HPLC-ESI-IT-MS/MS Analysis and Biological Activity of Triterpene Glycosides from the Colombian Marine Sponge Ectyoplasia ferox

Abstract
:1. Introduction
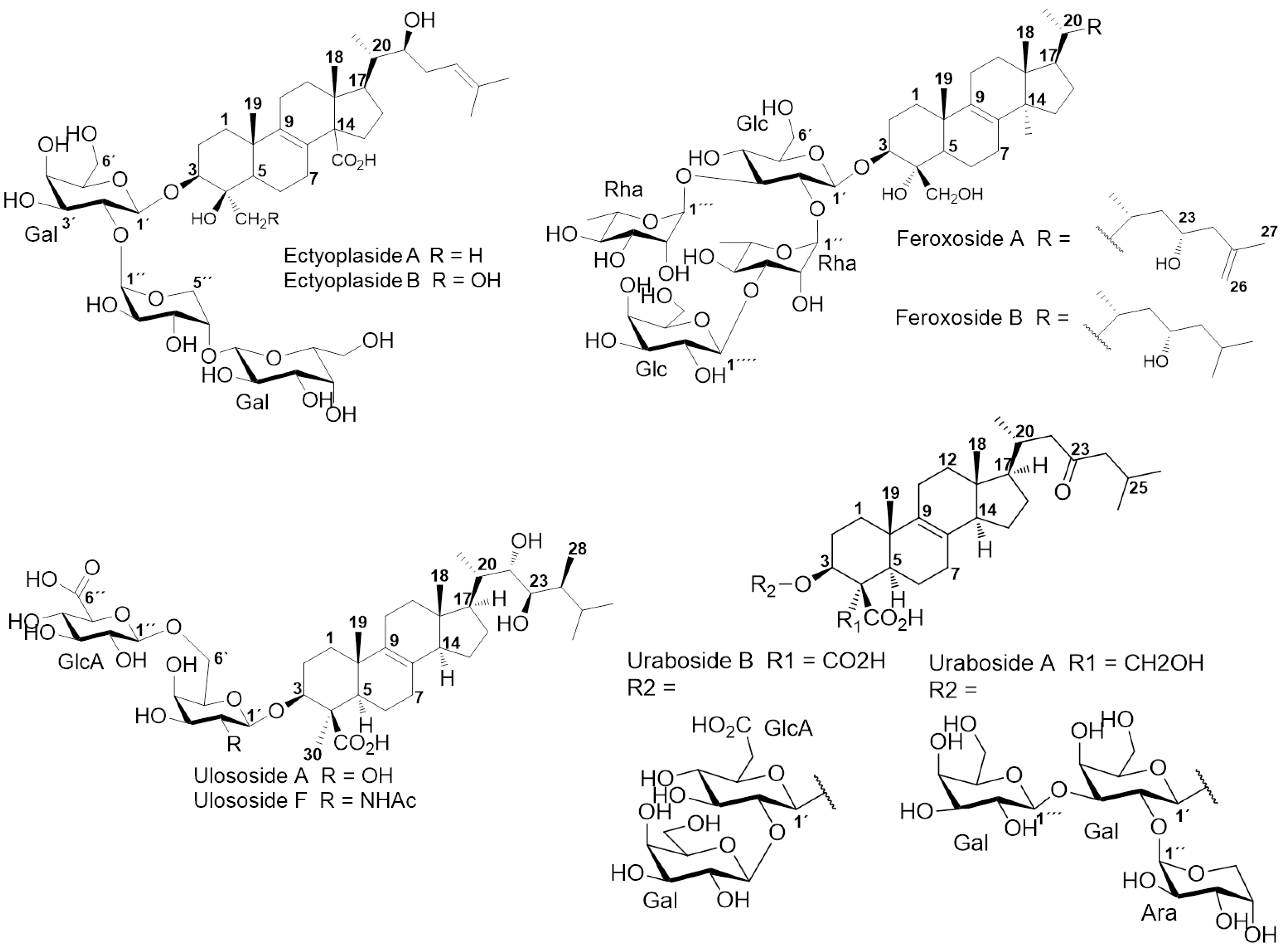
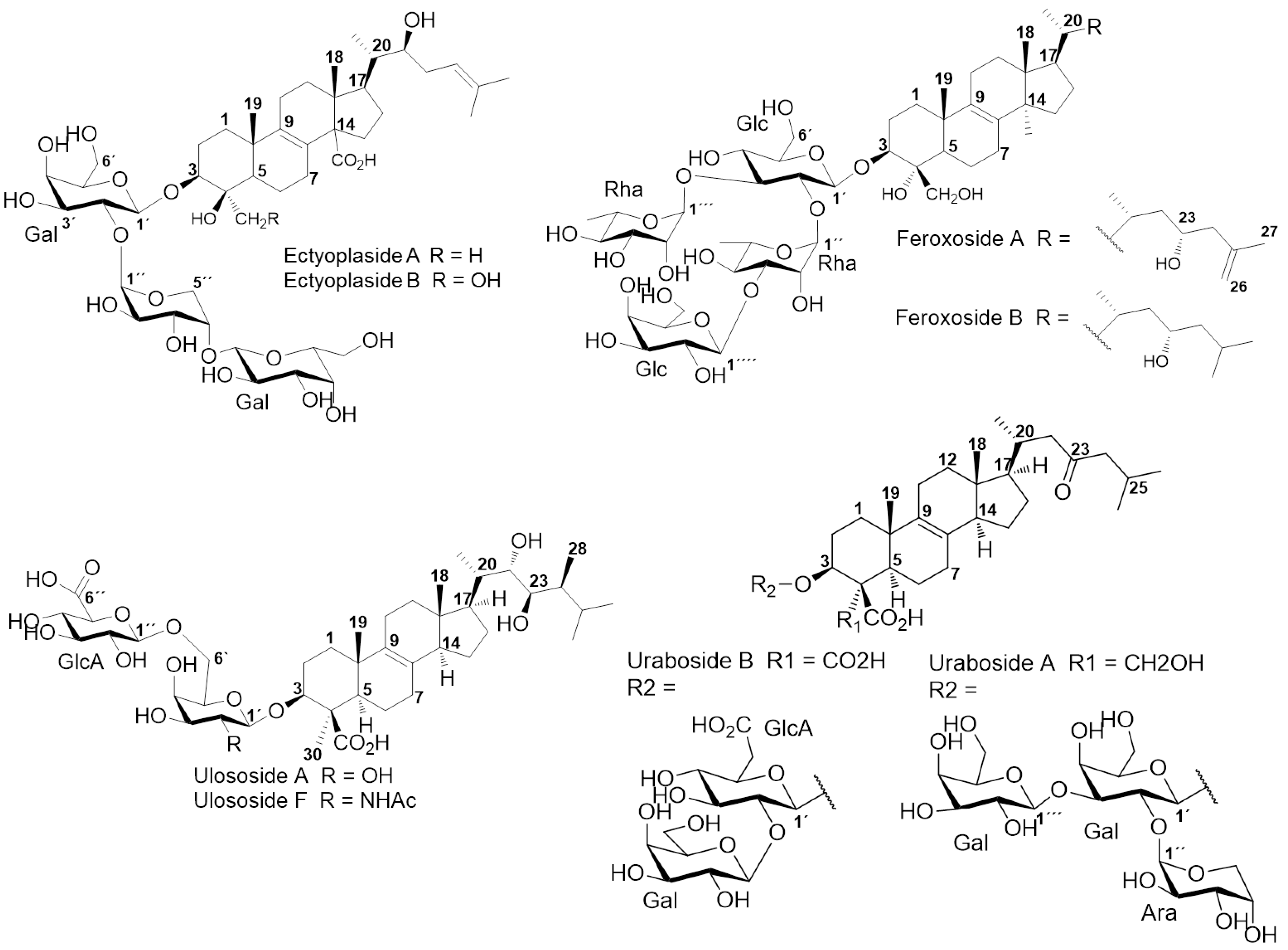
2. Results and Discussion
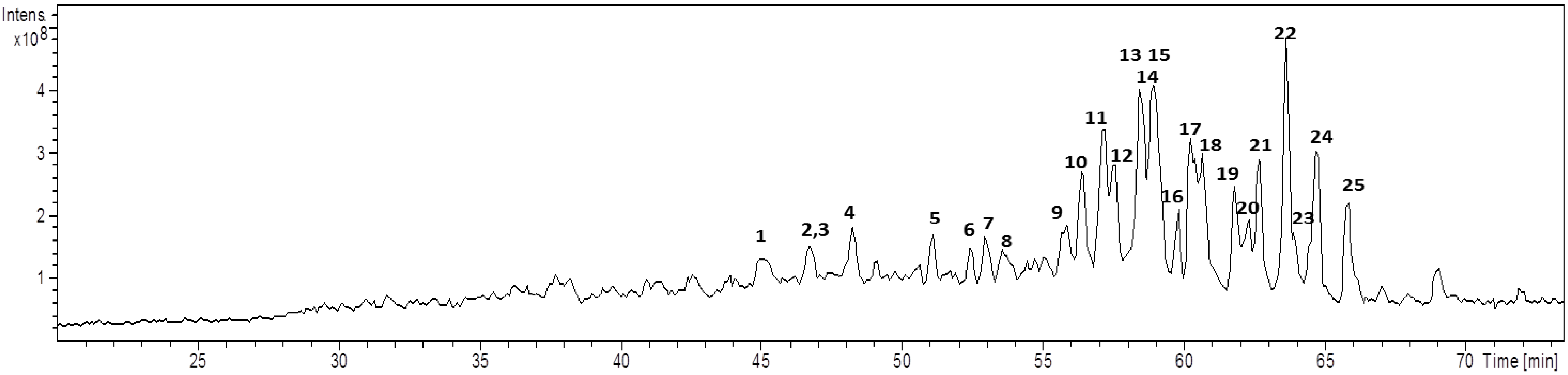
2.1. Chemical Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
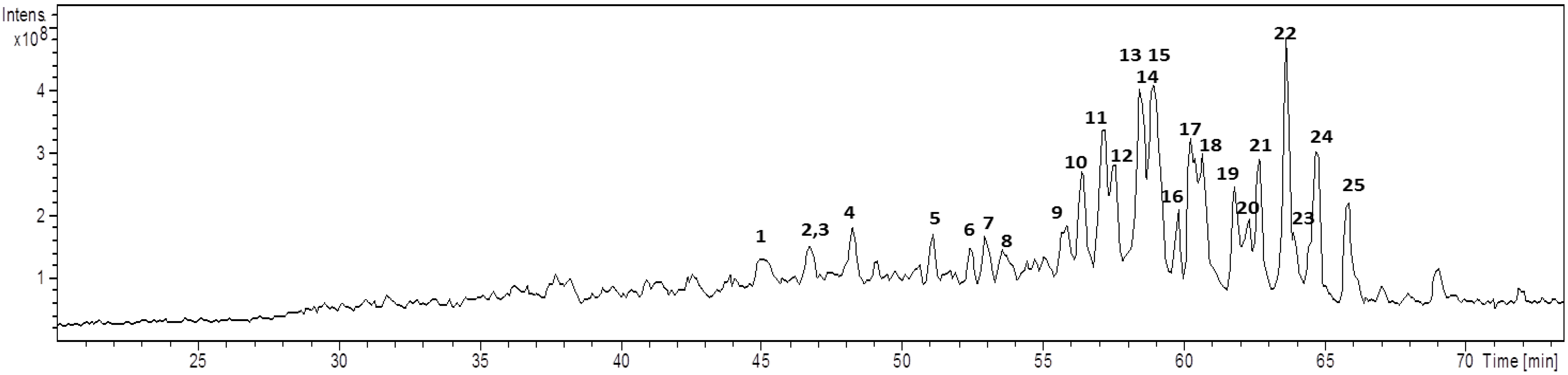
| Cmp | Rt (min) | m/z [M + Na]+ | MW | Diagnostic ions (m/z) a | Aglycone core b | Glycan core |
|---|---|---|---|---|---|---|
