Marine Natural Products Acting on the Acetylcholine-Binding Protein and Nicotinic Receptors: From Computer Modeling to Binding Studies and Electrophysiology

 ,
, 
Abstract
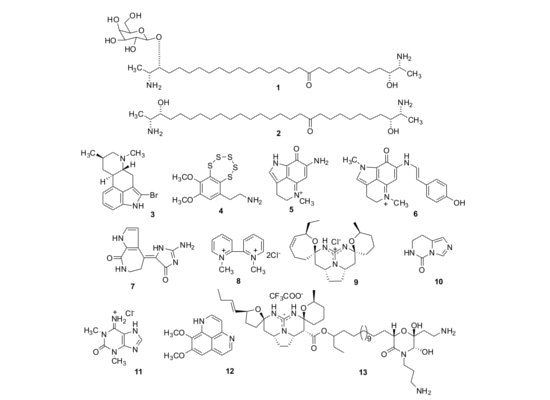
:
1. Introduction
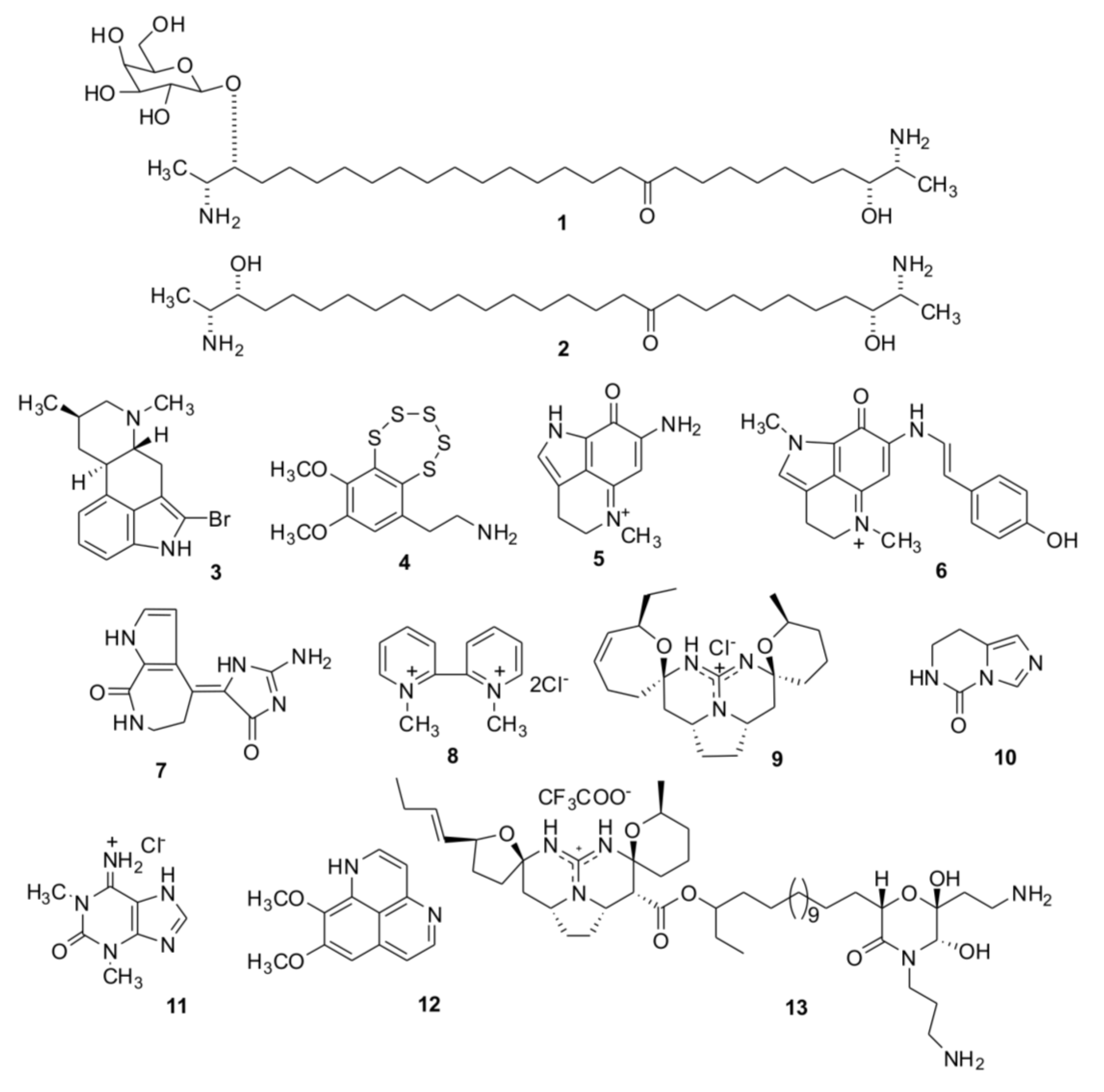
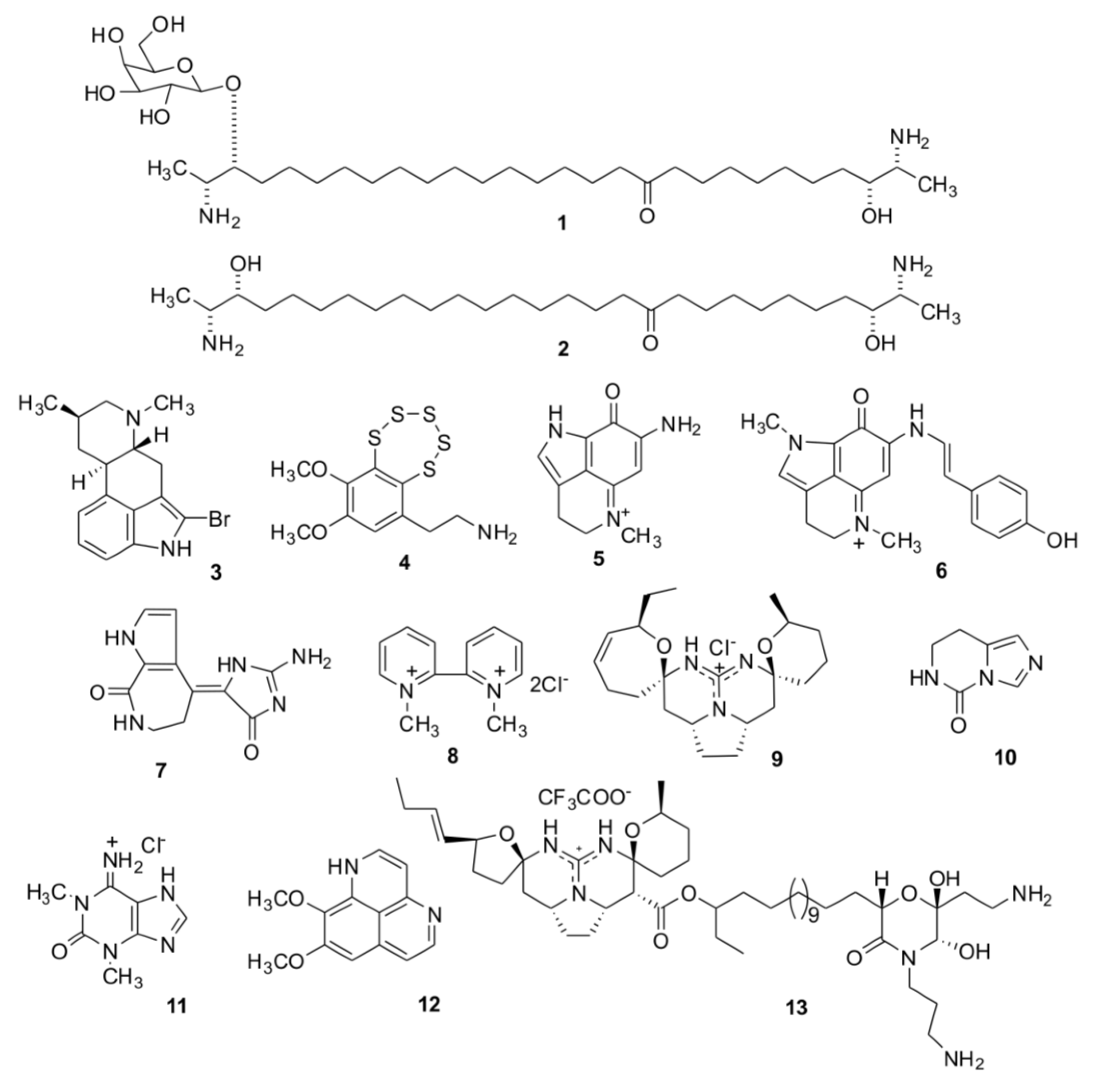
2. Results and Discussion
2.1. Isolation of Individual Compounds
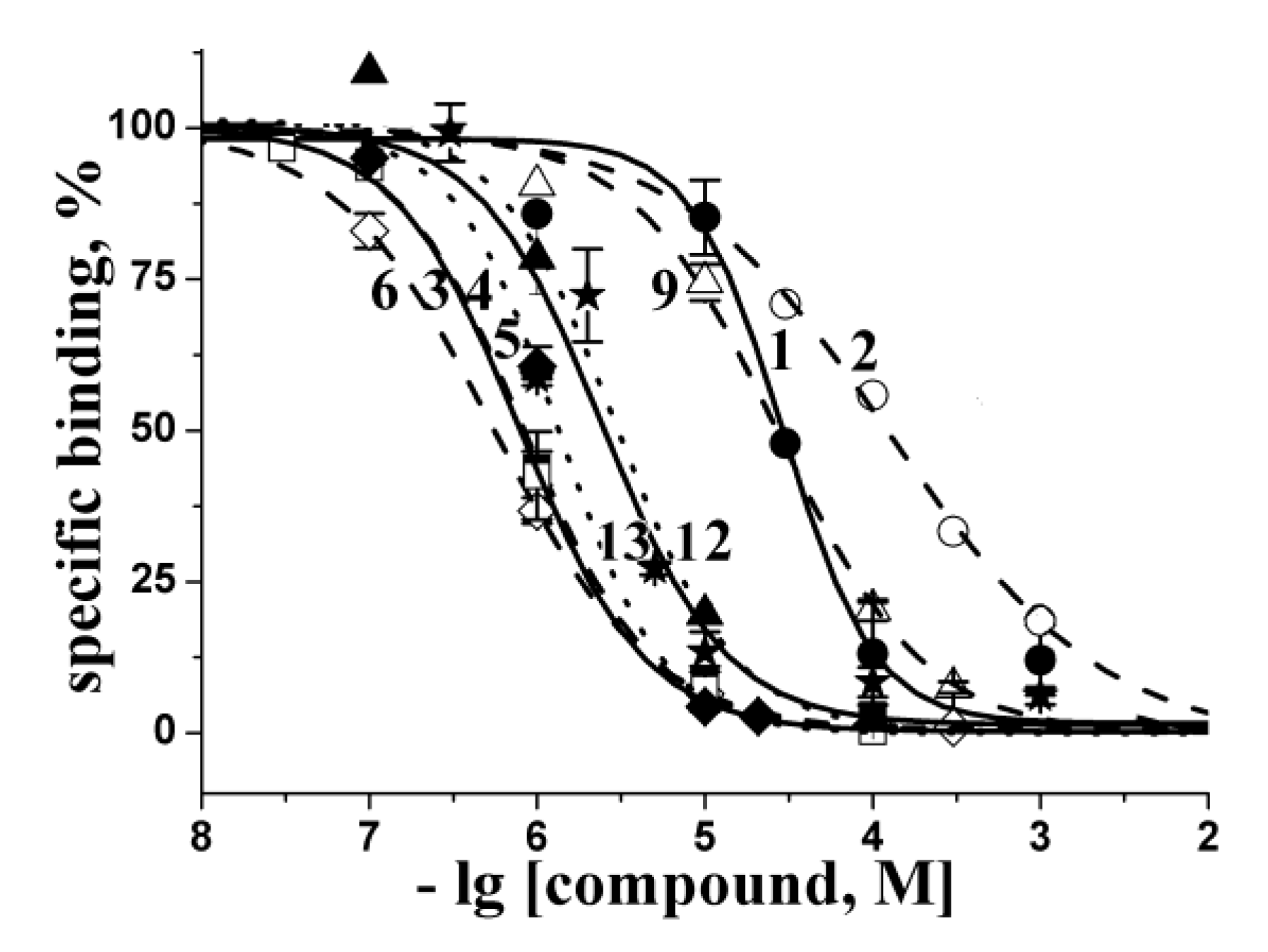
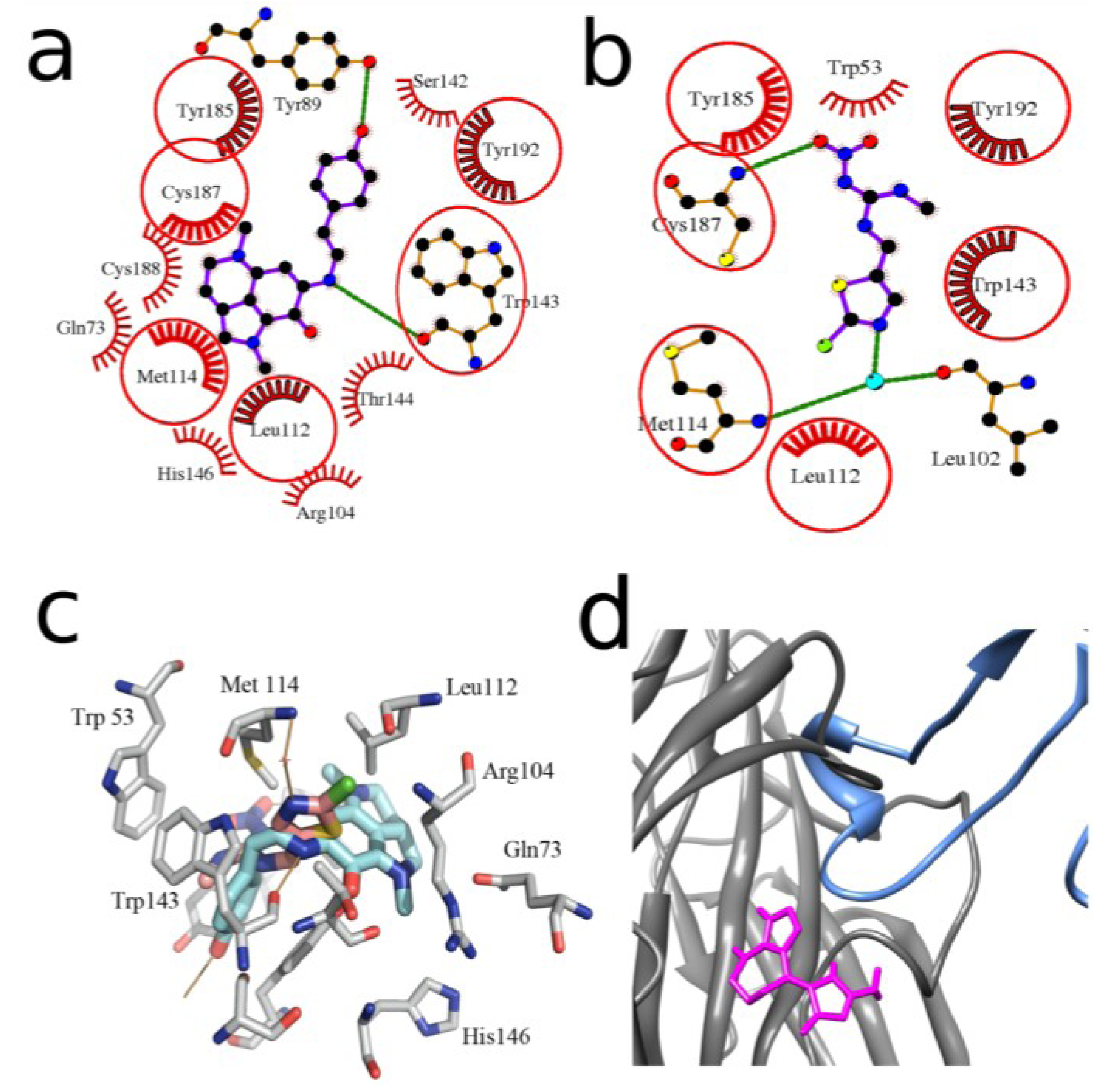
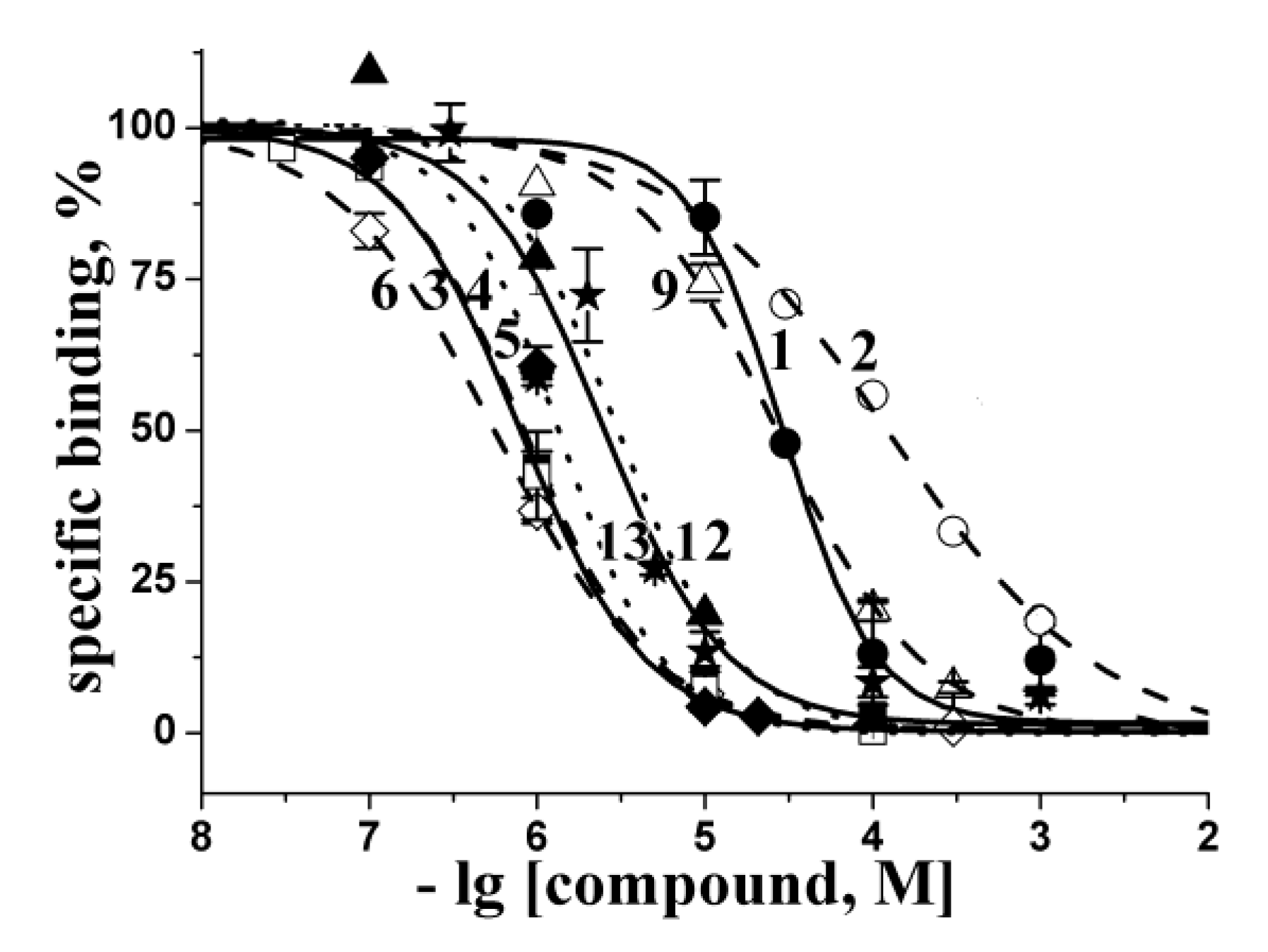
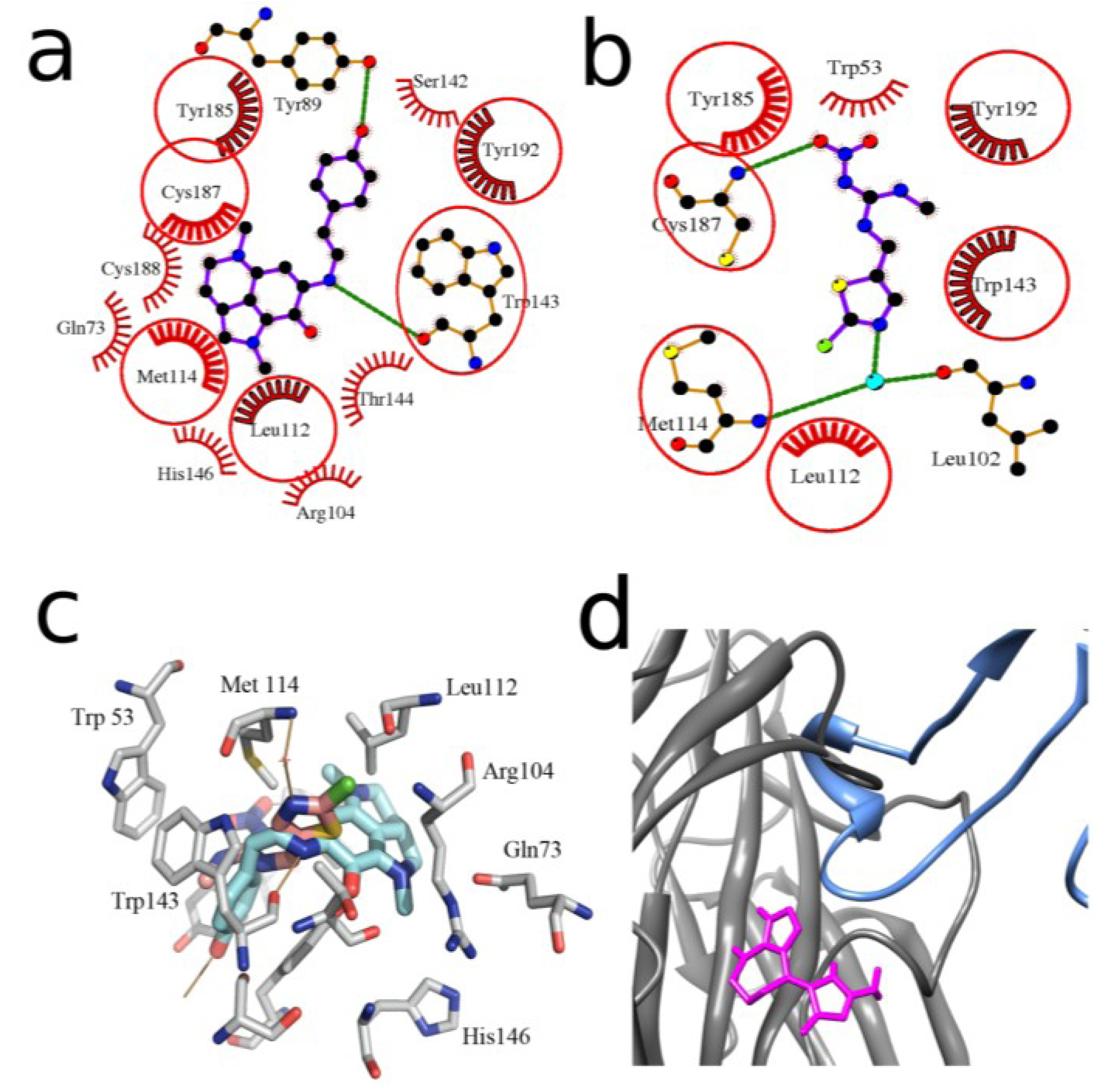
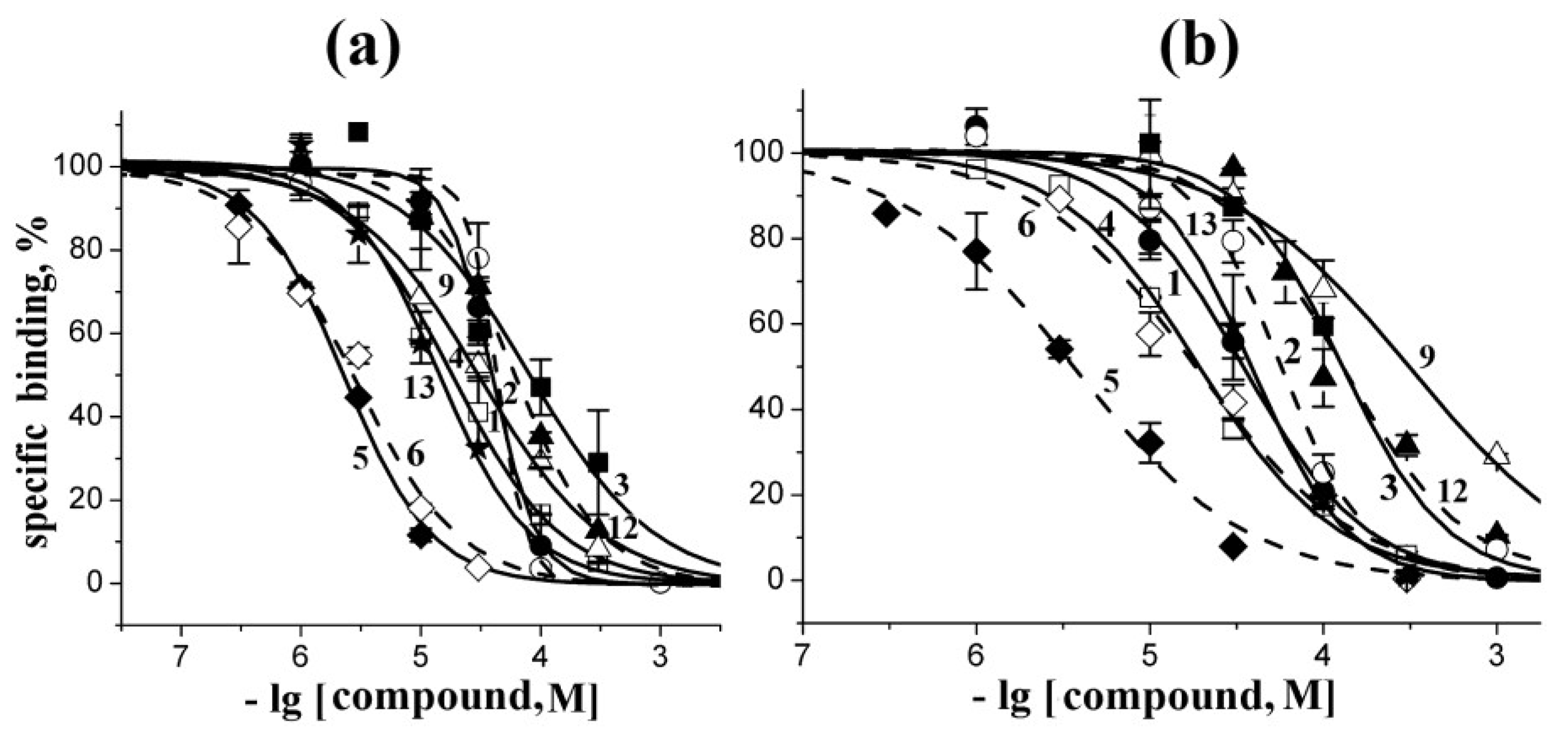
2.2. Docking to AChBP and Analysis of Binding Parameters in Competition with [125I]-αBgt

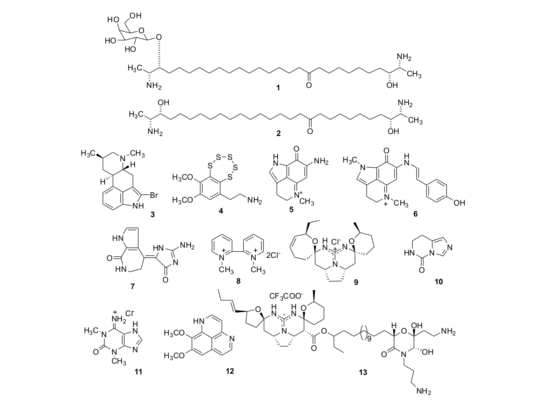{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Theoretical calculated Ki values (μM) for interaction with L. stagnalis AChBP | Experimental Ki values (μM) at interaction with L. stagnalis AChBP from radioligand assay | ||
|---|---|---|---|---|
| A | B | C | ||
| 1 * | n.d. | n.d. | 39 | 28 ± 4 |
| 2 * | n.d. | n.d. | 39 | 130 ± 10 |
| 3 | 0.98 | 1.7 0.64 ** | 2.0 | 0.83 ± 0.04 |
| 4 | 1.6 | 0.97 | 0.75 | 0.79 ± 0.03 |
| 5 | 2.2 | 0.46 | 5.8 | 1.3 ± 0.1 |
| 6 | 0.05 | 0.04 | 0.97 | 0.55 ± 0.01 |
| 7 | 8.2 | 0.79 0.50 ** | 1.7 | >1000 |
| 8 | 33 | 12 | 310 | 540 ± 60 |
| 9 | 0.08 | 0.47 | 0.40 | 27 ± 2 |
| 10 | 170 | 120 | n.d. 320 ** | >1000 |
| 11 | 150 | 83 11 ** | 350 | >1000 |
| 12 | 1.5 | 5.3 | 19 | 3.0 ± 0.4 |
| 13 * | 0.53 | 0.43 | 0.11 | 2.5 ± 0.4 |
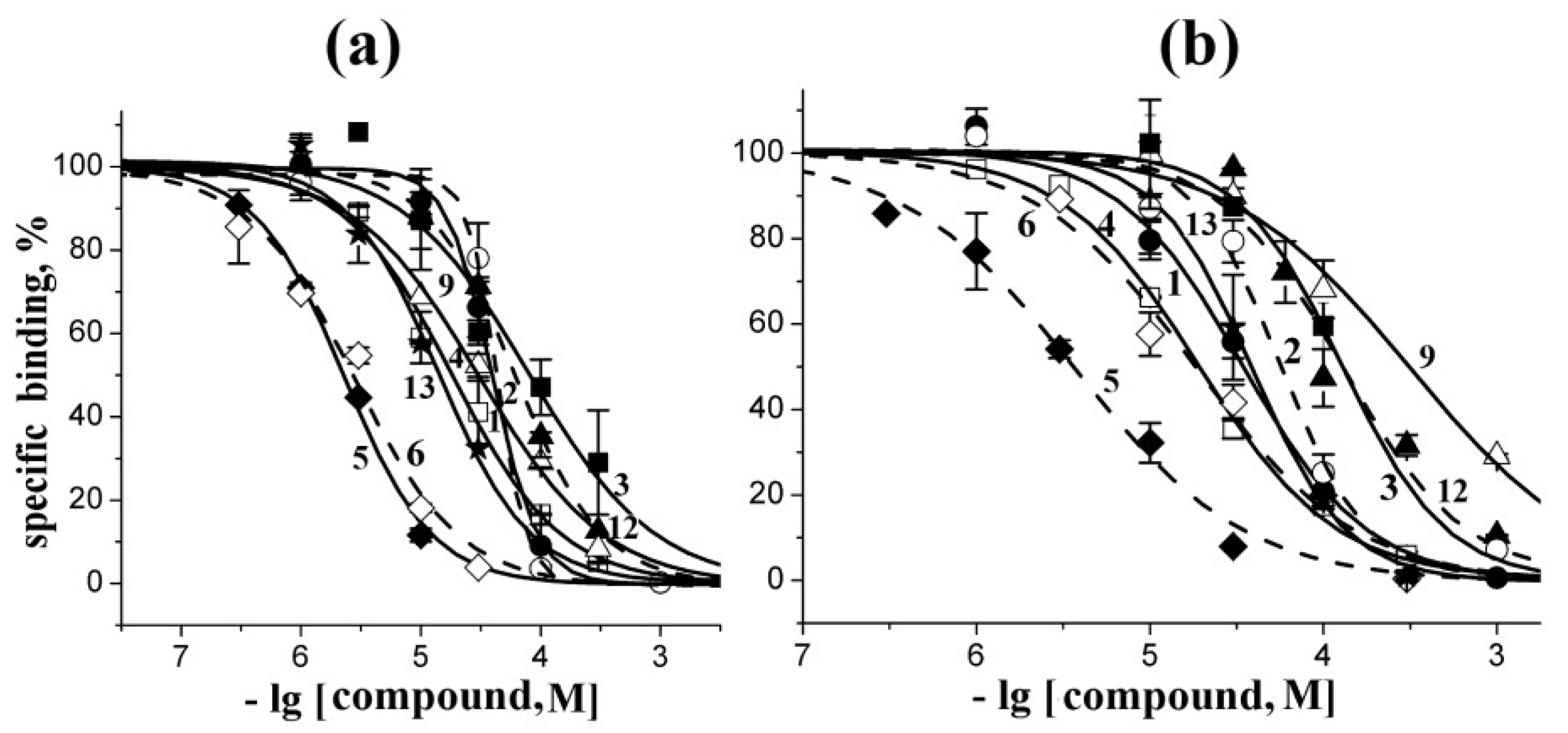
2.3. Analysis of Competition with [125I]-αBgt for Interactions with the Torpedo Californica and α7 nAChRs
| Compounds | Ki Values (μM) on… | |
|---|---|---|
| T. californica nAChR | human α7 nAChR | |
| 1 | 22 ± 2 | 33 ± 2 |
| 2 | 24 ± 2 | 56 ± 4 |
| 3 | 44 ± 7 | 130 ± 8 |
| 4 | 10 ± 1 | 19 ± 1 |
| 5 | 1.30 ± 0.05 | 3.6 ± 0.3 |
| 6 | 1.60 ± 0.17 | 18 ± 2 |
