Exploring the Chemodiversity and Biological Activities of the Secondary Metabolites from the Marine Fungus Neosartorya pseudofischeri

Abstract
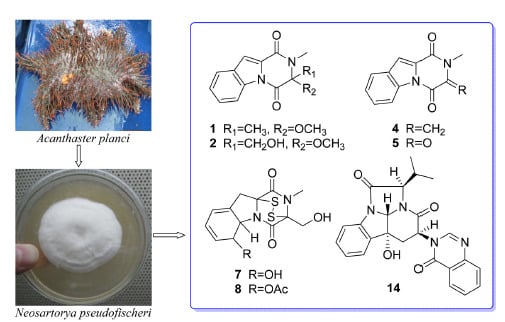
:
1. Introduction
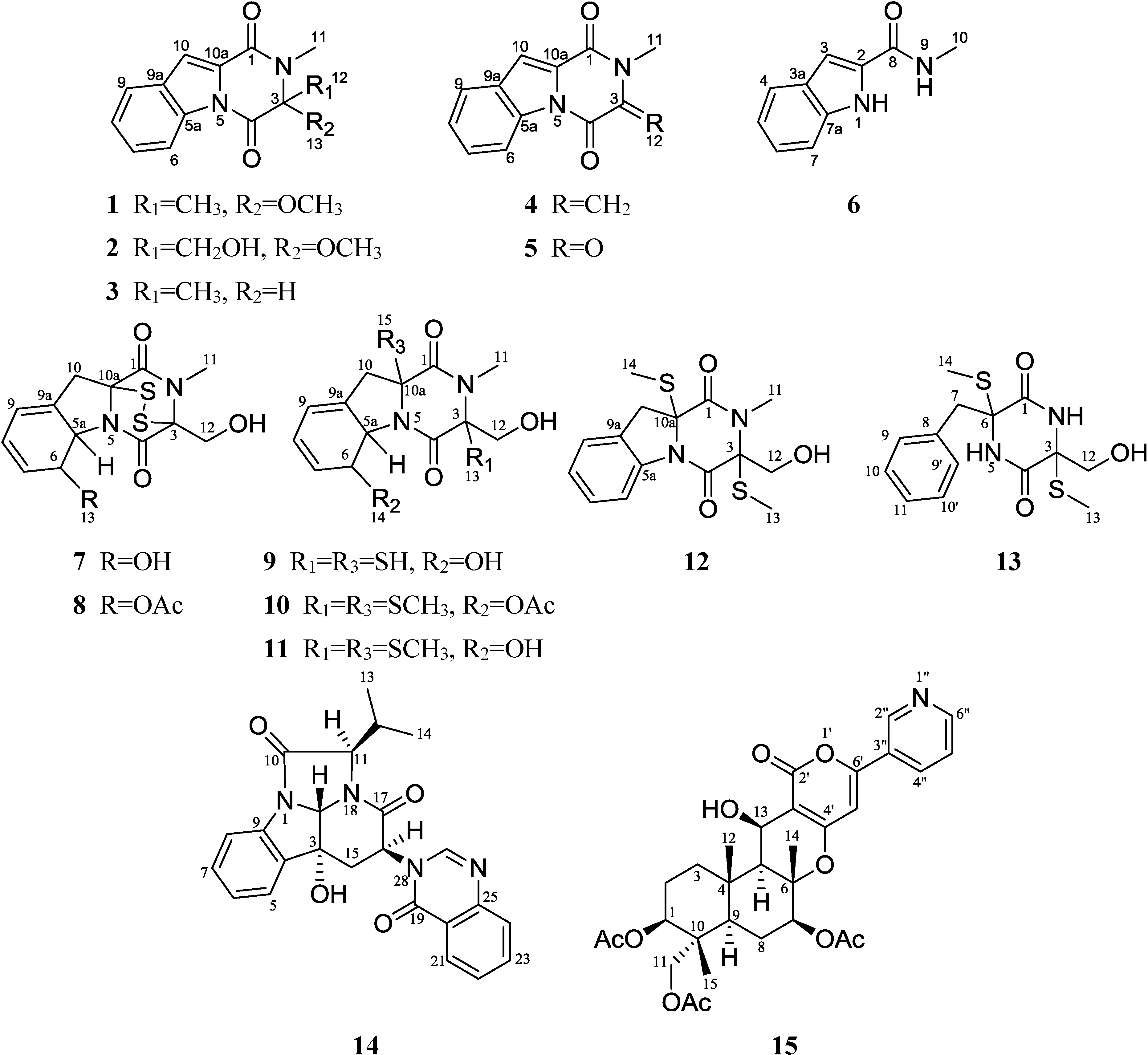
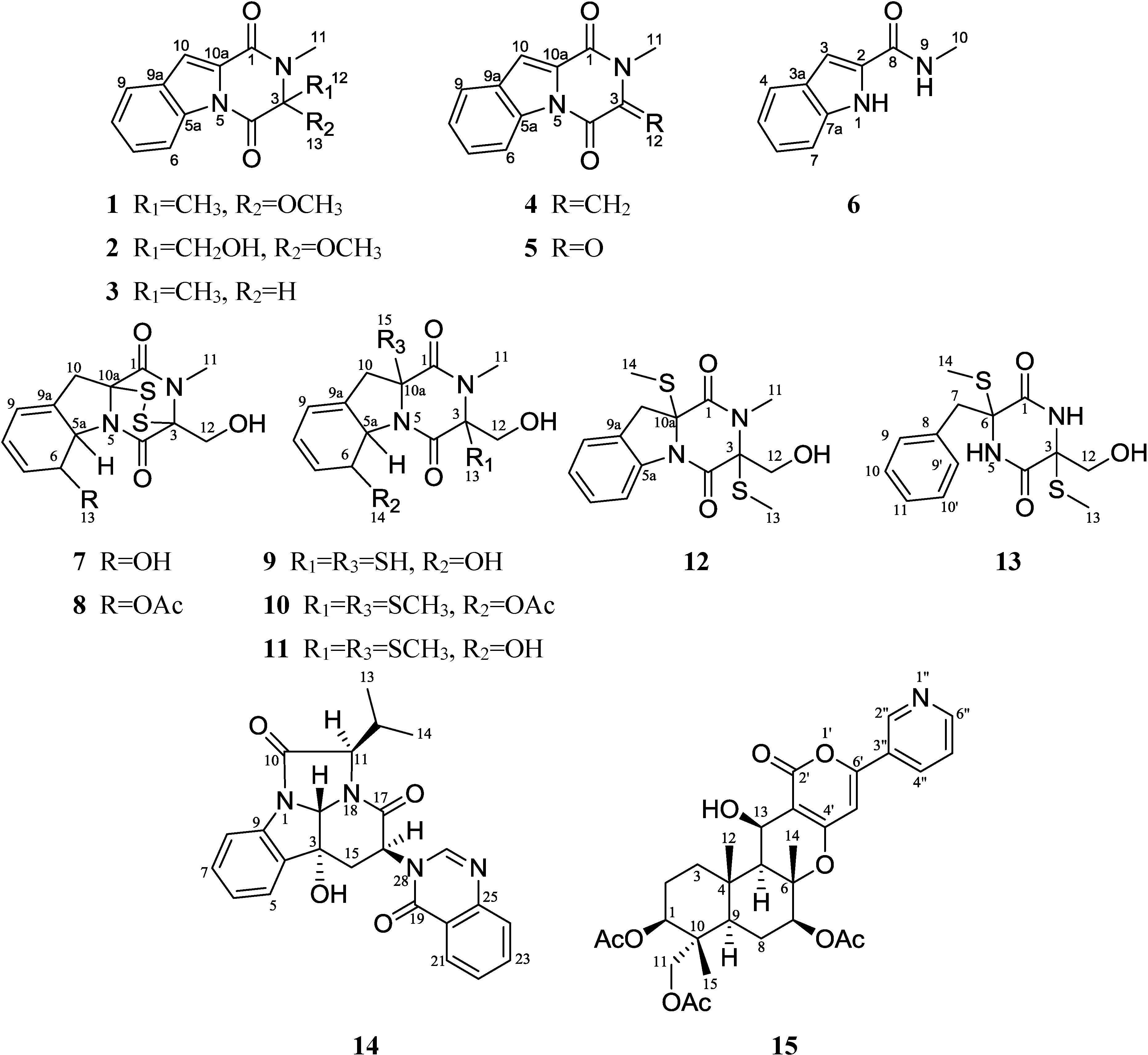
2. Results and Discussion
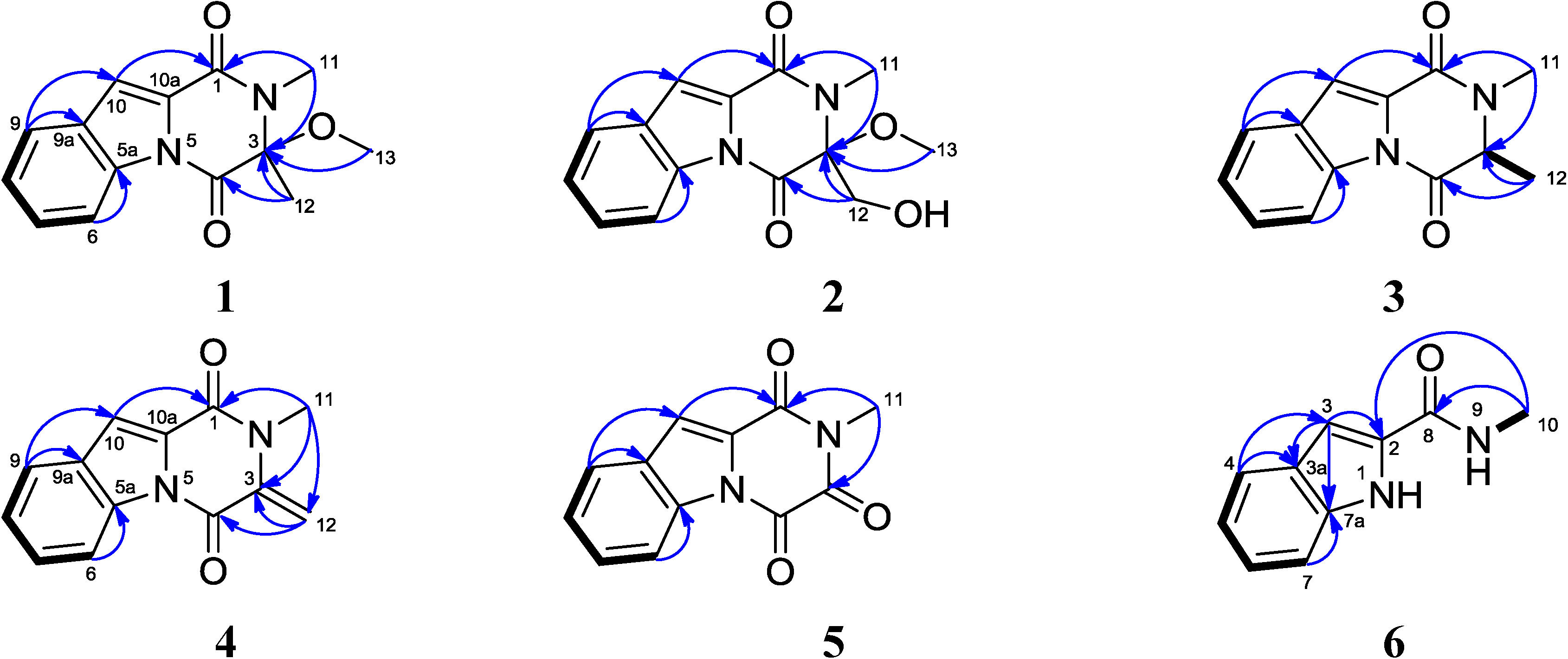
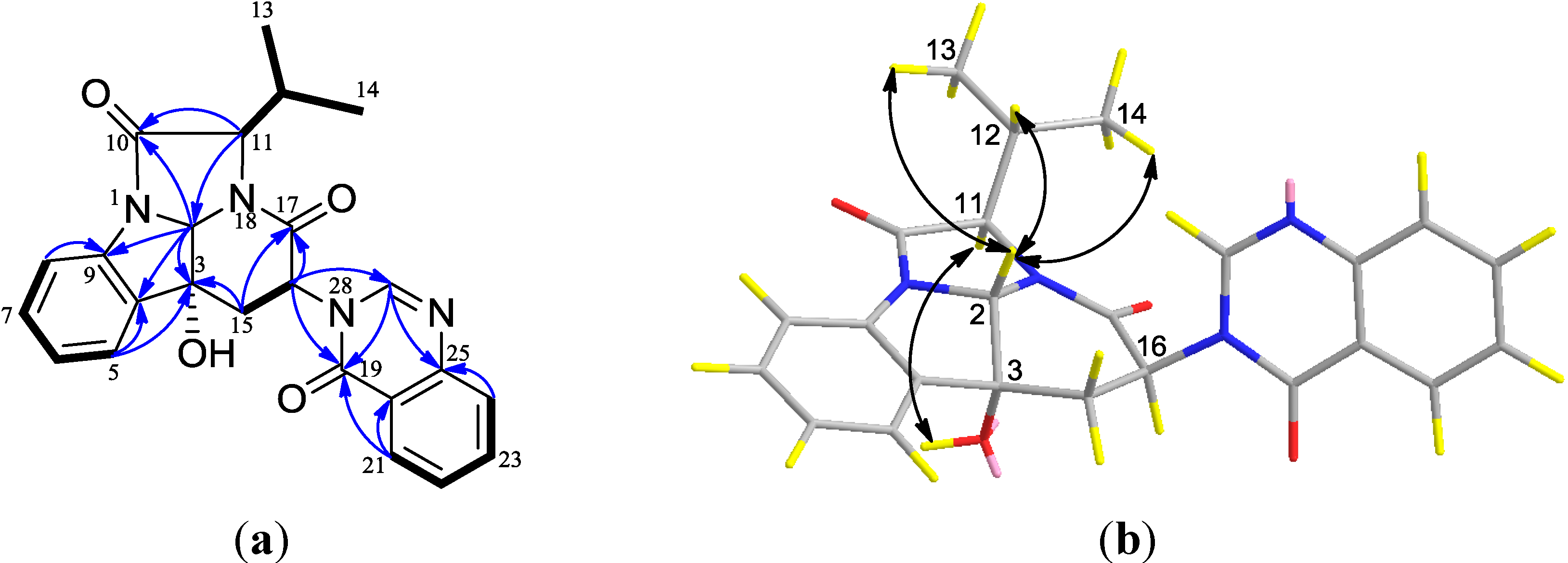
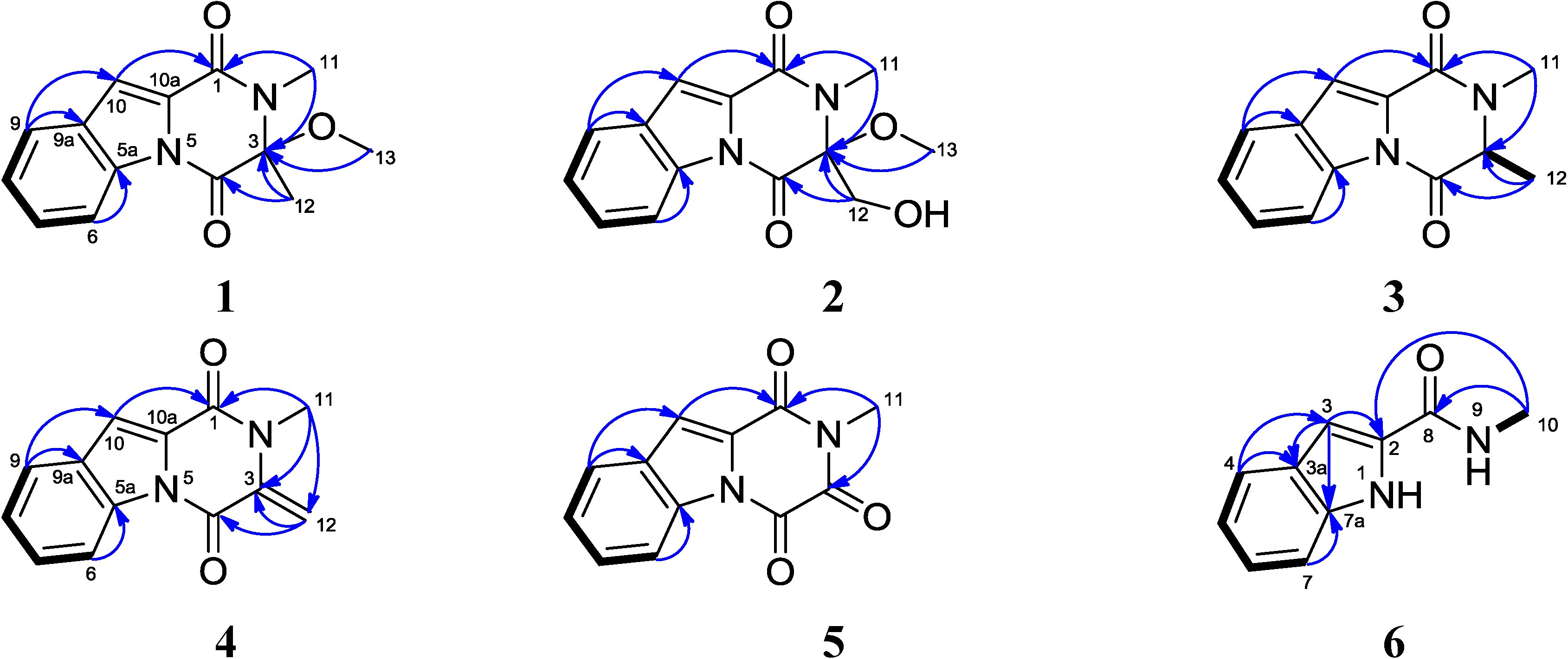
2.1. Structural Elucidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| δC | Type | δH, mult., (J in Hz) | δC | Type | δH, mult., (J in Hz) | δC | Type | δH, mult., (J in Hz) | |
| 1 | 156.7 | C | 158.5 | C | 156.2 | C | |||
| 2 | N | N | N | ||||||
| 3 | 90.7 | C | 93.6 | C | 60.2 | CH | 4.33, q (7.2) | ||
| 4 | 163.7 | C | 162.9 | C | 165.5 | C | |||
| 5 | N | N | N | ||||||
| 5a | 129.2 | C | 128.8 | C | 129.2 | C | |||
| 6 | 116.9 | CH | 8.49, dd (8.0, 0.8) | 116.6 | CH | 8.47, dd (7.6, 0.8) | 116.5 | CH | 8.43, d (8.0) |
| 7 | 128.2 | CH | 7.55, ddd (8.0, 8.0, 0.8) | 128.1 | CH | 7.49, ddd (7.6,7.6, 0.8) | 127.8 | CH | 7.51, dd (8.0, 8.0) |
| 8 | 125.8 | CH | 7.43, ddd (8.0, 8.0, 0.8) | 125.7 | CH | 7.26, ddd (7.6, 7.6, 0.8) | 125.4 | CH | 7.40, dd (8.0, 8.0) |
| 9 | 122.7 | CH | 7.72, dd (8.0, 0.8) | 122.5 | CH | 7.32, dd (7.6, 0.8) | 122.5 | CH | 7.70, d (8.0) |
| 9a | 134.8 | C | 134.5 | C | 134.8 | C | |||
| 10 | 114.9 | CH | 7.50, s | 115.0 | CH | 7.19, s | 114.1 | CH | 7.44, s |
| 10a | 127.8 | C | 127.5 | C | 128.5 | C | |||
| 11 | 26.6 | CH3 | 3.13, s | 26.5 | CH3 | 3.14, s | 31.8 | CH3 | 3.16, s |
| 12 | 25.3 | CH3 | 1.81, s | 64.9 | CH2 | 4.17, d (11.6);4.02, d (11.6) | 19.8 | CH3 | 1.71, d (7.2) |
| 13 | 52.0 | OCH3 | 3.20, s | 52.0 | OCH3 | 3.24, s | |||
| 12-OH | 2.04, brs | ||||||||
| Position | 4 a | 5 b | Position | 6 c | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | Type | δH, mult., (J in Hz) | δC | Type | δH, mult., (J in Hz) | δC | Type | δH, mult., (J in Hz) | ||
| 1 | 154.4 | C | 157.0 | C | 1 | NH | 11.04, brs | |||
| 2 | N | N | 2 | 132.8 | C | |||||
| 3 | 137.7 | C | 156.8 | C | 3 | 102.7 | CH | 7.07, s | ||
| 4 | 154.9 | C | 149.9 | C | 3a | 137.8 | C | |||
| 5 | N | N | 4 | 122.5 | CH | 7.61, d (8.0) | ||||
| 5a | 128.9 | C | 128.3 | C | 5 | 124.5 | CH | 7.22, dd (8.0, 8.0) | ||
| 6 | 117.0 | CH | 8.50, d (8.0) | 115.8 | CH | 8.32, d (8.0) | 6 | 120.9 | CH | 7.06, dd (8.0, 8.0) |
