Calcarides A–E, Antibacterial Macrocyclic and Linear Polyesters from a Calcarisporium Strain



Abstract
:1. Introduction
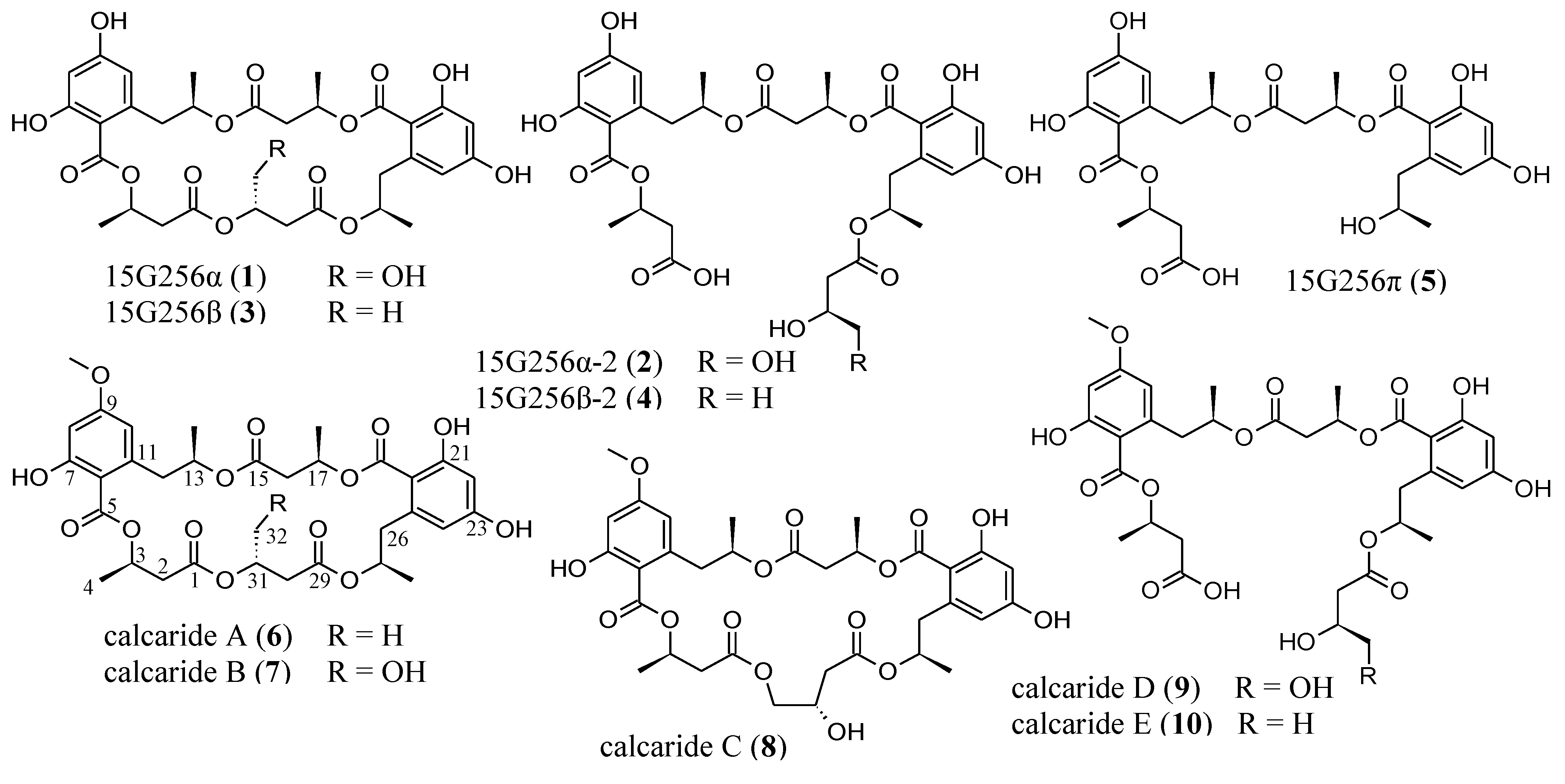
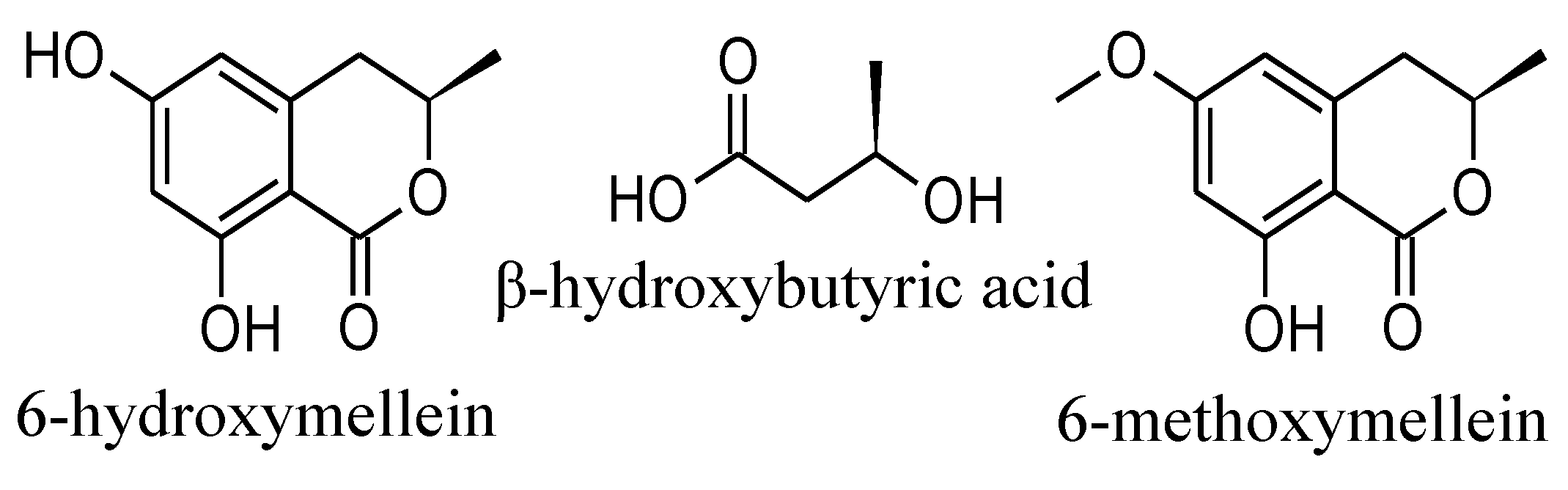
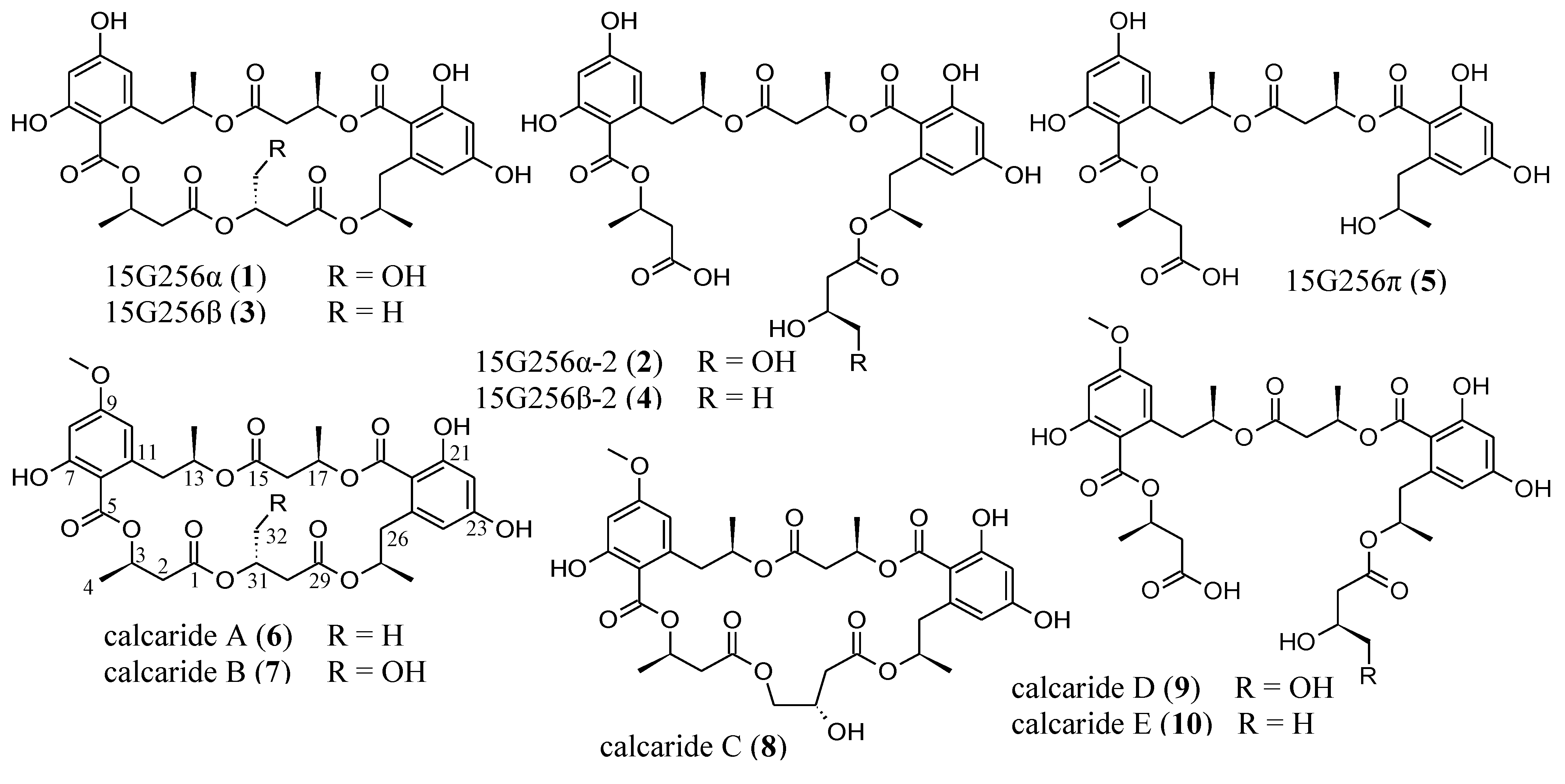
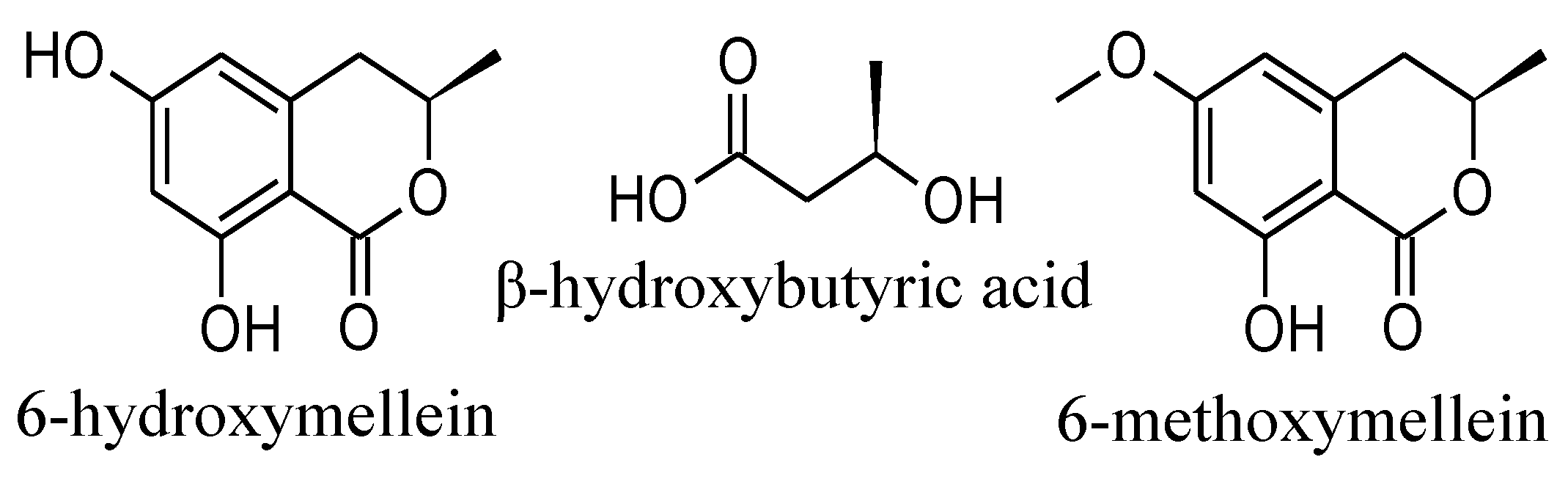
2. Results and Discussion
2.1. Isolation and Structure Elucidation

{kind=link}
{kind=link}
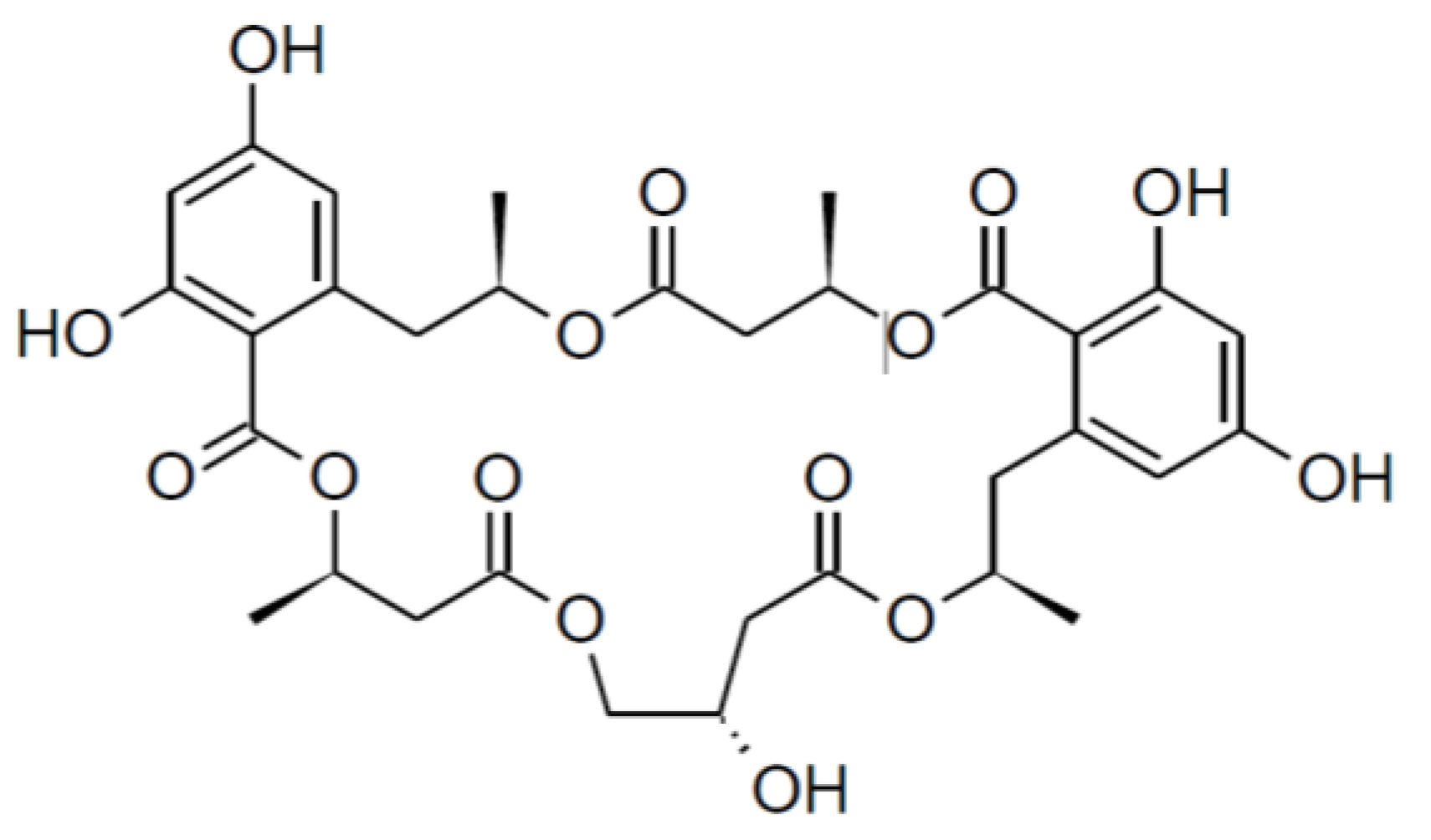{kind=link}
| Position | δC, Type | δH, Mult. (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 170.09, C | |||
| 2a | 40.6, CH2 | 2.91, dd (6.7, 16.3) | 2b, 3 | 1, 3, 4 |
| 2b | 2.74, dd (6.4, 16.3) | 2a, 3 | 1, 3, 4 | |
| 3 | 70.1, CH a | 5.56, m | 2a, 2b, 4 | 1, 2, 4, 5 |
| 4 | 20.0, CH3 | 1.450, d (6.4) | 3 | 1, 2, 3 |
| 5 | 171.0, C | |||
| 6 | 106.9, C | |||
| 7 | 165.5, C | |||
| 8 | 100.4, CH | 6.36, d (2.6) | 10 | 5, 6, 7, 9, 10 |
| 9 | 164.8, C | |||
| OCH3 | 55.8, CH3 | 3.82, s | 9 | |
| 10 | 111.9, CH | 6.44, d (2.6) | 8 | 5, 6, 8, 9, 12 |
| 11 | 142.9, C | |||
| 12a | 41.7, CH2 | 3.547, dd (6.8, 13.3) | 12b, 13 | 6, 10, 11, 13, 14 |
| 12b | 2.98, dd (7.5, 13.3) | 12a, 13 | 6, 10, 11, 13, 14 | |
| 13 | 73.1, CH | 5.05, m b | 12a, 12b, 14 | 11, 12, 14, 15 |
| 14 | 19.7, CH3 | 1.19, d (6.2) | 13 | 12, 13 |
| 15 | 170.12, C | |||
| 16a | 41.02, CH2 | 2.90, dd (6.3, 15.8) | 16b, 17 | 15, 17, 18 |
| 16b | 2.72, dd (7.1, 15.8) | 16a, 17 | 15, 17, 18 | |
| 17 | 70.0, CH a | 5.52, m | 16a, 16b, 18 | 15, 16, 18, 19 |
| 18 | 20.1, CH3 | 1.454, d (6.4) | 17 | 15, 16, 17 |
| 19 | 170.9, C | |||
| 20 | 105.6, C | |||
| 21 | 165.9, C | |||
| 22 | 102.6, CH | 6.30, d (2.5) | 24 | 19, 20, 21, 23, 24 |
| 23 | 163.1, C | |||