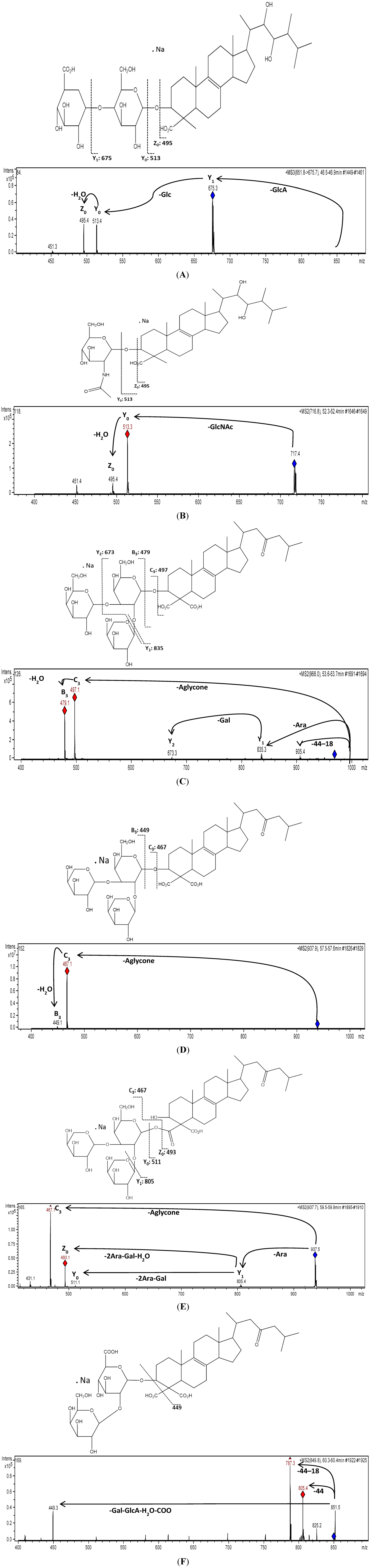
| 1 | 45.1 | 851 | 828 | 833, 513 [M − HexA − Hex + Na]+, 495 [M − HexA − Hex − H2O + Na]+, 452 | ULO-A | Hex-HexA- |
| 2 | 46.4 | 851 | 828 | 675 [M − HexA + Na]+, 513 [M − HexA − Hex + Na]+, 495 [M − HexA − Hex − H2O + Na]+, 451 | ULO-A | HexA-Hex- c |
| 3 | 46.6 | 985 | 962 | 967, 923, 853 [M − Pen + Na]+, 791, 497 [M − Aglycone + Na]+, 479 [M − Aglycone − H2O + Na]+ | AG-1 | Pen-Hex-Hex d |
| 4 | 48.0 | 851 | 828 | 833, 801, 513 [M − HexA − Hex + Na]+, 495 [M − HexA − Hex − H2O + Na]+ | ULO-A | HexA-Hex- |