| 7 | 128.2 | CH | 7.53, dd (8.0, 8.0) | 129.0 | CH | 7.63, d (8.0, 8.0) | 7 | 113.2 | CH | 7.58, d (8.0) |
| 8 | 125.5 | CH | 7.40, dd (8.0, 8.0) | 125.7 | CH | 7.47, d (8.0, 8.0) | 7a | 128.9 | C | |
| 9 | 122.8 | CH | 7.72, d (8.0) | 123.9 | CH | 7.88, d (8.0) | 8 | 163.0 | C | |
| 9a | 135.6 | C | 135.4 | C | 9 | NH | 7.83, brs | |||
| 10 | 115.3 | CH | 7.51, s | 116.3 | CH | 7.72, s | 10 | 26.4 | CH3 | 2.97, s |
| 10a | 127.6 | C | 127.7 | C | ||||||
| 11 | 29.6 | CH3 | 3.41, s | 26.6 | CH3 | 3.22, s | ||||
| 12 | 106.0 | CH2 | 6.15, s; 5.25, s | |||||||
| Position | 8 a | 9 b | ||||
|---|---|---|---|---|---|---|
| δC | Type | δH, mult., (J in Hz) | δC | Type | δH, mult., (J in Hz) | |
| 1 | 168.4 | C | 169.7 | C | ||
| 2 | N | N | ||||
| 3 | 78.2 | C | 78.6 | C | ||
| 4 | 165.9 | C | 168.1 | C | ||
| 5 | N | N | ||||
| 5a | 64.9 | CH | 5.36, d (14.1) | 70.6 | CH | 4.77, d (13.2) |
| 6 | 74.3 | CH | 5.82, d (14.1) | 72.7 | CH | 5.04, d (13.2) |
| 7 | 128.6 | CH | 5.93, m | 129.8 | CH | 5.93, m |
| 8 | 124.9 | CH | 5.93, m | 123.1 | CH | 5.88, m |
| 9 | 120.7 | CH | 5.60, d (7.5) | 120.7 | CH | 5.75, m |
| 9a | 131.7 | C | 130.0 | C | ||
| 10 | 41.4 | CH2 | 3.25, d (15.0); | CH2 | 3.28, d (16.2); | |
| 3.08, d (15.0) | 3.05, d (16.2) | |||||
| 10a | 77.6 | C | 77.2 | C | ||
| 11 | 29.0 | CH3 | 3.10, s | 28.9 | CH3 | 3.15, s |
| 12 | 62.4 | CH2 | 4.32, d (12.3) | 62.3 | CH2 | 4.39, d (12.0) |
| 4.00, d (12.3) | 4.06, d (12.0) | |||||
| 13 | 170.1 | COCH3 | ||||
| 21.4 | COCH3 | 2.17, s | ||||
| Position | 12 a | Position | 13 b | ||||
|---|---|---|---|---|---|---|---|
| δC | Type | δH, mult., (J in Hz) | δC | Type | δH, mult., (J in Hz) | ||
| 1 | 166.0 | C | 1 | 165.2 | C | ||
| 2 | N | 2 | NH | 8.95, brs | |||
| 3 | 71.7 | C | 3 | 65.8 | C | ||
| 4 | 161.8 | C | 4 | 165.0 | C | ||
| 5 | N | 5 | NH | 8.40, brs | |||
| 5a | 128.9 | C | 6 | 65.6 | C | ||
| 6 | 127.9 | CH | 8.03, d (8.0) | 7 | 43.3 | CH2 | 3.33, d (6.0); 3.31, d (6.0) |
| 7 | 126.2 | CH | 7.31, dd (8.0, 8.0) | 8 | 135.0 | C | |
| 8 | 125.2 | CH | 7.19, dd (8.0, 8.0) | 9/9′ | 130.0 | CH | 7.20, m |
| 9 | 118.1 | CH | 7.30, d (8.0) | 10/10′ | 127.6 | CH | 7.20, m |
| 9a | 140.6 | C | 11 | 126.5 | CH | 7.20, m | |
| 10 | 39.6 | CH2 | 4.50, d (12.0); 3.96, d (12.0) | 12 | 64. 8 | CH2 | 3.52, d (18.0); 3.00, d (18.0) |