| 24 | 113.3, CH | 6.35, d (2.5) | 22 | 19, 20, 22, 23, 26 |
| 25 | 143.3, C | |||
| 26a | 42.4, CH2 | 3.553, dd (5.7, 12.9) | 26b, 27 | 20, 24, 25, 27, 28 |
| 26b | 2.78, dd (8.6, 12.9) | 26a, 27 | 20, 24, 25, 27, 28 | |
| 27 | 72.5, CH | 5.01, m b | 26a, 26b, 28 | 25, 26, 28, 29 |
| 28 | 19.5, CH3 | 1.14, d (6.2) | 27 | 26, 27 |
| 29 | 170.2, C | |||
| 30a | 41.01, CH2 | 2.63 c | 30b, 31 | 29, 31, 32 |
| 30b | 2.63 c | 30a, 31 | 29, 31, 32 | |
| 31 | 68.4, CH | 5.28, m | 30a, 30b, 32 | 1, 29, 30, 32 |
| 32 | 19.9, CH3 | 1.27, d (6.3) | 31 | 29, 30, 31 |
| Calcaride A (6) | Calcaride B (7) | Calcaride C (8) | |
|---|---|---|---|
| Position | δH, Mult. (J in Hz) | δH, Mult. (J in Hz) | δH, Mult. (J in Hz) |
| 2a | 2.91, dd (6.7, 16.3) | 2.88, dd (7.7, 16.6) | 2.878, dd (8.1, 16.2) |
| 2b | 2.74, dd (6.4, 16.3) | 2.73, d (5.3, 16.6) | 2.79, dd (4.8, 16.2) |
| 3 | 5.56, m | 5.56, m | 5.59, m |
| 4 | 1.450, d (6.4) | 1.42, d (6.3) | 1.44, d (6.4) |
| 8 | 6.36, d (2.6) | 6.30, d (2.6) | 6.31, d (2.6) |
| OCH3 | 3.82, s | 3.75, s | 3.74, s a |
| 10 | 6.44, d (2.6) | 6.36, d (2.6) | 6.35, d (2.6) |
| 12a | 3.547, dd (6.8, 13.3) | 3.34 b | 3.35 c |
| 12b | 2.98, dd (7.5, 13.3) | 2.96, dd (6.4, 13.8) | 2.99, dd (6.6, 14.0) |
| 13 | 5.05, m d | 5.07, m | 5.16, m |
| 14 | 1.19, d (6.2) | 1.20, d (6.2) | 1.22, d (6.2) |
| 16a | 2.90, dd (6.3, 15.8) | 2.81, dd (7.6, 15.8) | 2.85, dd (6.6, 16.0) |
| 16b | 2.72, dd (7.1, 15.8) | 2.67, dd (5.7, 15.8) | 2.69, dd (6.6, 16.0) |
| 17 | 5.52, m | 5.49, m | 5.48, m |
| 18 | 1.454, d (6.4) | 1.40, d (6.4) | 1.39, d (6.3) |
| 22 | 6.30, d (2.5) | 6.22, s | 6.21, d (2.5) |
| 24 | 6.35, d (2.5) | 6.22, s | 6.23, d (2.5) |
