120.2 CH | 4.98 (d, 10.3) | 122.2 CH | − | 165.4 C |
| 4 | − | 143.9 C | − | 142.9 C | − | 137.5 C |
| 5 | 2.29 (m)
2.46 (m) | 31.5 CH2 | 2.26 (m)
2.42 (m) | 36.0 CH2 | − | 204.7 C |
| 6 | 2.81 (m)
2.91 (m) | 34.6 CH2 | 1.63 (m)
2.20 (m) | 26.3 CH2 | 6.60 (d, 15.8) | 148.2 CH |
| 7 | − | 213.5 C | 3.45 (d, 11.0) | 62.9 CH | 6.21 (d, 15.8) | 121.5 CH |
| 8 | − | 78.6 C | − | 74.8 C | − | 166.4 C |
| 9 | 1.87 (m)
1.98 (m) | 39.5 CH2 | 2.20 (m)
2.62 (m) | 43.3 CH2 | 3.74 (s) | 51.9 CH3 |
| 10 | 2.04 (m)
2.18 (m) | 22.0 CH2 | 5.53 (ddd, 15.8, 10.3, 4.8) | 123.1 CH | 1.30 (m)
1.82 (m) | 25.4 CH2 |
| 11 | 4.72 (br t, 7.6) | 123.9 CH | 5.82 (d, 15.8) | 138.2 CH | 1.32 (m)
1.45 (m) | 30.1 CH2 |
| 12 | − | 135.2 C | − | 82.0 C | 1.30 (m) | 22.7 CH2 |
| 13 | 2.04 (m) | 35.9 CH2 | 2.10 (m)
1.38 (m) | 41.7 CH2 | 0.87 (t, 7.6) | 13.9 CH3 |
| 14 | 2.29 (m)
2.46 (m) | 25.7 CH2 | 2.20 (m)
2.62 (m) | 21.5 CH2 | 1.87 (s) | 11.0 CH3 |
| 15 | − | 123.1 C | − | 123.5 C | 1.73 (br s) | 8.5 CH3 |
| 16 | − | 174.9 C | − | 175.0 C | | |
| 17 | 1.81 (s) | 9.0 CH3 | 1.84 (s) | 8.9 CH3 | | |
| 18 | 1.94 (br s) | 18.3 CH3 | 1.76 (br s) | 15.2 CH3 | | |
| 19 | 1.31 (s) | 29.0 CH3 | 1.32 (s) | 23.7 CH3 | | |
| 20 | 1.56 (br s) | 15.6 CH3 | 1.63 (s) | 24.4 CH3 | | |
| 21 | | | − | 170.0 C | | |
| 22 | | | 2.05 (s) | 22.2 CH3 | | |
3.3. Extraction and Separation
Frozen soft coral (4 kg, total wet weight) was chopped into small pieces and extracted with ethyl acetate at room temperature (4 L × 5). The combined ethyl acetate extracts were concentrated in vacuo to a brown gum. The dried EtOAc-soluble material (250 g) was subjected to gravity chromatography in a silica gel column (6 × 120 cm) using n-hexane-EtOAc gradient separated into eight fractions.
Fraction 4 (2.2 g) eluted with n-hexane–EtOAc (6:1) was subjected to silica gel column separation to afford 4 (100 mg). The remaining sub-fraction 4 was collected and re-purified by HPLC using MeOH/H2O (8:2 v/v) to afford 3 (15 mg). Fraction 5 (75.3 mg) was purified by Sephadex LH-20 using hexane-CHCl3-MeOH (7:4:0.5) followed by reverse phase HPLC in MeOH/H2O (7:3 v/v) to afford 1 (17 mg), and 2 (20 mg).
7-Keto-8α-hydroxy-deepoxysarcophine (
1): Colorless oil;
![Marinedrugs 12 01977 i003]()
= +15.0 (
c 0.03, CHCl
3);
1H NMR and
13C NMR data, see
Table 1; HR-EI-MS [M]
+ m/z 332.1935 (calc. 332.1988, C
20H
28O
4).