| 10a | 70.8 | C | 13 | 12.8 | CH3 | 2.11, s | |
| 11 | 28.9 | CH3 | 3.20, s | 14 | 13.5 | CH3 | 2.29, s |
| 12 | 63.9 | CH2 | 3.62, d (16.8); 3.51, d (16.8) | ||||
| 13 | 14.5 | CH3 | 2.32, s | ||||
| 14 | 13.8 | CH3 | 2.24, s | ||||
| Position | δC | Type | δH, mult., (J in Hz) |
|---|---|---|---|
| 1 | N | ||
| 2 | 84.8 | CH | 5.95, s |
| 3 | 74.8 | C | |
| 4 | 135.2 | C | |
| 5 | 124.7 | CH | 7.43, brd (7.6) |
| 6 | 126.4 | CH | 7.15, dd (7.6, 7.6) |
| 7 | 130.9 | CH | 7.35, dd (7.6, 7.6) |
| 8 | 115.6 | CH | 7.53, d (7.6) |
| 9 | 138.5 | C | |
| 10 | 171.4 | C | |
| 11 | 70.3 | CH | 4.43, d (8.4) |
| 12 | 30.1 | CH | 2.35, dqq (8.4, 6.4, 6.4) |
| 13 | 20.1 | CH3 | 1.17, d (6.4) |
| 14 | 19.1 | CH3 | 1.21, d (6.4) |
| 15 | 36.5 | CH2 | 2.49, dd (15.2, 5.2); 3.23, dd (15.2, 4.8) |
| 16 | 56.8 | CH | 5.11, dd (5.2, 4.8) |
| 17 | 163.6 | C | |
| 18 | N | ||
| 19 | 161.7 | C | |
| 20 | 121.4 | C | |
| 21 | 127.4 | CH | 8.20, dd (7.6, 0.8) |
| 22 | 127.8 | CH | 7.50, dd (7.6, 7.6) |
| 23 | 134.9 | CH | 7.71, ddd (7.6, 7.6, 0.8) |
| 24 | 126.1 | CH | 7.64, d (7.6) |
| 25 | 144.4 | C | |
| 26 | N | ||
| 27 | 147.5 | CH | 8.61, s |
| 28 | N | ||
| 3-OH | 4.09, brs |
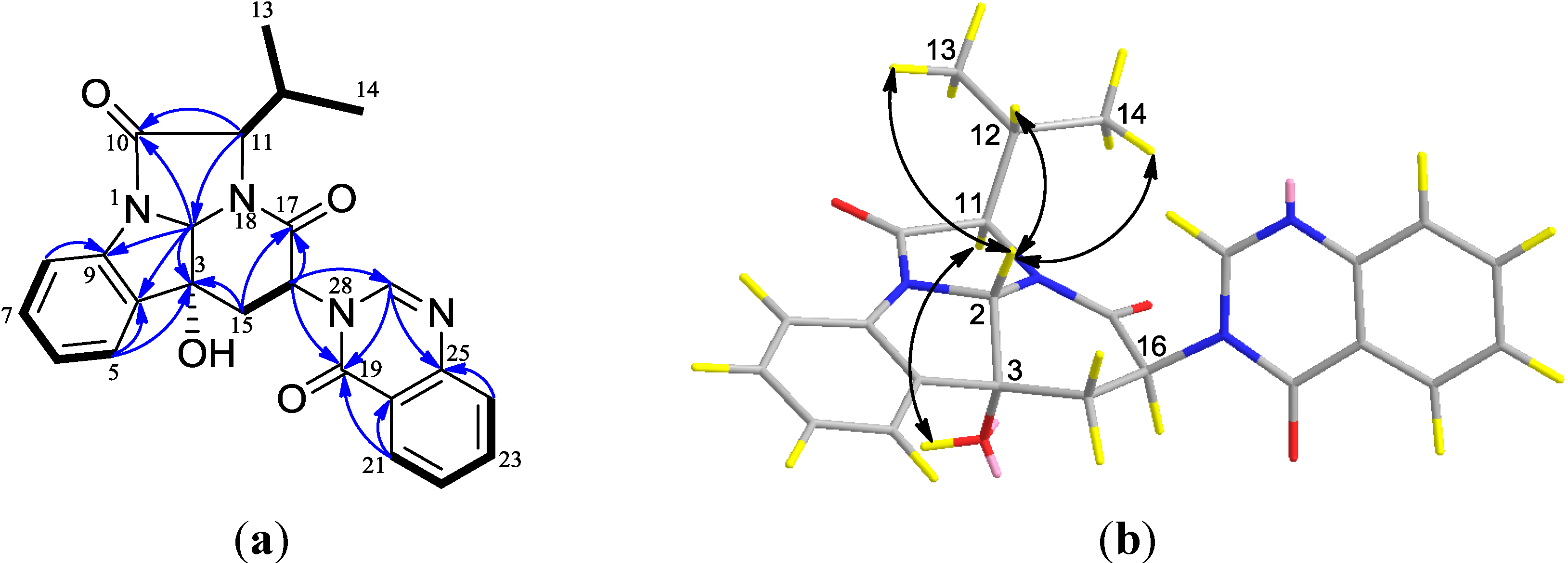
| Position | In CDCl3 | In Acetone-d6 | |||
|---|---|---|---|---|---|
| δC | Type | δH, mult., (J in Hz) | δC | δH, mult., (J in Hz) | |
| 1 | 73.5 | CH | 5.00, dd (10.8, 4.8) | 74.5 | 4.99, dd (10.8, 4.8) |
| 2 | 22.7 | CH2 | 1.83, m; 1.90, m | 23.7 | 1.85, m; 1.89, m |
| 3 | 36.2 | CH2 | 1.37, m; 2.16, m | 36.8 | 1.49, m; 2.17, ddd (12.6, 4.8, 4.8) |
| 4 | 37.9 | C | 38.8 | ||
| 5 | 54.7 | CH | 1.53, d (4.2) | 55.1 | 1.67, d (4.8) |
| 6 | 83.3 | C | 83.9 | ||
| 7 | 77.7 | CH | 4.78, dd (12.0, 4.8) | 79.0 | 4.81, dd (11.4, 4.8) |
| 8 | 25.2 | CH2 | 1.63, ddd (12.0, 12.0, 11.4); 1.78, dd (11.4, 4.8) | 26.0 | 1.74, m; 1.83, ddd (10.8, 5.4, 5.4) |
| 9 | 45.4 | CH | 1.58, d (12.0) | 46.2 | 1.72, d (12.0) |
| 10 | 40.3 | C | 41.3 | ||
| 11 | 64.8 | CH2 | 3.77, d (11.4); 3.70, d (11.4) | 65.6 | 3.76, d (12.0); 3.72, d (12.0) |
| 12 | 17.4 | CH3 | 1.43, s | 17.9 | 1.53, s |
| 13 | 60.1 | CH | 4.99, d (4.2) | 60.5 | 5.00, d (4.8) |
| 14 | 16.2 | CH3 | 1.69, s | 16.8 | 1.76, s |
| 15 | 13.2 | CH3 | 0.83, s | 13.6 | 0.93, s |
| 2′ | 163.6 | C | 163.3 | ||
| 3′ | 103.3 | C | 104.2 | ||
| 4′ | 162.0 | C | 162.7 | ||