| 26a | 3.553, dd (5.7, 12.9) | 3.32 b | 3.31 c |
| 26b | 2.78, dd (8.6, 12.9) | 2.77, dd (8.1, 13.3) | 2.885, dd (6.9, 13.4) |
| 27 | 5.01, m d | 4.98, m | 5.03, m |
| 28 | 1.14, d (6.2) | 1.11, d (6.2) | 1.16, d (6.2) |
| 30a | 2.63 e | 2.68, dd (5.1, 16.7) | 2.51, dd (6.0, 15.3) |
| 30b | 2.63 e | 2.64, dd (8.1, 16.7) | 2.43, dd (7.2, 15.3) |
| 31 | 5.28, m | 5.28, m | 4.17, m |
| 32a | 1.27, d (6.3) | 3.65, dd (4.5, 11.9) | 4.12, dd (4.8, 11.1) |
| 32b | 3.58, dd (5.1, 11.9) | 4.08, dd (5.5, 11.1) |
| Calcaride A (6) | Calcaride B (7) | Calcaride C (8) | Calcaride D (9) | Calcaride E (10) | |
|---|---|---|---|---|---|
| Position | δC | δC | δC | δC | δC |
| 1 | 170.09 | 171.5 a | 171.6 | 173.9 b | 175.9 b |
| 2 | 40.6 | 41.0 | 41.2 | 41.8 | 43.3 |
| 3 | 70.1 c | 70.2 | 70.5 | 70.8 | 71.5 |
| 4 | 20.0 | 20.2 | 20.2 | 20.1 | 20.3 |
| 5 | 171.0 | 171.13 | 171.53 d | 171.5 e | 171.56 f |
| 6 | 106.9 | 108.7 | 108.2 | 107.2 | 107.3 |
| 7 | 165.5 | 164.6 | 165.08 g,h | 166.0 i | 166.0 j |
| 8 | 100.4 | 100.7 | 100.7 | 100.8 | 100.8 |
| 9 | 164.8 | 164.9 | 165.0 h | 165.0 i | 165.0 j |
| OCH3 | 55.8 | 55.8 | 55.8 k | 55.9 | 55.9 |
| 10 | 111.9 | 111.5 | 111.3 | 112.8 | 112.8 |
| 11 | 142.9 | 142.6 | 142.7 | 143.2 | 143.3 |
| 12 | 41.7 | 41.8 | 41.7 | 43.2 | 43.4 |
| 13 | 73.1 | 73.6 | 73.2 | 73.1 | 73.2 |
| 14 | 19.7 | 20.0 | 20.0 | 20.52 | 20.6 |
| 15 | 170.12 | 171.37 a | 171.3 d | 171.2 | 171.2 |
| 16 | 41.02 | 41.6 | 41.6 l | 41.8 | 41.8 |
| 17 | 70.0 c | 70.4 | 70.3 | 70.0 | 70.0 |
| 18 | 20.1 | 20.2 | 20.1 | 19.9 | 19.9 |
| 19 | 170.9 | 171.07 | 171.48 d | 171.6 e | 171.62 f |
| 20 | 105.6 | 106.7 | 107.3 | 105.8 | 105.7 |