| 7 | >1000 | >1000 |
| 8 | >1000 | >1000 |
| 9 | 17 ± 1 | 310 ± 28 |
| 10 | >1000 | >1000 |
| 11 | >1000 | >1000 |
| 12 | 34 ± 1 | 120 ± 15 |
| 13 | 8.0 ± 1.0 | 38 ± 2 |
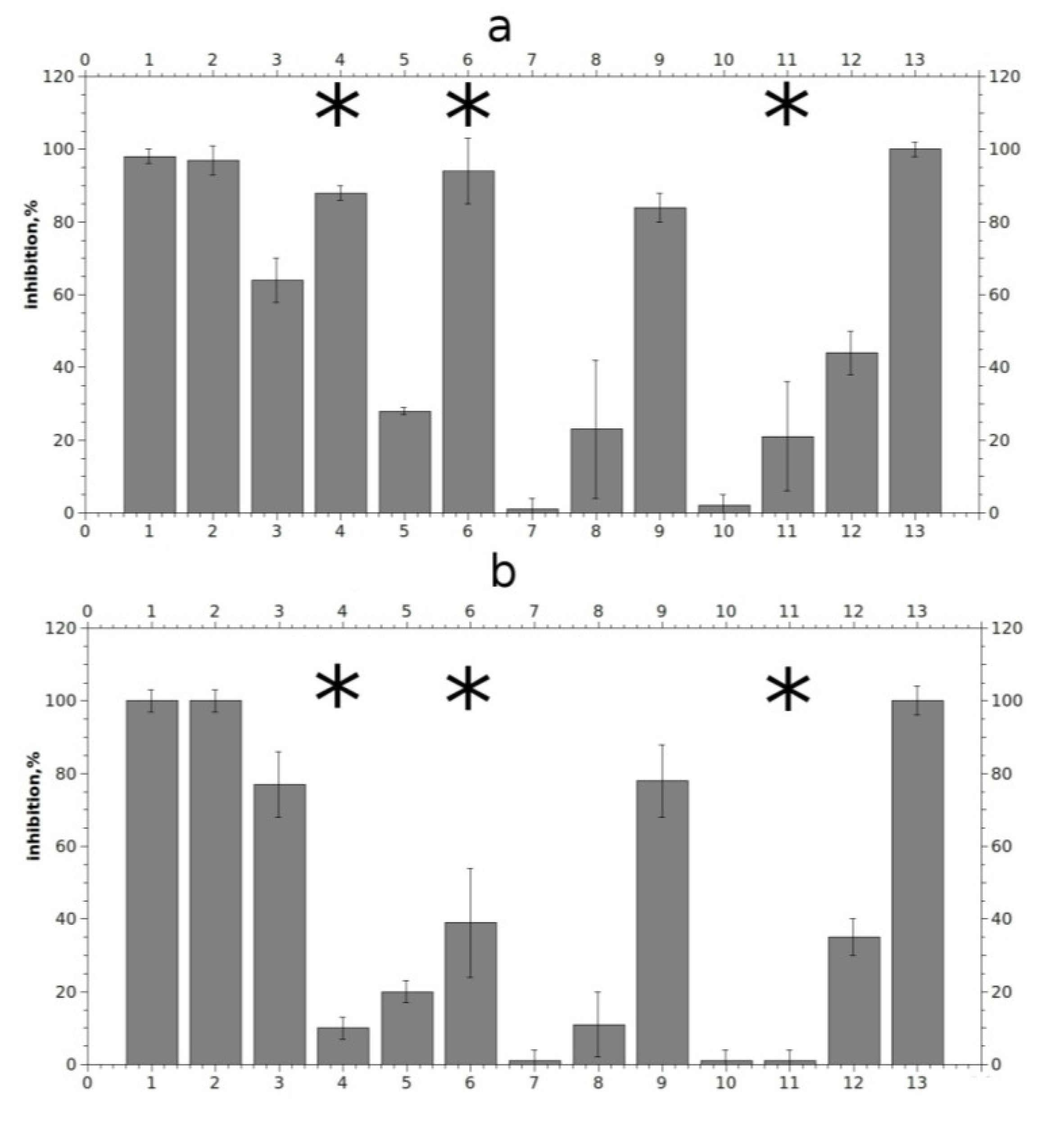
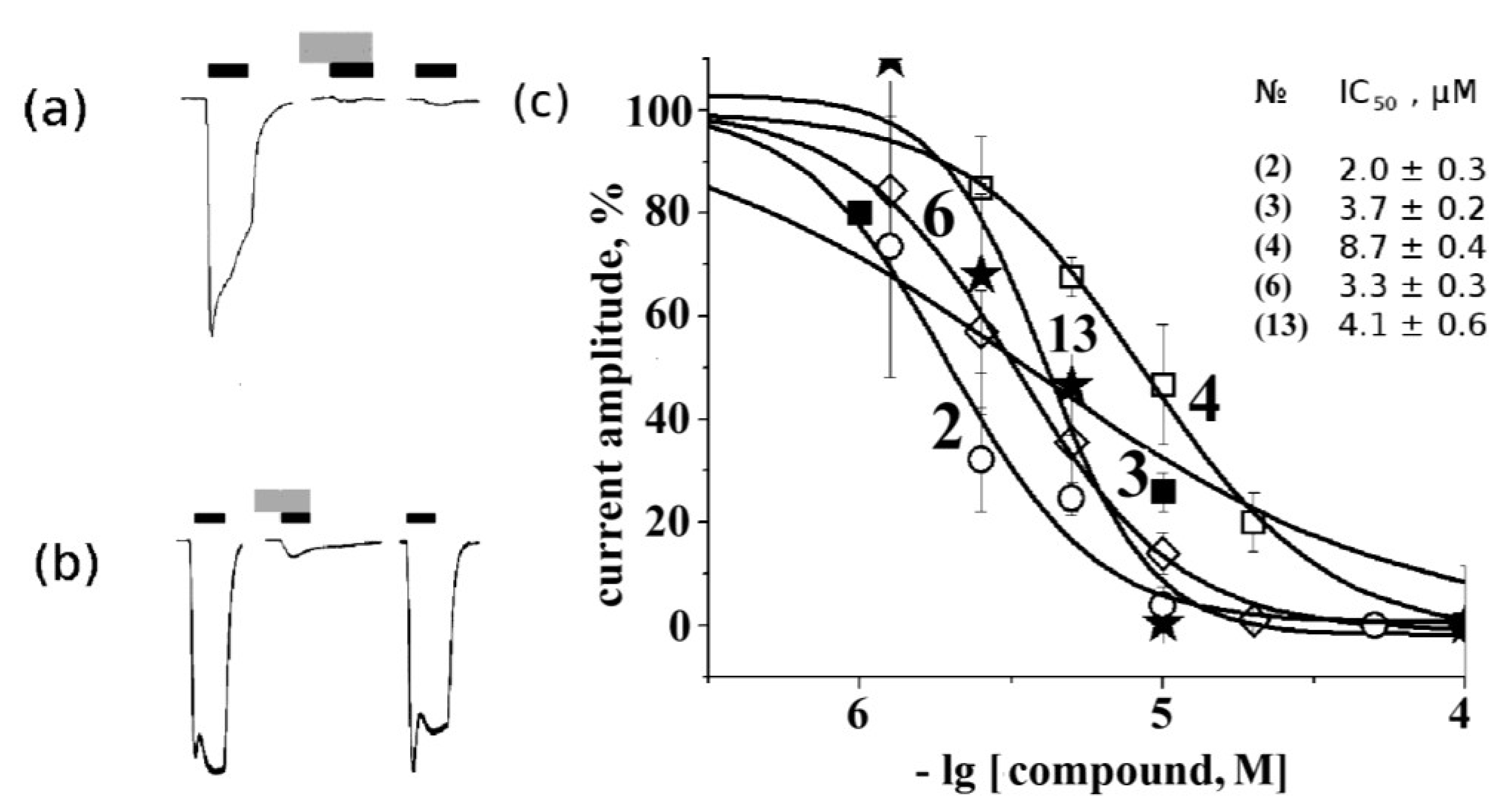
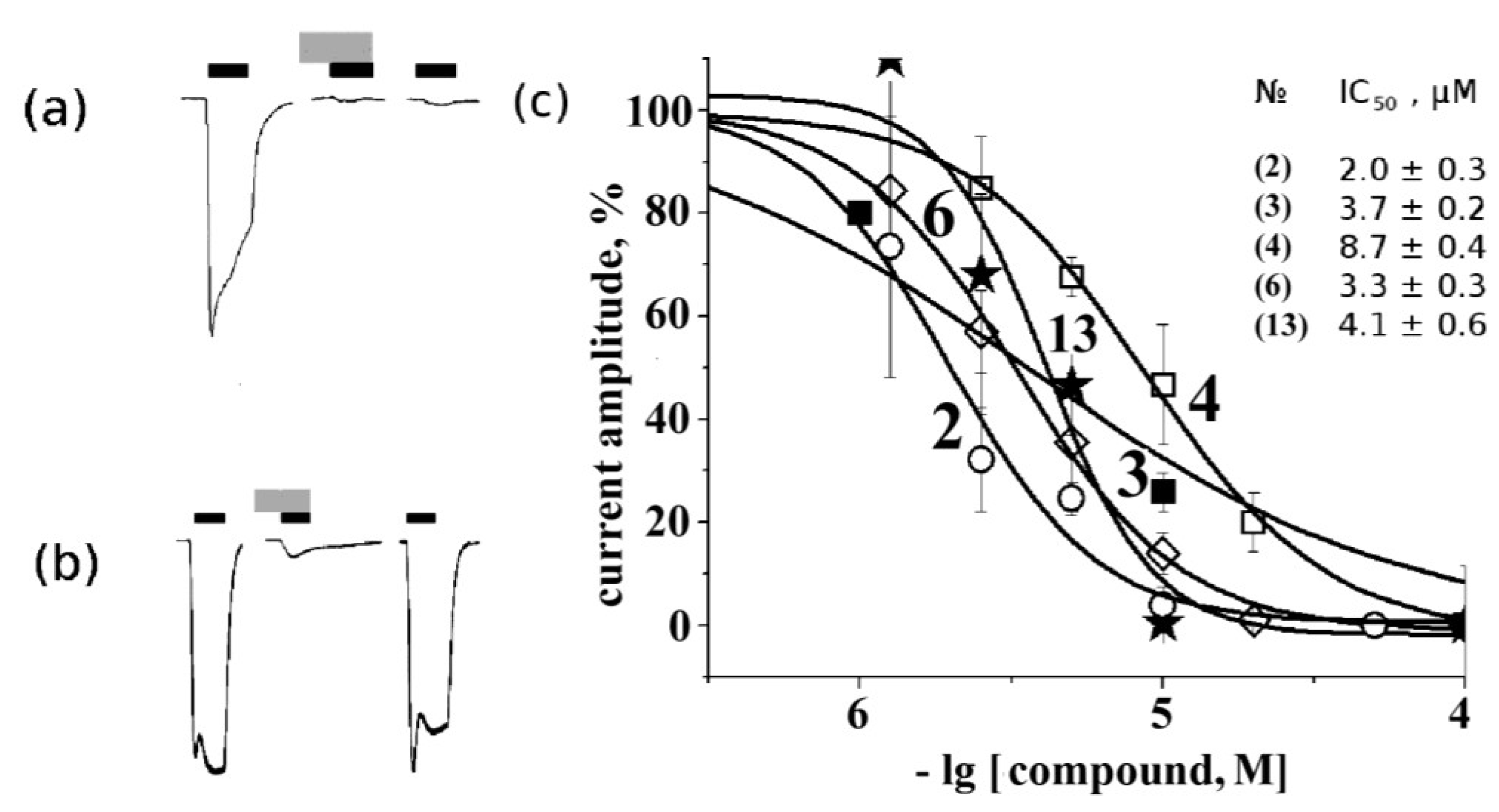
2.4. Electrophysiological Analysis of Effects on Functional Activity of Nicotinic Receptors

3. Experimental Section
3.1. Isolation of Individual Compounds
3.2. Computer Modeling
3.3. Radioligand Assay
3.4. Electrophysiology Measurements
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Changeux, J.P. The nicotinic acetylcholine receptor: The founding father of the pentameric ligand-gated ion channel superfamily. J. Biol. Chem. 2012, 287, 40207–40215. [Google Scholar] [CrossRef]
- Kalamida, D.; Poulas, K.; Avramopoulou, V.; Fostieri, E.; Lagoumintzis, G.; Lazaridis, K.; Sideri, A.; Zouridakis, M.; Tzartos, S.J. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J. 2007, 274, 3799–3845. [Google Scholar] [CrossRef]
- Tsetlin, V.; Hucho, F. Nicotinic acetylcholine receptors at atomic resolution. Curr. Opin. Pharmacol. 2009, 9, 306–310. [Google Scholar] [CrossRef]
- Sine, S.M.; Engel, A.G. Recent advances in Cys-loop receptor structure and function. Nature 2006, 440, 448–455. [Google Scholar] [CrossRef]
- Prickaerts, J.; van Goethem, N.P.; Chesworth, R.; Shapiro, G.; Boess, F.G.; Methfessel, C.; Reneerkens, O.A.; Flood, D.G.; Hilt, D.; Gawryl, M.; et al. EVP-6124, a novel and selective α7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of α7 nicotinic acetylcholine receptors. Neuropharmacology 2012, 62, 1099–1110. [Google Scholar]
- Chang, C.C. Looking back on the discovery of α-bungarotoxin. J. Biomed. Sci. 1999, 6, 368–375. [Google Scholar]
- Tsetlin, V. Snake venom α-neurotoxins and other ‘three-finger’ proteins. Eur. J. Biochem. 1999, 264, 281–286. [Google Scholar] [CrossRef]