| 5 | 50.7 | 837 | 814 | 661 [M − HexA + Na]+, 675 [M − Hex + Na]+, 499 [M − HexA − Hex + Na]+, 435 | AG-2 | HexA, Hex e |
| 6 | 52.3 | 716 | 693 | 684, 513 [M − HexNAc + Na]+, 495 [M − HexNAc − H2O + Na]+, 451 | ULO-A | HexNAc- |
| 7 | 53.0 | 819 | 796 | 687 [M − pen + Na]+, 525 [M − Pen − Hex + Na]+, 479 | AG-3 | Pen-Hex-R |
| 8 | 53.6 | 967 | 944 | 905 [M − CO2 − H2O + Na]+, 835 [M − Pen + Na]+, 673 [M − Pen − Hex + Na]+, 497 [M − Aglycone + Na]+ | URA-B | Pen-Hex-Hex d |
| 9 | 54.0 | 951 | 928 | 819 [M − Pen + Na]+, 497 [M − Aglycone + Na]+ | AG-4 | Pen-Hex-Hex d |
| 10 | 56.1 | 979 | 956 | 497 [M − Aglycone + Na]+ | AG-5 | Pen-Hex-Hex d |
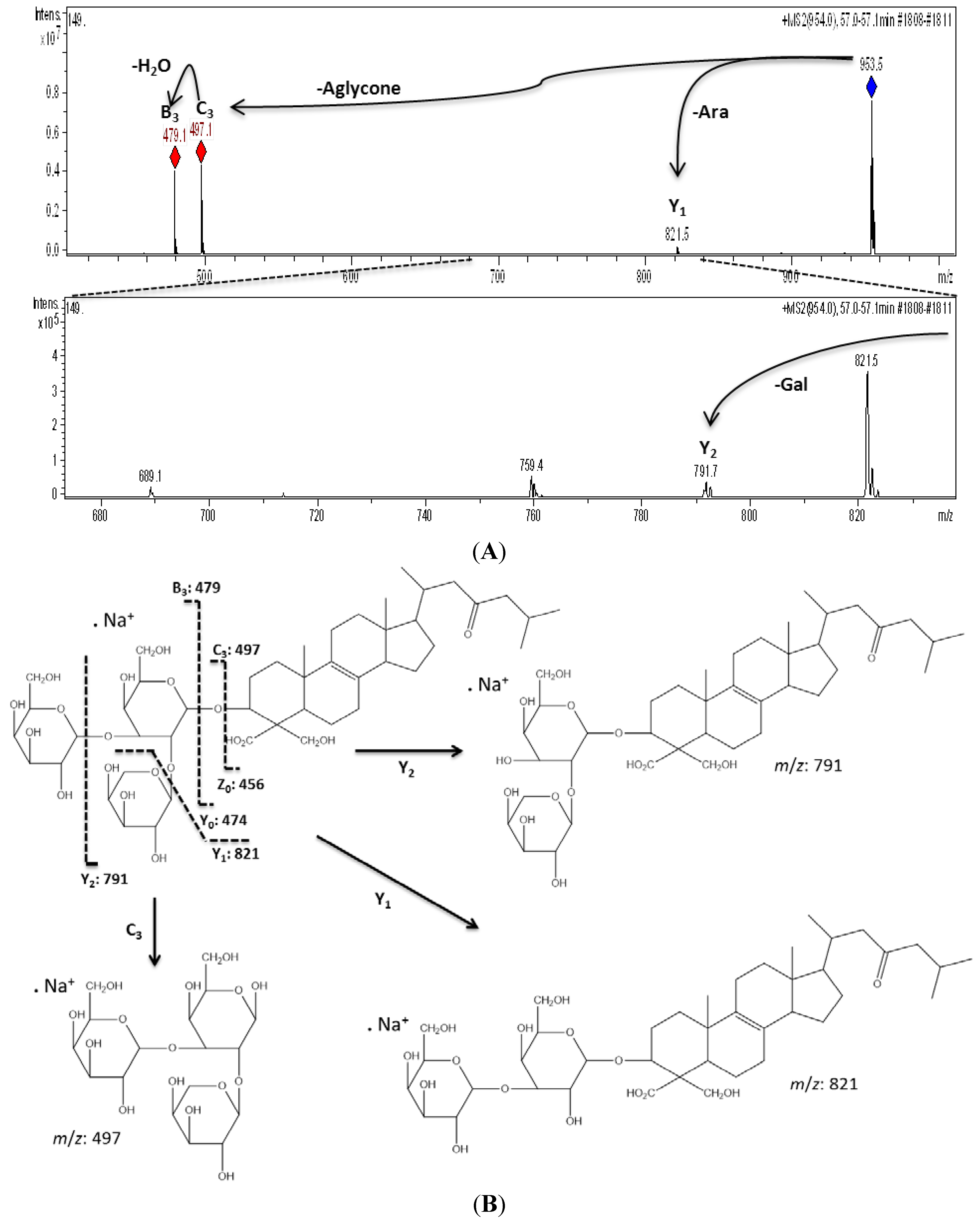
| 11 | 57.0 | 953 | 930 | 821 [M − Pen + Na]+, 791 [M − Hex + Na]+, 497 [M − Aglycone + Na]+, 479 [M − Aglycone − H2O + Na]+ | URA-A | Pen-Hex-Hex d |
| 12 | 57.6 | 937 | 914 | 875 [M − CO2 − H2O + Na]+, 805 [M − Pen + Na]+, 467 [M − Aglycone + Na]+, 449 [M − Aglycone − H2O + Na]+ | URA-B | Pen-Pen-Hex- |