| 5′ | 99. 9 | CH | 6.48, s | 100.1 | 6.71, s |
| 6′ | 156.4 | C | 158.1 | ||
| 2″ | 145.4 | CH | 9.05, s | 147.7 | 9.08, s |
| 3″ | 127.9 | C | 128.5 | ||
| 4″ | 134.3 | CH | 8.20, d (8.4) | 133.8 | 8.24, ddd (7.8, 1.8, 1.8) |
| 5″ | 124.2 | CH | 7.51, brd ( 8.4) | 124.7 | 7.53, dd (7.8, 4.8) |
| 6″ | 149.8 | CH | 8.72, s | 152.2 | 8.69, d (4.8) |
| 1-O-CO-CH3 | 170.5 | C | 170.6 | ||
| 7-O-CO-CH3 | 170.0 | C | 170.3 | ||
| 11-O-CO-CH3 | 170.9 | C | 170.8 | ||
| 1-O-CO-CH3 | 21.1 | CH3 | 2.07, s | 21.1 | 2.02, s |
| 7-O-CO-CH3 | 21.2 | CH3 | 2.15, s | 21.2 | 2.10, s |
| 11-O-CO-CH3 | 20.8 | CH3 | 2.03, s | 20.7 | 2.00, s |
| 13-OH | OH | 3.06, brs | 2.90, brs | ||
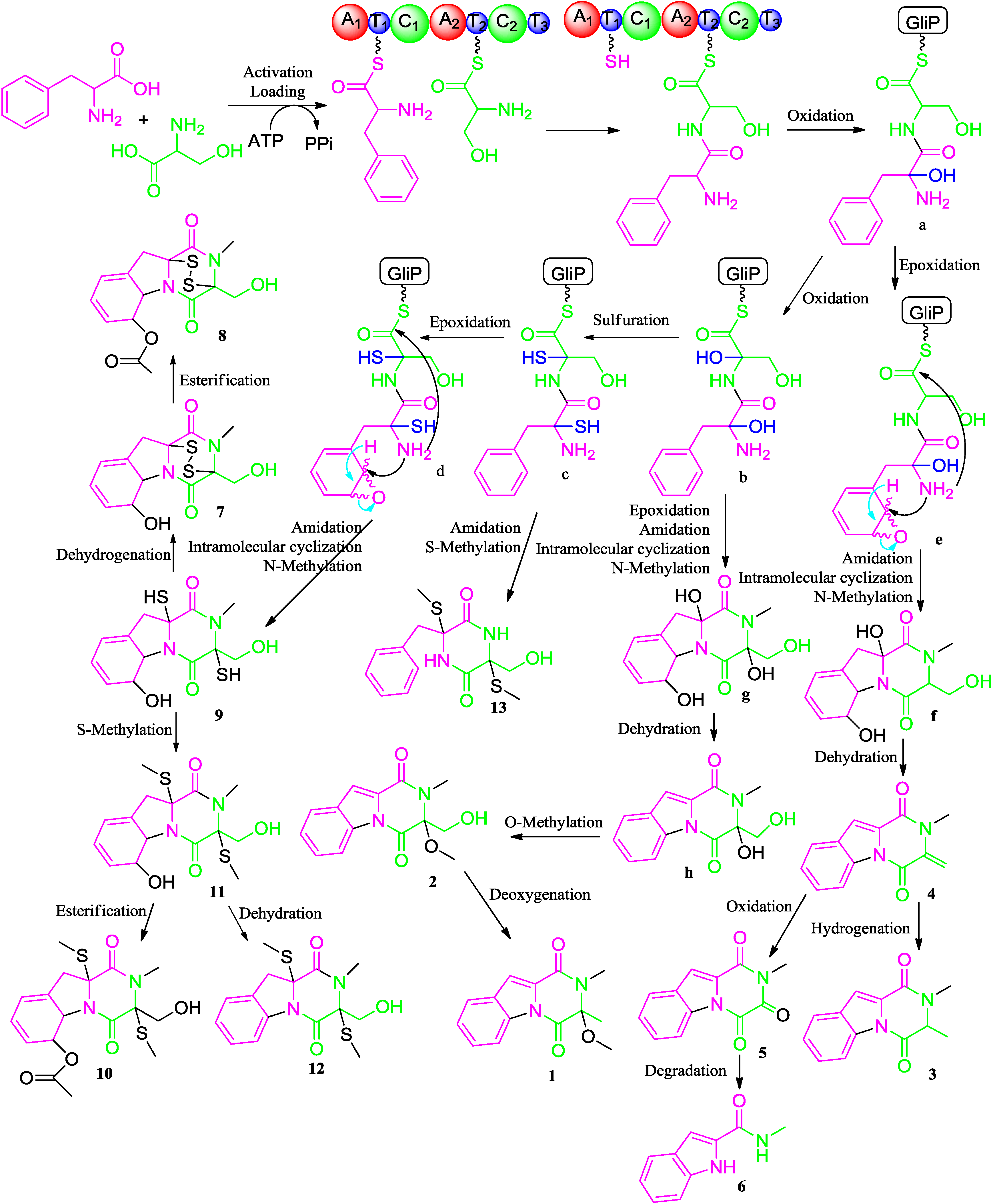
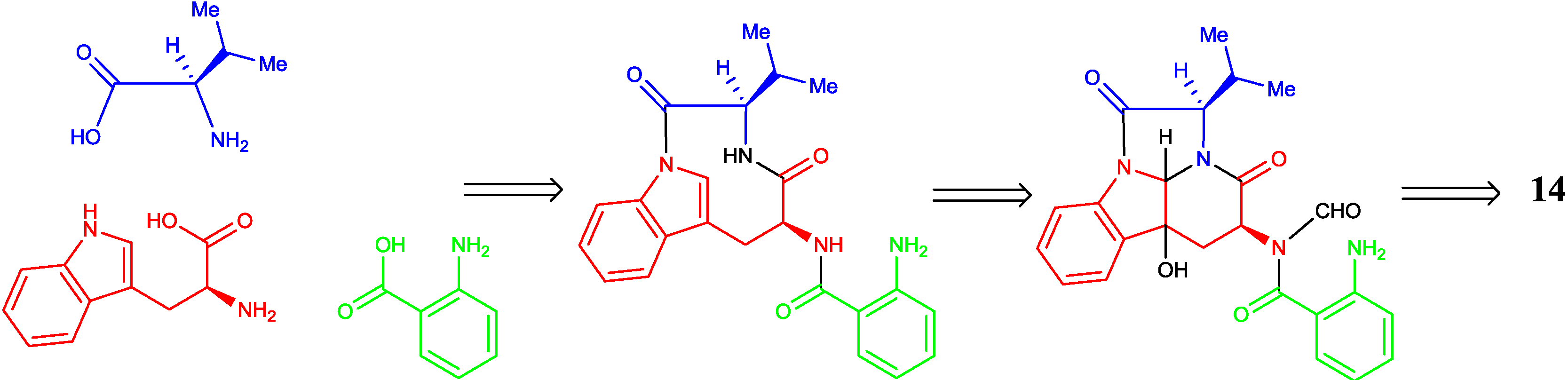
2.2. Proposed Biosynthetic Pathway
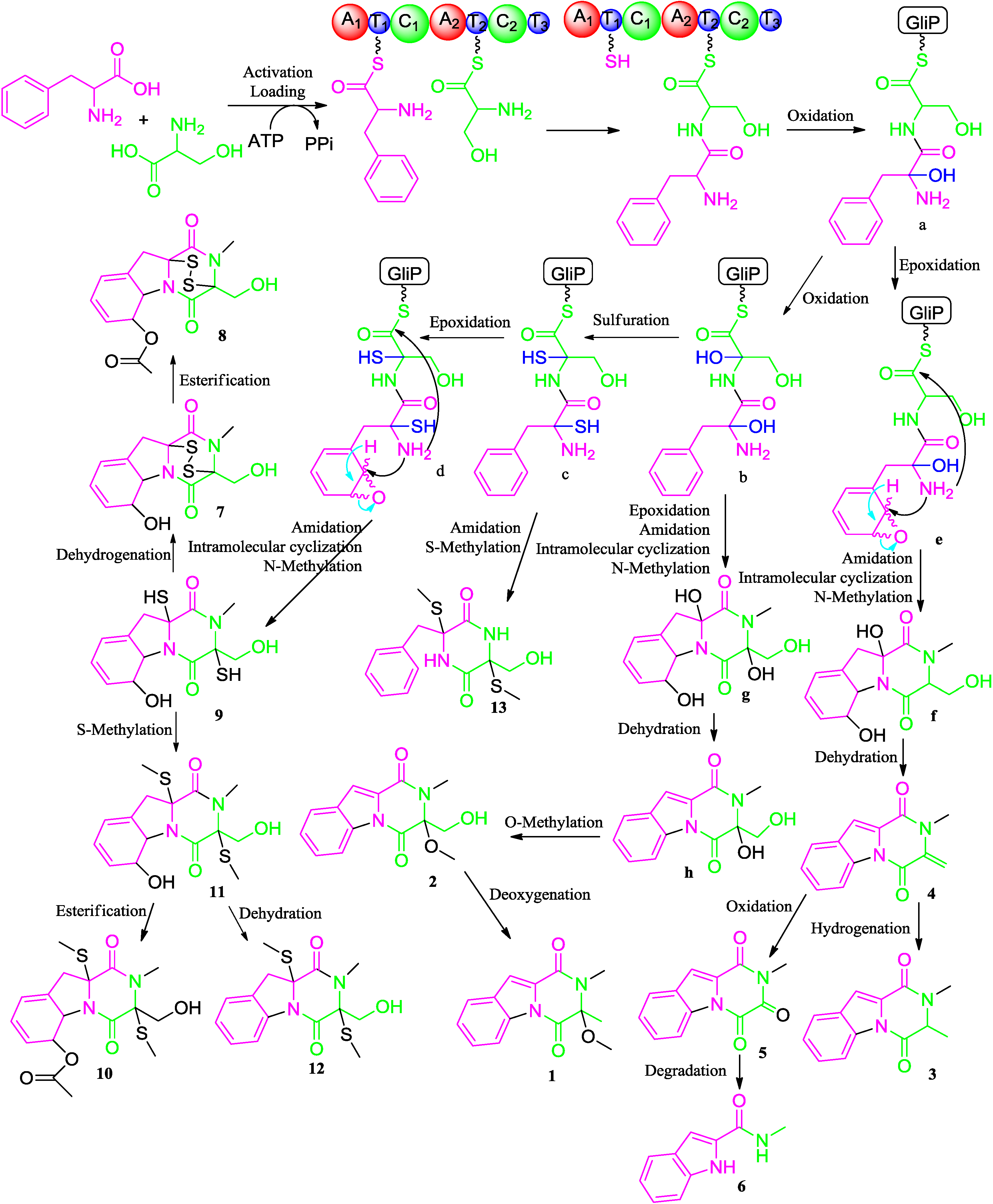
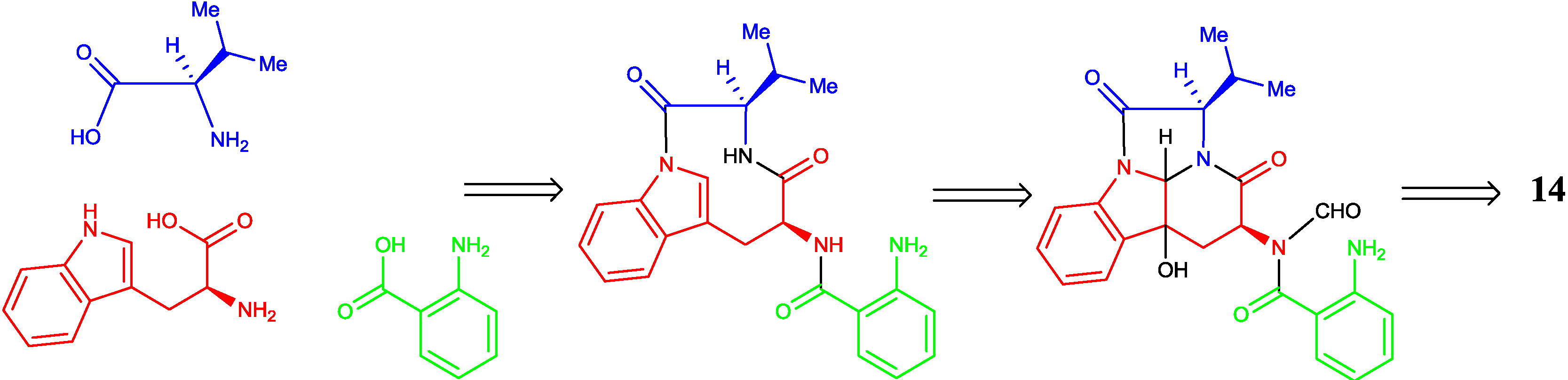
2.3. Biological Activity
| Compound | Staphylococcus aureus (ATCC29213) | Staphylococcus aureus (R3708) | Escherichia coli (ATCC25922) |
|---|---|---|---|
| 4 | 283.11 | 70.70 | >1,132 |
| 7 | 12.20 | 1.53 | 24.53 |
| 8 | 86.91 | 21.73 | >695.65 |
| 9 | 48.78 | 1.52 | 97.56 |
| Vancomycin | 0.84 | 2.01 | |
| Ampicillin sodium | 8.07 | 129.24 | 6.73 |
| Compound | Cell line | ||
|---|---|---|---|
| 293 | HCT-116 | RKO | |
| 4 | 30.10 ± 0.90 | 10.34 ± 1.41 | 33.56 ± 1.22 |
| 5 | >50 | >50 | >50 |
| 7 | 1.58 ± 0.03 | 1.24 ± 0.38 | 0.80 ± 0.20 |
| 8 | 4.49 ± 0.24 | 0.89 ± 0.04 | 1.24 ± 0.18 |
| 9 | 1.26 ± 0.04 | 0.43 ± 0.04 | 0.41 ± 0.07 |
| 10 | >50 | >50 | >50 |
| 11 | 16.39 ± 0.38 | 8.59 ± 0.96 | 10.32 ± 0.04 |
| 12 | >50 | >50 | >50 |
| 13 | >50 | >50 | >50 |
| 5-Fluorouracil | 2.04 ± 0.22 | 45.86 ± 4.58 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Strain and Culture Method
3.3. Extraction and Isolation
3.4. Antibacterial Activity Assay
3.5. Cytotoxicity Assay
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, Y.; Zheng, J.K.; Liu, P.P.; Wang, W.; Zhu, W.M. Three new compounds from Aspergillus terreus PT06-2 grown in a high salt medium. Mar. Drugs 2011, 9, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Li, D.Y.; Hua, H.M.; Ma, E.L.; Li, Z.L. Caryophyllene sesquiterpenes from the marine-derived fungus Ascotricha sp. ZJ-M-5 by the one strain−many compounds strategy. J. Nat. Prod. 2014, 77, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- James, W.; Suzanne, M.S.; David, J.N.; Karina, M.Z. Tetramic