- Kini, R.M.; Doley, R. Structure, function and evolution of three-finger toxins: Mini proteins with multiple targets. Toxicon 2010, 56, 855–867. [Google Scholar] [CrossRef]
- Halai, R.; Craik, D.J. Conotoxins: Natural product drug leads. Nat. Prod. Rep. 2009, 26, 526–536. [Google Scholar] [CrossRef]
- Teichert, R.W.; Olivera, B.M. Natural products and ion channel pharmacology. Future Med. Chem. 2010, 2, 731–744. [Google Scholar] [CrossRef]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef]
- Bourne, Y.; Radic, Z.; Aráoz, R.; Talley, T.T.; Benoit, E.; Servent, D.; Taylor, P.; Molgó, J.; Marchot, P. Structural determinants in phycotoxins and AChBP conferring high affinity binding and nicotinic AChR antagonism. Proc. Natl. Acad. Sci. USA 2010, 107, 6076–6081. [Google Scholar] [CrossRef]
- Liao, J.W.; Kang, J.J.; Liu, S.H.; Jeng, C.R.; Cheng, Y.W.; Hu, C.M.; Tsai, S.F.; Wang, S.C.; Pang, V.F. Effects of cartap on isolated mouse phrenic nerve diaphragm and its related mechanism. Toxicol. Sci. 2000, 55, 453–459. [Google Scholar] [CrossRef]
- Tsuneki, H.; You, Y.; Toyooka, N.; Sasaoka, T.; Nemoto, H.; Dani, J.A.; Kimura, I. Marine alkaloids(−)-pictamine and (−)-lepadin B block neuronal nicotinic acetylcholine receptors. Biol. Pharm. Bull. 2005, 28, 611–614. [Google Scholar] [CrossRef]
- Rucktooa, P.; Smit, A.B.; Sixma, T.K. Insight in nAChR subtype selectivity from AChBP crystal structures. Biochem. Pharmacol. 2009, 78, 777–787. [Google Scholar] [CrossRef]
- Yakel, J.L. Gating of nicotinic ACh receptors: Latest insights into ligand binding and function. J. Physiol. 2010, 588, 597–602. [Google Scholar] [CrossRef]
- Dutertre, S.; Ulens, C.; Büttner, R.; Fish, A.; van Elk, R.; Kendel, Y.; Hopping, G.; Alewood, P.F.; Schroeder, C.; Nicke, A.; et al. AChBP-targeted α-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J. 2007, 26, 3858–3867. [Google Scholar] [CrossRef]