| 13 | 58.2 | 921 | 898 | 789 [M − Pen + Na]+, 657 [M − 2 Pen + Na]+, 467 [M − Aglycone + Na]+, 449 [M − Aglycone − H2O + Na]+ | AG-4 | Pen-Pen-Hex- |
| 14 | 58.4 | 1097 | 1074 | 965 [M − Pen + Na]+, 921 [M − HexA + Na]+, 833 [M − 2 Pen + Na]+, 657 [M − 2 Pen − HexA + Na]+ | AG-4 | Pen-Pen-HexA-Hex- |
| 15 | 58.6 | 1111 | 1088 | 965 [M − dHex + Na]+, 833 [M − dHex − Pen + Na]+, 657 [M − dHex − Pen − HexA + Na]+, 495 [M − dHex − Pen − HexA − Hex + Na]+ | AG-4 | dHex-HexA-Pen-Hex- |
| 16 | 59.7 | 937 | 914 | 805 [M − Pen + Na]+, 511 [M − 2 Pen − Hex + Na]+, 493 [M − 2 Pen − Hex − H2O + Na]+, 467 [M − Aglycone + Na]+, 431 | URA-B | Pen-Pen-Hex- |
| 17 | 60.1 | 965 | 942 | 833 [M − Pen + Na]+, 789 [M − Pen − COO + Na]+, 657 [M − Pen − HexA + Na]+ | AG-4 | Pen-HexA-Hex- |
| 18 | 60.3 | 849 | 826 | 805 [M − CO2 + Na]+, 787 [M − COO − H2O + Na]+, 449 [M − COO − HexA − Hex − H2O + Na]+ | URA-B | Hex-HexA f |
| 19 | 61.6 | 803 | 780 | 641 [M − Hex + Na]+, 465 [M − Hex − HexA + Na]+ | AG-6 | Hex-HexA f |