7β-Chloro-8α-hydroxy-12-acetoxy-deepoxysarcophine (
2): Colorless oil;
![Marinedrugs 12 01977 i003]()
= −20.1 (
c 0.01, CHCl
3);
1H NMR and
13C NMR data, see
Table 1; HR-FAB-MS [M + H]
+ m/z 411.1943 (calc. 411.1938, C
22H
32ClO
5).
(
E)-Methyl-3-(5-butyl-1-hydroxy-2,3-dimethyl-4-oxocyclopent-2-enyl)acrylate(
3): Colorless oil;
![Marinedrugs 12 01977 i003]()
= +1.2 (
c 0.01, CHCl
3);
1H NMR and
13C NMR data, see
Table 1; HR-EI-MS [M]
+ m/z 266.1552 (calc. 266.1518 C
15H
22O
4).
(+)-Sarcophine (
4):
![Marinedrugs 12 01977 i003]()
= +95.0 (
c 0.5, CHCl
3); lit.
![Marinedrugs 12 01977 i003]()
= +92 (
c 1.0, CHCl
3) [
15].
3.4. Cell Culture
Cell-culture material was purchased from Cambrex BioScience (Copenhagen, Denmark) and chemicals from Sigma-Aldrich (St. Louis, MO, USA), except where noted otherwise. Human hepatocarcinoma and breast adenocarcinoma cell lines, HepG2 and MCF-7 respectively, purchased from the American type culture collection (ATCC, Manassas, VA, USA) were used to evaluate the cytotoxic effect of the isolated metabolites. Cells are routinely cultured in Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 units/mL penicillin G sodium, 100 units/mL streptomycin sulphate and 250 ng/mL amphotericin B. Cells are maintained at sub-confluency in humidified air at 37 °C containing 5% CO2. For sub-culturing, monolayer cells are harvested after trypsin/EDTA treatment. Cells for bioassays were assayed at 75% confluence. Test compounds were dissolved in dimethyl sulphoxide (DMSO) and diluted to begin with a concentration of 100 μg/mL and serially diluted to a final concentration of 12.5 μg/mL. Assays were run in triplicate unless noted otherwise.
3.5. Anti-Tumor Activity
Compound cytotoxicity against HepG2 and MCF-7 cell lines was assayed via a 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide (MTT) spectrophotometric cell-viability assay. The MTT assay is based on the ability of active mitochondrial dehydrogenase enzyme of living cells to cleave the tetrazolium ring of the yellow MTT to form dark-blue insoluble formazan crystals that are largely impermeable to cell membranes, resulting in its accumulation within healthy cells. With loss of cell viability, formazan crystals leak into the media. Cell viability is monitored and quantified spectrophotometrically from formazan crystals extracted from the cells [
16]. Briefly, cells are diluted in serum-free media to 5.0 × 10
4 cells/well based on turbidity measurements, plated into flat-bottom 96-well microplates, and incubated for 48 h with 20 μL of test sample. After incubation, media is removed and 40 μL MTT (0.5 mg MTT/ ml 0.9% aqueous NaCl) is added into each well and then incubated for an additional 4 h. MTT crystals are solubilized by adding 180 µL of acidified isopropanol to each well and plates are agitated at room temperature, followed by absorbance measurements at 570 nm using a microplate ELISA reader (BMG LABTECH, Ortenberg, Germany). Triplicate repeats were performed for each concentration and the average was calculated. Data were expressed as the percentage of relative viability compared with the untreated cells and with the vehicle control. Cytotoxicity was reported as a relative viability compared with the untreated control. Percent relative viability was determined based cell viability: (Absorbance of treated cells/absorbance of control cells) × 100. Half maximal inhibitory concentrations (IC
50) were calculated based on dose-response curves.

 and
and 



 ) and HMBC (
) and HMBC ( 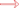 ) correlations of 1–3.