acid analogues produced by coculture of Saccharopolyspora erythraea with Fusarium pallidoroseum. J. Nat. Prod. 2014, 77, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.; Olivier, S.; Nadine, B.; Michel, M.; Katia, G.; Jean-Luc, W. De novo production of metabolites by fungal co-culture of Trichophyton rubrum and Bionectria ochroleuca. J. Nat. Prod. 2013, 76, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Wei, H.J.; Zhu, T.J.; Li, D.H.; Lin, Z.J.; Gu, Q.Q. Three new cytochalasins from the marine-derived fungus Spicaria elegans KLA03 by supplementing the cultures with l- and d-Tryptophan. Chem. Biodivers. 2011, 8, 887–894. [Google Scholar] [CrossRef]
- Christopher, T.W.; Stuart, W.H.; Brian, D.A.; Gao, X.; Tang, Y. Short pathways to complexity generation: Fungal peptidyl alkaloid multicyclic scaffolds from anthranilate building blocks. ACS Chem. Biol. 2013, 8, 1366–1382. [Google Scholar] [CrossRef] [PubMed]
- Jeremy, B.; Nida, M.; Whittney, N.B.; Lacey, H.; Lindsey, N.S.; Tina, M.; Dennis, E.K.; Betty, B.; Alberto, van O.; Bill, B. Epigenetic tailoring for the production of anti-infective cytosporones from the marine fungus Leucostoma persoonii. Mar. Drugs 2012, 10, 762–774. [Google Scholar] [CrossRef]
- Teigo, A.; Takashi, Y.; Yoshiteru, O. Aromatic polyketide productionin cordyceps indigotica, an entomopathogenic fungus, induced by exposure to a histone deacetylase inhibitor. Org. Lett. 2012, 14, 2006–2009. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Jiang, W.H.; Liang, W.L.; Huang, J.X.; Mo, Y.F.; Ding, Y.Q.; Lam, C.K.; Qian, X.J.; Zhu, X.F.; Lan, W.J. Induced marine fungus Chondrostereum sp. as a means of producing new sesquiterpenoids chondrosterins I and J by using glycerol as the carbon source. Mar. Drugs 2014, 12, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Chen, T.; Xie, Y.L.; Chen, W.D.; Zhu, X.F.; Lan, W.J. Isolation and structural elucidation of chondrosterins F-H from the marine fungus Chondrostereum sp. Mar. Drugs 2013, 11, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Xie, Y.L.; Xie, Z.L.; Chen, Y.; Lam, C.K.; Lan, W.J. Chondrosterins A–E, triquinane-type sesquiterpenoids from soft coral-associated fungus Chondrostereum sp. Mar. Drugs 2012, 10, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Lan, W.J.; Lam, C.K.; Yang, F.; Zhu, X.F. Hirsutane sesquiterpenoids from the marine- derived fungus Chondrostereum sp. Chem. Biodivers. 