| 21 | 165.9 | 165.4 | 165.14 g | 166.2 | 166.3 |
| 22 | 102.6 | 102.8 | 102.7 | 102.7 | 102.7 |
| 23 | 163.1 | 163.5 | 163.4 | 163.5 | 163.6 |
| 24 | 113.3 | 113.3 | 112.6 | 113.8 | 113.9 |
| 25 | 143.3 | 143.2 | 143.1 | 143.7 | 143.7 |
| 26 | 42.4 | 42.3 | 41.5 l | 43.5 | 43.6 |
| 27 | 72.5 | 73.5 | 73.6 | 72.7 | 72.6 |
| 28 | 19.5 | 19.5 | 19.8 | 20.49 | 20.6 |
| 29 | 170.2 | 171.43 a | 171.9 | 172.7 | 172.5 |
| 30 | 41.01 | 36.7 | 40.4 | 39.9 | 45.1 |
| 31 | 68.4 | 72.7 | 67.1 | 69.9 | 65.4 |
| 32 | 19.9 | 63.6 | 68.5 | 66.4 | 23.0 |
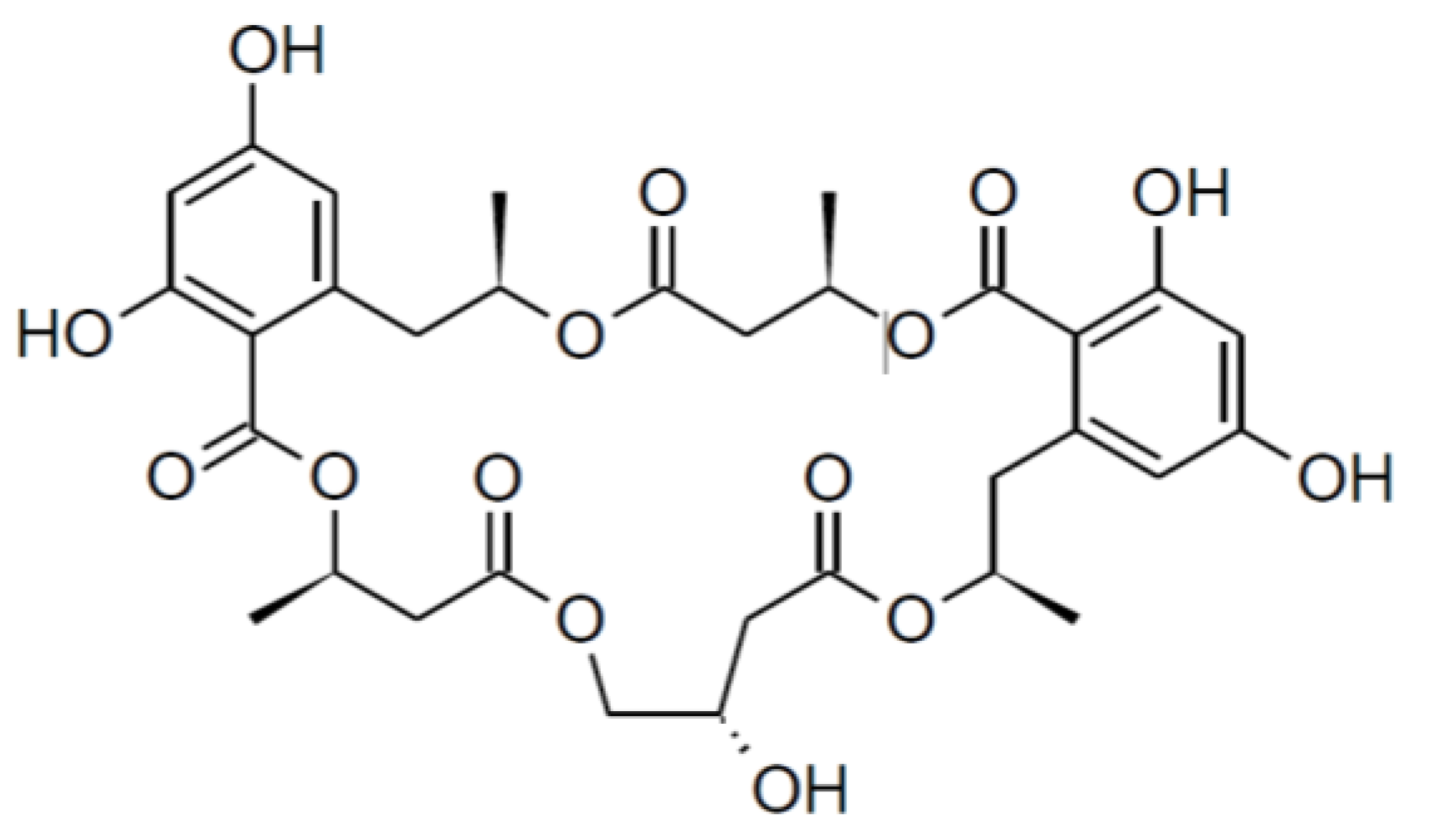
| Calcaride D (9) | Calcaride E (10) | |
|---|---|---|
| Position | δH, Mult. (J in Hz) | δH, Mult. (J in Hz) |
| 2a | 2.767, dd (7.5, 15.8) | 2.72, dd (7.8, 15.4) |
| 2b | 2.69, dd (5.3, 15.8) | 2.61, dd (5.9, 15.4) a |
| 3 | 5.57, m | 5.58, m |
| 4 | 1.43, d (6.4) | 1.43, d (6.3) |
| 8 | 6.33, s | 6.33, s |
| OCH3 | 3.77, s | 3.77, s |
| 10 | 6.33, s | 6.33, s |
| 12a | 3.29 b | 3.35, dd (3.5, 13.6) a |
| 12b | 2.91, dd (9.7, 13.7) | 2.87, dd (9.8, 13.6) |
| 13 | 5.22, m | 5.22, m |
| 14 | 1.25, d (6.2) | 1.26, d (6.3) |
| 16a | 2.66, dd (7.2, 15.7) | 2.65, dd (7.2, 15.6) |
| 16b | 2.62, dd (5.8, 15.7) | 2.62, dd (6.0, 15.6) |
| 17 | 5.47, m | 5.47, m |
| 18 | 1.30, d (6.4) | 1.30, d (6.3) |
| 22 | 6.203, d (2.6) | 6.19, d (2.5) |
| 24 | 6.197, d (2.6) | 6.18, d (2.5) |
| 26a | 3.17, dd (4.0, 13.4) | 3.20, dd (3.8, 13.5) |
| 26b | 2.772, dd (9.2, 13.4) | 2.74, dd (9.5, 13.5) |
| 27 | 5.15, m | 5.14, m |
| 28 | 1.23, d (6.2) | 1.23, d (6.2) |
| 30a | 2.47, dd (5.2, 15.4) | 2.36, dd (6.9, 14.7) |
| 30b | 2.27, dd (7.9, 15.4) | 2.26, dd (6.5, 14.7) |