) correlations of 1–3.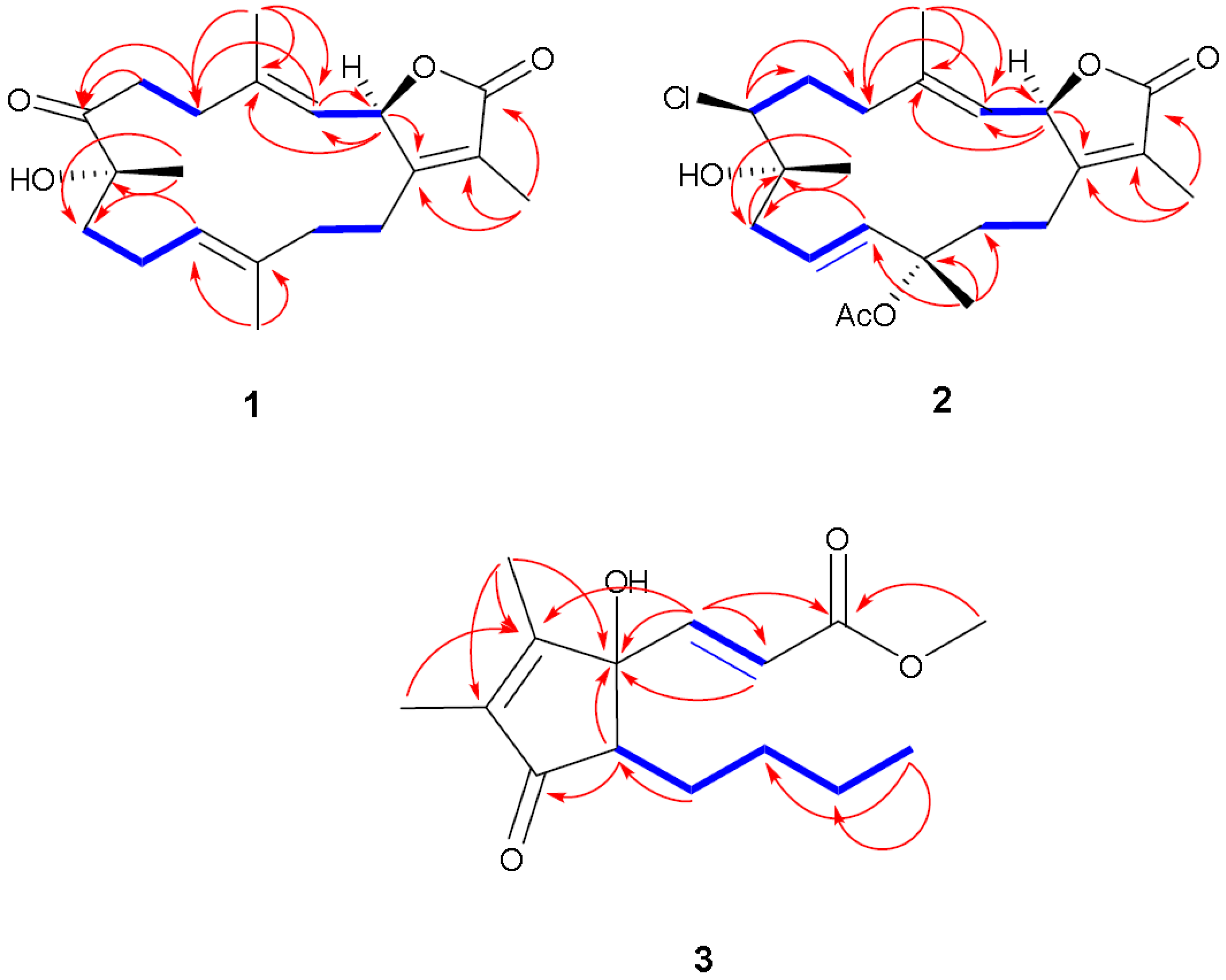

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 = +15.0 (c 0.03, CHCl3); 1H NMR and 13C NMR data, see Table 1; HR-EI-MS [M]+ m/z 332.1935 (calc. 332.1988, C20H28O4).
= +15.0 (c 0.03, CHCl3); 1H NMR and 13C NMR data, see Table 1; HR-EI-MS [M]+ m/z 332.1935 (calc. 332.1988, C20H28O4).