2011, 8, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.J.; Zhao, Y.; Xie, Z.L.; Liang, L.Z.; Shao, W.Y.; Zhu, L.P.; Yang, D.P.; Zhu, X.F.; Li, H.J. Novel sorbicillin analogues from the marine fungus Trichoderma sp. associated with the seastar Acanthaster planci. Nat. Prod. Commun. 2012, 7, 1337–1340. [Google Scholar] [PubMed]
- Zhao, Y.; Li, S.Q.; Li, H.J.; Lan, W.J. Lanostane triterpenoids from the fungus Ceriporia lacerate. Chem. Nat. Compd. 2013, 49, 653–656. [Google Scholar] [CrossRef]
- Xie, Z.L.; Li, H.J.; Wang, L.Y.; Liang, W.L.; Liu, W.; Lan, W.J. Trichodermaerin, a new lactone from the marine fungus Trichoderma erinaceum associated with the sea star Acanthaster planci. Nat. Prod. Commun. 2013, 8, 67–68. [Google Scholar] [PubMed]
- Lan, W.J.; Liu, W.; Liang, W.L.; Xu, Z.; Le, X.; Xu, J.; Lam, C.K.; Yang, D.P.; Li, H.J.; Wang, L.Y. Pseudaboydins A and B: Novel isobenzofuranone derivatives from marine fungus Pseudallescheria boydii associated with starfish Acanthaster planci. Mar. Drugs 2014, 12, 4188–4199. [Google Scholar]
- Peterson, S.W. Neosartorya pseudofischeri sp. nov. and its relationship to other species in Aspergillus section Fumigati. Mycol. Res. 1992, 96, 547–554. [Google Scholar]
- Yukihiro, A.; Hideaki, K.; Rie, O.; Arika, Y.; Hiroshi, M.; Hiroyuki, O. Azaspirene: A novel angiogenesis inhibitor containing a 1-oxa-7-azaspiro[4.4]non-2-ene-4,6-dione skeleton produced by the fungus Neosartorya sp. Org. Lett. 2002, 4, 2845–2848. [Google Scholar]
- Marco, M.; Anna, A.; Veronique, M.; Angela, B.; Alessio, C.; Laetitia, M.Y.B.; Maurizio, V.; Alexander, K.; Robert, K.; Antonio, E. Fischerindoline, a pyrroloindole sesquiterpenoid isolated from Neosartorya pseudofischeri, with in vitro growth inhibitory activity in human cancer cell lines. Tetrahedron 2013, 69, 7466–7470. [Google Scholar]
- John, R.J.; James, B.B. Gliotoxin. X. dethiogliotoxin and related compounds. J. Am. Chem. Soc. 1953, 75, 2103–2109. [Google Scholar]
- Ajay, K.B.; Das, K.G.; Funke, P.T.; Irene, K.; Shukla, O.P.; Khanchandani, K.S.; Suhadolnik, R.J. Biosynthetic studies on gliotoxin using stable isotopes and mass spectral methods. J. Am. Chem. Soc. 1968, 90, 1038–1041. [Google Scholar] [CrossRef]