| 31 | 3.94, m | 4.02, m |
| 32a | 3.44, dd (4.9, 11.2) | 1.05, d (6.2) |
| 32b | 3.40, dd (5.7, 11.2) |
2.2. Antibacterial Activity
| Compound | S. epidermidis MIC [µM] | X. campestris MIC [µM] | P. acnes MIC [µM] |
|---|---|---|---|
| 15G256α (1) | 12.9 (±3.6) | 30.8 (±3.1) | >100 |
| 15G256α-2 (2) | >150 | >150 | >200 |
| 15G256β (3) | 16.9 (±0.6) | 14.9 (±7.5) | >200 |
| 16G256β-2 (4) | >150 | >150 | >100 |
| 15G256π (5) | >150 | >150 | 14.1 (±1.8) |
| calcaride A (6) | 68.8 (±3.7) | 5.5 (±1.3) | >200 |
| calcaride B (7) | 52.3 (±2.3) | 22.6 (±9.2) | >200 |
| calcaride C (8) | 29.6 (±1.6) | 61.4 (±12.7) | >100 |
| calcaride D (9) | >150 | >150 | >200 |
| calcaride E (10) | 104.3 (±7.8) | >150 | >100 |
| chloramphenicol | 4.5 | 1.0 | 0.5 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Cultivation
3.4. Isolation Procedure
3.5. Antibacterial Assays
4. Conclusions
Acknowledgments
Abbreviations
| NMR | nuclear magnetic resonance |
| HRESIMS | high-resolution electrospray ionization mass spectrometry |
| HPLC | high-performance liquid chromatography |
| MS | mass spectrometry |
| ESIMS | electrospray ionization mass spectrometry |
| DEPT | distortionless enhancement by polarization transfer |
| COSY | correlation spectroscopy |
| HSQC | heteronuclear single-quantum coherence |
| HMBC | heteronuclear multiple bond correlation |
| Mult | multiplicity |
| ESI | electrospray ionization; calcd, calculated |
| SI | |
| OD600 | optical density at 600 nm |
| DMSO | dimethyl sulfoxide |
| PYG | medium, peptone yeast glucose medium |
| DSMZ | German collection of microorganisms and cell cultures |
Conflicts of Interest