- Ulens, C.; Akdemir, A.; Jongejan, A.; van Elk, R.; Bertrand, S.; Perrakis, A.; Leurs, R.; Smit, A.B.; Sixma, T.K.; Bertrand, D.; et al. Use of acetylcholine binding protein in the search for novel α7 nicotinic receptor ligands. In silico docking, pharmacological screening, and X-ray analysis. J. Med. Chem. 2009, 52, 2372–2383. [Google Scholar] [CrossRef]
- Akdemir, A.; Rucktooa, P.; Jongejan, A.; van Elk, R.; Bertrand, S.; Sixma, T.K.; Bertrand, D.; Smit, A.B.; Leurs, R.; de Graaf, C.; et al. Acetylcholine binding protein (AChBP) as template for hierarchical in silico screening procedures to identify structurally novel ligands for the nicotinic receptors. Bioorg. Med. Chem. 2011, 19, 6107–6119. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Denisenko, V.A.; Stonik, V.A.; Milgrom, Y.M.; Rashkes, Y.V. Rhizochalin, a novel secondary metabolite of mixed biosynthesis from the sponge Rhizochalina incrustata. Tetrahedron Lett. 1989, 30, 6581–6584. [Google Scholar] [CrossRef]
- Molinski, T.F.; Makarieva, T.N.; Stonik, V.A. (−)-Rhizochalin is a dimeric enantiomorphic (2R)-sphingolipid. Absolute configuration on pseudo-C2V-symmetric bic-2-amino-3-alkanols by CD. Angew. Chem. Int. Ed. Engl. 2000, 39, 4076–4079. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Ilyin, S.G.; Stonik, V.A.; Lyssenko, K.A.; Denisenko, V.A. Pibocin, the first ergoline marine alkaloid from the far-eastern ascidian Eudistoma sp. Tetrahedron Lett. 1999, 40, 1591–1594. [Google Scholar] [CrossRef]
- Guzii, A.G.; Makarieva, T.N.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Dyshlovoy, S.A.; Krasokhin, V.B.; Stonik, V.A. Monanchocidin: A new apoptosis-inducing polycyclic guanidine alkaloid from the marine sponge Monanchora pulchra. Org. Lett. 2010, 12, 4292–4295. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Stonik, V.A.; Dmitrenok, A.S.; Grebnev, B.B.; Isakov, V.V.; Rebachyk, N.M. Varacin and three new marine antimicrobial polysulfides from the Far-Eastern ascidian Polycitor sp. J. Nat. Prod. 1995, 58, 254–258. [Google Scholar] [CrossRef]