- Johnson, J.R.; Hasbrouck, R.B.; Dutcher, J.D.; Bruce, W.F. Gliotoxin. V. The Structure of certain indole derivatives related to gliotoxin. J. Am. Chem. Soc. 1945, 67, 423. [Google Scholar] [CrossRef]
- Ali, M.S.; Shannon, J.S.; Taylor, A. Isolation and structures of 1,2,3,4-tetrahydro-l,4-dioxopyrazino[l,2-a]-indoles from cultures of Penicillium terlikowskii. J. Chem. Soc. C 1968, 2044–2048. [Google Scholar] [CrossRef]
- Mourad, K. Gliotoxin: Uncommon 1H couplings and revised 1H and 13C-NMR assignments. J. Nat. Prod. 1990, 53, 717–719. [Google Scholar] [CrossRef]
- Katharine, R.W.; Joseline, R.; Ang, K.H.; Karen, T.; Jennifer, E.C.; James, M.; Phillip, C. Assessing the trypanocidal potential of natural and semi-synthetic diketopiperazines from two deep water marine-derived fungi. Bioorg. Med. Chem. 2010, 18, 2566–2574. [Google Scholar] [CrossRef]
- Didier, V.D.; Junji, I.; Kazuro, S.; Hong, Y.; Hideo, T.; Satoshi, O. Inhibition of farnesyl-protein transferase by gliotoxin and acetylgliotoxin. J. Antibiot. 1992, 45, 1802–1805. [Google Scholar] [CrossRef]
- Daniel, H.S.; Nicole, R.; Thorsten, H.; Peter, H.; Axel, A.B.; Christian, H. Transannular disulfide formation in gliotoxin biosynthesis and its role in self-resistance of the human pathogen Aspergillus fumigates. J. Am. Chem. Soc. 2010, 132, 10136–10141. [Google Scholar] [CrossRef] [PubMed]
- Afiyatullov, S.S.; Kalinovskii, A.I.; Pivkin, M.V.; Dmitrenok, P.S.; Kuznetsova, T.A. Alkaloids from the marine isolate of the fungus Aspergillus fumigatus. Chem. Nat. Compd. 2005, 41, 236–238. [Google Scholar] [CrossRef]
- Kirby, G.W.; Robins, D.J.; Sefton, M.A.; Talekar, R.R. Biosynthesis of bisdethiobis(methylthio) gliotoxin, a new metabolite of Gliocladium Deliquescens. J. Chem. Soc. Perkin Trans Ⅰ. 1980, 119–121. [Google Scholar] [CrossRef]
- Zhao, W.Y.; Zhu, T.J.; Han, X.X.; Fan, G.T.; Liu, H.B.; Zhu, W.M.; Gu, Q.Q. A new gliotoxin analogue from a marine-derived fungus Aspergillus fumigatus Fres. Nat. Prod. Res. 2009, 23, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Atsuki, O.; Kaoru, I.; Masaki, O.; Hiroshi, T.; Satoshi, O.; Tohru, N. Total synthesis of pyripyropene A. Tetrahedron 2011, 67, 8195–8203. [Google Scholar] [CrossRef]