References
- Sutton, B.C. Hyphomycetes from Manitoba and Saskatchewan, Canada; Commonwealth Mycological Institute: Kew, Surrey, UK, 1973; pp. 1–143. [Google Scholar]
- Somrithipol, S.; Jones, E.B.G. Calcarisporium phaeopodium sp. nov., a new hyphomycete from Thailand. Sydowia 2006, 58, 133–140. [Google Scholar]
- Ji, L.L.; Song, Y.C.; Tan, R.X. A potent feed preservative candidate produced by Calcarisporium sp., an endophyte residing in stargrass (Cynodon dactylon). J. Appl. Microbiol. 2004, 96, 352–358. [Google Scholar]
- Kramer, C.L.; Pady, S.M.; Rogerson, C.T.; Ouye, L.G. Kansas aeromycology II. Materials, methods, and general results. Trans. Kans. Acad. Sci. 1956, 62, 184–199. [Google Scholar]
- Watson, P. Calcarisporium arbuscula living as an endophyte in apparently healthy sporophores of Russula and Lactarius. Trans. Brit. Mycol. Soc. 1955, 38, 409–414. [Google Scholar] [CrossRef]
- Schlingmann, G.; Milne, L.; Carter, G.T. Isolation and identification of antifungal polyesters from the marine fungus Hypoxylon oceanicum LL-15G256. Tetrahedron 2002, 58, 6825–6835. [Google Scholar] [CrossRef]
- Breinholt, J.; Jensen, G.W.; Nielsen, R.I.; Olsen, C.E.; Frisvad, J.C. Antifungal macrocyclic polylactones from Penicillium verruculosum. J. Antibiot. 1993, 46, 1101–1108. [Google Scholar] [CrossRef]
- Ito, M.; Maruhashi, M.; Sakai, N.; Mizoue, K.; Hanada, K. NG-011 and NG-012, novel potentiators of nerve growth factor. I. Taxonomy, isolation, and physico-chemical and biological properties. J. Antibiot. 1992, 45, 1559–1565. [Google Scholar] [CrossRef]