- Utkina, N.K.; Makarchenko, A.E.; Denisenko, V.A.; Dmitrenok, P.S. Zyzzyanone A, a novel pyrrolo[3,2-f]indole alkaloid from the Australian marine sponge Zyzzya fuliginosa. Tetrahedron Lett. 2004, 45, 7491–7494. [Google Scholar] [CrossRef]
- Radisky, D.C.; Radisky, E.S.; Barrows, L.R.; Copp, B.R.; Kramer, R.A.; Ireland, C.M. Novel cytotoxic topoisomerase-II inhibiting pyrroloiminoquinones from Fijian sponges of the genus Zyzzya. J. Am. Chem. Soc. 1993, 115, 1632–1638. [Google Scholar] [CrossRef]
- Carney, J.R.; Scheuer, P.J.; Kelly-Borges, M. Makaluvamine-G, a cytotoxic pigment from an Indonesian sponge Histodermella sp. Tetrahedron 1993, 49, 8483–8486. [Google Scholar] [CrossRef]
- Utkina, N.K.; Fedoreev, S.A.; Maximov, O.B. Nitrogen-containing metabolites from marine sponge Acanthella cartery. Chem. Nat. Comp. 1984, 20, 511–512. [Google Scholar] [CrossRef]
- Sharma, G.M.; Buyer, J.S.; Pomerantz, M.W. Characterization of a yellow compound isolated from the marine sponge Phakellia flabellata. J. Chem. Soc. Chem. Commun. 1980, 10, 435–436. [Google Scholar]
- Vagias, C.; Tsitsimpikow, C.; Rapti, T.; Roussis, V. 1,1-Dimethyl-[2,2′]bipyridyldium salt from bivalve Callista chione. Nat. Prod. Lett. 2000, 14, 425–428. [Google Scholar]
- Braekman, J.C.; Daloze, D.; Tavares, R.; Hajdu, E.; van Soest, R.W.M. Novel polycyclic guanidine alkaloids from two marine sponges of the genus Monanchora. J. Nat. Prod. 2000, 63, 193–196. [Google Scholar] [CrossRef]
- Santalova, E.A.; Denisenko, V.A.; Stonik, V.A. Dibromotyrosine and histamine derivatives from the tropical marine sponge Aplysina sp. Nat. Prod. Commun. 2010, 5, 377–382. [Google Scholar]