- Mootz, H.D.; Marahiel, M.A. Biosynthetic systems for nonribosomal peptide antibiotic assembly. Curr. Opin. Chem. Biol. 1997, 1, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Balibar, C.J.; Walsh, C.T. GliP, a multimodular nonribosomal peptide synthetase in Aspergillus fumigatus, makes the diketopiperazine scaffold of gliotoxin. Biochemistry 2006, 45, 15029–15038. [Google Scholar] [CrossRef] [PubMed]
- Amnat, E.; Anake, K.; Céline, B.; Véronique, M.; Leka, M.; Florence, L.; Artur, S.; Robert, K.; Werner, H. Secondary metabolites from a culture of the fungus Neosartorya pseudofischeri and their In vitro activity in human cancer cells. Planta Med. 2012, 78, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Jiao, R.H.; Xu, S.; Liu, J.Y.; Ge, H.M.; Ding, H.; Xu, C.; Zhu, H.L.; Tan, R.X. Chaetominine, a cytotoxic alkaloid produced by endophytic Chaetomium sp. IFB-E015. Org. Lett. 2006, 8, 5709–5712. [Google Scholar] [CrossRef] [PubMed]
- Bryan, K.S.; Yeung, Y.N.; Robin, B.K.; John, R.C.; Wesley, Y.Y.; Paul, J.S.; Michelle, K.B. The Kapakahines, cyclic peptides from the marine sponge Cribrochalina olemda. J. Org. Chem. 1996, 61, 7168–7173. [Google Scholar]
- Casey, J.T.; O’Cleirigh, C.; Walsh, P.K.; O’Shea, D.G. Development of a robust microtiter plate-based assay method for assessment of bioactivity. J. Microbiol. Methods 2004, 58, 327–334. [Google Scholar] [CrossRef] [PubMed]
- GraphPad Prism 6, GraphPad Software, Inc.: La Jolla, CA, USA, 2014.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, W.-L.; Le, X.; Li, H.-J.; Yang, X.-L.; Chen, J.-X.; Xu, J.; Liu, H.-L.; Wang, L.-Y.; Wang, K.-T.; Hu, K.-C.; et al. Exploring the Chemodiversity and Biological Activities of the Secondary Metabolites from the Marine Fungus Neosartorya pseudofischeri. Mar. Drugs 2014, 12, 5657-5676. https://doi.org/10.3390/md12115657
Liang W-L, Le X, Li H-J, Yang X-L, Chen J-X, Xu J, Liu H-L, Wang L-Y, Wang K-T, Hu K-C, et al. Exploring the Chemodiversity and Biological Activities of the Secondary Metabolites from the Marine Fungus Neosartorya pseudofischeri. Marine Drugs. 2014; 12(11):5657-5676. https://doi.org/10.3390/md12115657
Chicago/Turabian StyleLiang, Wan-Ling, Xiu Le, Hou-Jin Li, Xiang-Ling Yang, Jun-Xiong Chen, Jun Xu, Huan-Liang Liu, Lai-You Wang, Kun-Teng Wang, Kun-Chao Hu, and et al. 2014. "Exploring the Chemodiversity and Biological Activities of the Secondary Metabolites from the Marine Fungus Neosartorya pseudofischeri" Marine Drugs 12, no. 11: 5657-5676. https://doi.org/10.3390/md12115657