- Roy, K.; Chatterjee, S.; Deshmukh, S.K.; Vijayakumar, E.K.S.; Ganguli, B.N.; Fehlhaber, H.-W. Orbuticin, a new secondary metabolite from Acremonium butyri. J. Antibiot. 1996, 49, 1186–1187. [Google Scholar] [CrossRef]
- Kondo, H.; Kurama, M.; Nakajima, S.; Osada, K.; Ookura, A.; Suda, H. Estrogenic BE-26263 and its manufacture with Scedosporium apiospermum. Jpn. Kokai Tokkyo Koho JP 05032658, 1993. [Google Scholar]
- Arai, M.; Tomoda, H.; Okuda, T.; Wang, H.; Tabata, N.; Masuma, R.; Yamaguchi, Y.; Omura, S. Funicone-related compounds, potentiators of antifungal miconazole activity, produced by Talaromyces flavus FKI-0076. J. Antibiot. 2002, 55, 172–180. [Google Scholar] [CrossRef]
- Zhou, Z.-Y.; Liu, R.; Jiang, M.-Y.; Zhang, L.; Niu, Y.; Zhu, Y.-C.; Dong, Z.-J.; Liu, J.-K. Two new cleistanthane diterpenes and a new isocoumarine from cultures of the basidiomycete Albatrellus confluens. Chem. Pharm. Bull. 2009, 57, 975–978. [Google Scholar] [CrossRef]
- Ito, M.; Tsuchida, Y.; Mizoue, K.; Hanada, K. NG-011 and NG-012, novel potentiators of nerve growth factor. II. The structure determination of NG-011 and NG-012. J. Antibiot. 1992, 45, 1566–1572. [Google Scholar] [CrossRef]
- Silber, J.; Ohlendorf, B.; Labes, A.; Näther, C.; Imhoff, J.F. Calcaripeptides A–C, cyclodepsipeptides from a Calcarisporium strain. J. Nat. Prod. 2013, 76, 1461–1467. [Google Scholar] [CrossRef]
- Stevens, R.B. Mycology Guidebook; University of Washington Press: Seattle, WA, USA, 1974; pp. 1–703. [Google Scholar]
- Buckingham, J. Dictionary of Natural Products on DVD; Version 21:1; CRC Press Taylor and Francis Group: London, UK, 2012. [Google Scholar]