- Ciminiello, P.; Costantino, V.; Fattorusso, E.; Magno, S.; Mangoni, A.; Pansini, M. Chemistry of Verongida sponges. Constituents of the Caribbean sponge Aplysina fistularis forma fulva. J. Nat. Prod. 1994, 57, 705–712. [Google Scholar] [CrossRef]
- Jeong, S.-J.; Inagaki, M.; Higuchi, R.; Miyamoto, T.; Ono, M.; Kuwano, M.; van Soest, R.W.M. 1,3-Dimethylisoguaninium, an antiangiogenic purine analog from the sponge Amphimedon paraviridis. Chem. Pharm. Bull. Chem, 51, 731–733. [Google Scholar]
- Shubina, L.K.; Kalinovsky, A.I.; Fedorov, S.A.; Radchenko, O.S.; Denisenko, V.A.; Dmitrenok, P.S.; Dyshlovoy, S.A.; Krasokhin, V.B.; Stonik, V.A. Aaptamine alkaloids from the Vietnamese sponge Aaptos sp. Nat. Prod. Commun. 2009, 4, 1085–1088. [Google Scholar]
- Nakamura, H.; Kobayashi, J.; Ohizumi, H. Isolation and structure of aaptamine a novel heteroaromatic substance possessing alpha-blocking activity from the sea sponge Aaptos aaptos. Tetrahedron Lett. 1982, 23, 5555–5558. [Google Scholar] [CrossRef]
- Celie, P.H.; van Rossum-Fikkert, S.E.; van Dijk, W.J.; Brejc, K.; Smit, A.B.; Sixma, T.K. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 2004, 41, 907–914. [Google Scholar] [CrossRef]
- Shahsavar, A.; Kastrup, J.S.; Nielsen, E.O.; Kristensen, J.L.; Gajhede, M.; Balle, T. Crystal structure of Lymnaea stagnalis AChBP complexed with the potent nAChR antagonist DHβE suggests a unique mode of antagonism. PLoS One 2012, 7, e40757. [Google Scholar]
- Bourne, Y.; Talley, T.T.; Hansen, S.B.; Taylor, P.; Marchot, P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J. 2005, 24, 1512–1522. [Google Scholar] [CrossRef]
- Ihara, M.; Okajima, T.; Yamashita, A.; Oda, T.; Hirata, K.; Nishiwaki, H.; Morimoto, T.; Akamatsu, M.; Ashikawa, Y.; Kuroda, S.; et al. Crystal structures of Lymnaea stagnalis AChBP in complex with neonicotinoid insecticides imidacloprid and clothianidin. Invertebr. Neurosci. 2008, 8, 71–81. [Google Scholar] [CrossRef]
- Wonnacott, S.; Barik, J. Nicotinic ACh Receptors; Tocris Bioscience Scientific Review Series Tocris Reviews NO. 28; Tocris Bioscience: Bristol, UK, 2007. [Google Scholar]
- Kasheverov, I.E.; Zhmak, M.N.; Vulfius, C.A.; Gorbacheva, E.V.; Mordvintsev, D.Y.; Utkin, Y.N.; van Elk, R.; Smit, A.B.; Tsetlin, V.I. α-Conotoxin analogs with additional positive charge show increased selectivity towards Torpedo californica and some neuronal subtypes of nicotinic acetylcholine receptors. FEBS J. 2006, 273, 4470–4481. [Google Scholar] [CrossRef]
- Kasheverov, I.E.; Zhmak, M.N.; Fish, A.; Rucktooa, P.; Khruschov, A.Yu.; Osipov, A.V.; Ziganshin, R.H.; D’hoedt, D.; Bertrand, D.; Sixma, T.K.; et al. Interaction of α-conotoxin ImII and its analogs with nicotinic receptors and acetylcholine-binding proteins: Additional binding sites on Torpedo receptor. J. Neurochem. 2009, 111, 934–944. [Google Scholar] [CrossRef]
- Kasheverov, I.E.; Zhmak, M.N.; Khruschov, A.Y.; Tsetlin, V.I. Design of new α-conotoxins: From computer modeling to synthesis of potent cholinergic compounds. Mar. Drugs 2011, 9, 1698–1714. [Google Scholar] [CrossRef]
- Kasheverov, I.E.; Utkin, Y.N.; Tsetlin, V.I. Naturally occurring and synthetic peptides acting on nicotinic acetylcholine receptors. Curr. Pharm. Des. 2009, 15, 2430–2452. [Google Scholar] [CrossRef]
- Utkin, Y.N.; Kukhtina, V.V.; Kryukova, E.V.; Chiodini, F.; Bertrand, D.; Methfessel, C.; Tsetlin, V.I. “Weak toxin” from Naja kaouthia is a nontoxic antagonist of α7 and muscle-type nicotinic acetylcholine receptors. J. Biol. Chem. 2001, 276, 15810–15815. [Google Scholar] [CrossRef]
- Lyukmanova, E.N.; Shulepko, M.A.; Buldakova, S.L.; Kasheverov, I.E.; Shenkarev, Z.O.; Reshetnikov, R.V.; Filkin, S.Y.; Kudryavtsev, D.S.; Ojomoko, L.O.; Kryukova, E.V.; et al. Water-soluble LYNX1 residues important for interaction with muscle-type and/or neuronal nicotinic receptors. J. Biol. Chem. 2013, 288, 15888–15899. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar]