- Walle, T.; Ta, N.; Kawamori, T.; Wen, X.; Tsuji, P.A.; Walle, U.K. Cancer chemopreventive properties of orally bioavailable flavonoids - methylated versus unmethylated flavones. Biochem. Pharmacol. 2007, 73, 1288–1296. [Google Scholar] [CrossRef]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef]
- Li, J.W.-H.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar]
- Grüschow, S.; Rackham, E.J.; Goss, R.J.M. Diversity in natural product families is governed by more than enzyme promiscuity alone: establishing control of the pacidamycin portfolio. Chem. Sci. 2011, 2, 2182–2186. [Google Scholar] [CrossRef]
- Wickerham, L.J. Taxonomy of Yeasts; US Dept. of Agriculture: Washington, DC, USA, 1951; pp. 1–56. [Google Scholar]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Silber, J.; Ohlendorf, B.; Labes, A.; Erhard, A.; Imhoff, J.F. Calcarides A–E, Antibacterial Macrocyclic and Linear Polyesters from a Calcarisporium Strain. Mar. Drugs 2013, 11, 3309-3323. https://doi.org/10.3390/md11093309
Silber J, Ohlendorf B, Labes A, Erhard A, Imhoff JF. Calcarides A–E, Antibacterial Macrocyclic and Linear Polyesters from a Calcarisporium Strain. Marine Drugs. 2013; 11(9):3309-3323. https://doi.org/10.3390/md11093309
Chicago/Turabian StyleSilber, Johanna, Birgit Ohlendorf, Antje Labes, Arlette Erhard, and Johannes F. Imhoff. 2013. "Calcarides A–E, Antibacterial Macrocyclic and Linear Polyesters from a Calcarisporium Strain" Marine Drugs 11, no. 9: 3309-3323. https://doi.org/10.3390/md11093309
APA StyleSilber, J., Ohlendorf, B., Labes, A., Erhard, A., & Imhoff, J. F. (2013). Calcarides A–E, Antibacterial Macrocyclic and Linear Polyesters from a Calcarisporium Strain. Marine Drugs, 11(9), 3309-3323. https://doi.org/10.3390/md11093309