- Brams, M.; Pandya, A.; Kuzmin, D.; van Elk, R.; Krijnen, L.; Yakel, J.L.; Tsetlin, V.; Smit, A.B.; Ulens, C. A structural and mutagenic blueprint for molecular recognition of strychnine and d-tubocurarine by different Cys-loop receptors. PLoS Biol. 2011, 9, e1001034. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kudryavtsev, D.; Makarieva, T.; Utkina, N.; Santalova, E.; Kryukova, E.; Methfessel, C.; Tsetlin, V.; Stonik, V.; Kasheverov, I. Marine Natural Products Acting on the Acetylcholine-Binding Protein and Nicotinic Receptors: From Computer Modeling to Binding Studies and Electrophysiology. Mar. Drugs 2014, 12, 1859-1875. https://doi.org/10.3390/md12041859
Kudryavtsev D, Makarieva T, Utkina N, Santalova E, Kryukova E, Methfessel C, Tsetlin V, Stonik V, Kasheverov I. Marine Natural Products Acting on the Acetylcholine-Binding Protein and Nicotinic Receptors: From Computer Modeling to Binding Studies and Electrophysiology. Marine Drugs. 2014; 12(4):1859-1875. https://doi.org/10.3390/md12041859
Chicago/Turabian StyleKudryavtsev, Denis, Tatyana Makarieva, Natalia Utkina, Elena Santalova, Elena Kryukova, Christoph Methfessel, Victor Tsetlin, Valentin Stonik, and Igor Kasheverov. 2014. "Marine Natural Products Acting on the Acetylcholine-Binding Protein and Nicotinic Receptors: From Computer Modeling to Binding Studies and Electrophysiology" Marine Drugs 12, no. 4: 1859-1875. https://doi.org/10.3390/md12041859
APA StyleKudryavtsev, D., Makarieva, T., Utkina, N., Santalova, E., Kryukova, E., Methfessel, C., Tsetlin, V., Stonik, V., & Kasheverov, I. (2014). Marine Natural Products Acting on the Acetylcholine-Binding Protein and Nicotinic Receptors: From Computer Modeling to Binding Studies and Electrophysiology. Marine Drugs, 12(4), 1859-1875. https://doi.org/10.3390/md12041859