| 20 | 62.2 | 805 | 782 | 743, 673 [M − Pen + Na]+, 611 [M − HexA − H2O + Na]+ | URA-A | Pen-HexA e |
| 21 | 62.7 | 803 | 780 | 641 [M − Hex + Na]+, 627 [M − HexA + Na]+, 465 [M − Hex − HexA + Na]+ | AG-4 | Hex-HexA f |
| 22 | 63.5 | 805 | 782 | 643 [M − Hex + Na]+, 467 [M − Hex − HexA + Na]+ | AG-7 | Hex-HexA f |
| 23 | 63.9 | 817 | 794 | 641 [M − HexA + Na]+, 479 [M − HexA − Hex + Na]+ | AG-8 | HexA-Hex c |
| 24 | 64.5 | 761 | 738 | 629 [M − Pen + Na]+, 467 [M − Pen − Hex + Na]+ | AG-7 | Pen-Hex- |
| 25 | 65.8 | 787 | 764 | 655 [M − Pen + Na]+, 479 [M − Pen − HexA + Na]+ | AG-8 | Pen-HexA- |
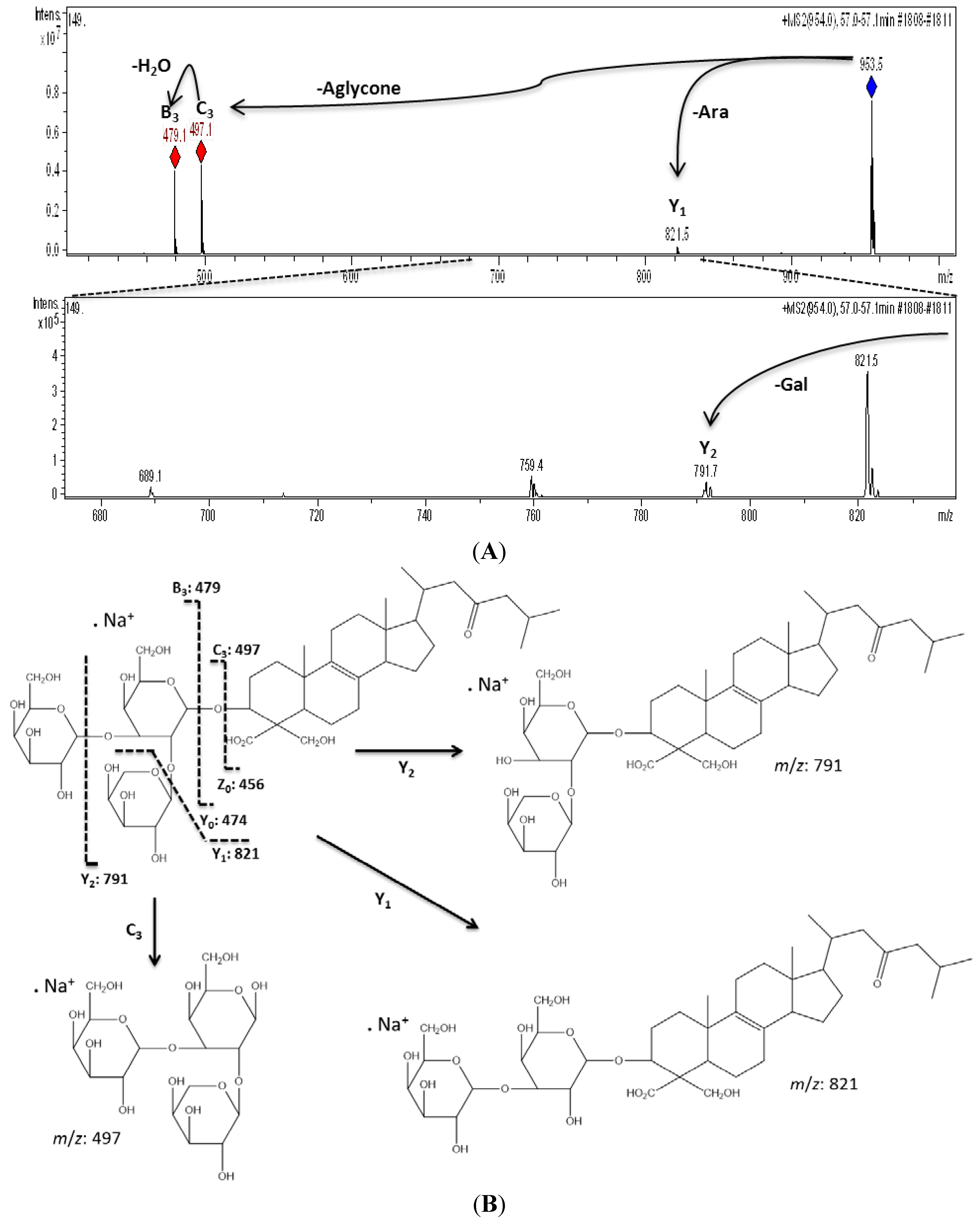
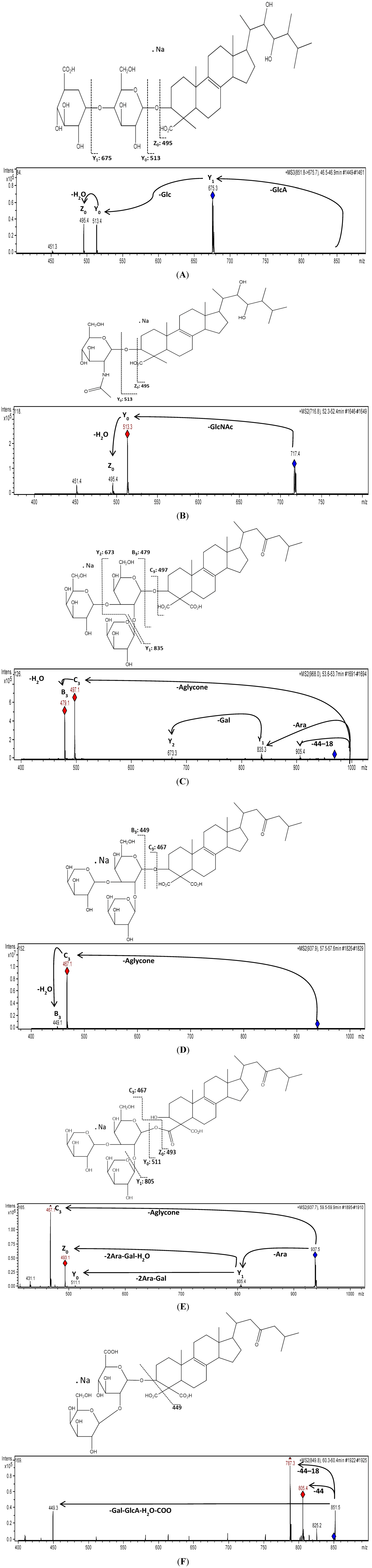
2.2. Biological Analysis
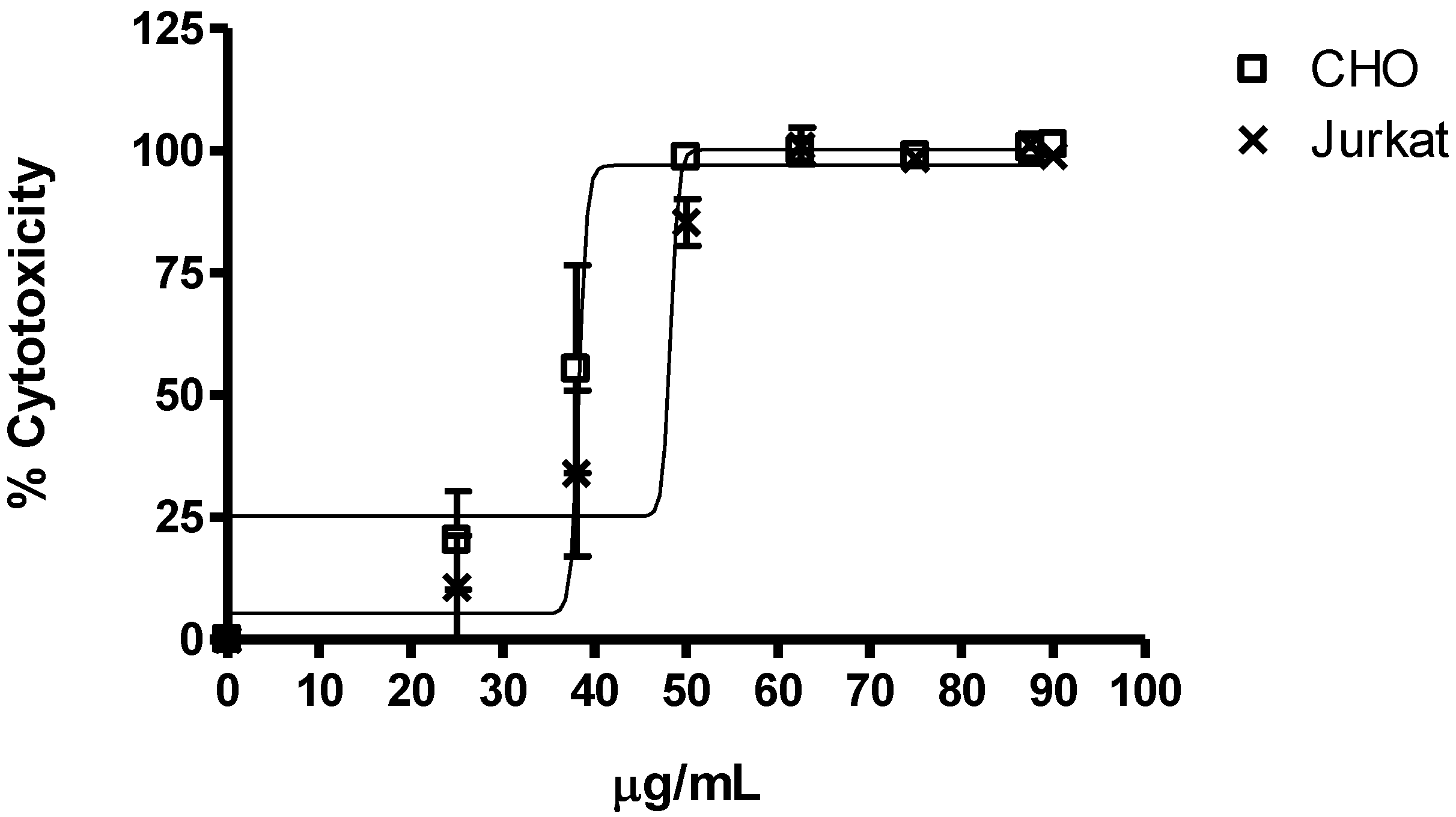
2.2.1. Cytotoxicity
| Treatment µg/mL | CHO cell line | Jurkat cell line | Hemolytic activity |
|---|---|---|---|
| Solvent (PBS 5%–10%) | 0.97 ± 0.01 | 0.98 ± 0.01 | 0.31 ± 0.03 |
| 0 | 0.90 ± 0.02 | 0.97 ± 0.00 | 0.00 ± 0.00 |
| 25 | 0.9 ± 0.06 | 0.83 ± 0.15 | - |
| 50 | 0.04 ± 0.01 | 0.35 ± 0.01 | 0.43 ± 0.16 |
| 100 | 0,00 ± 0.00 | 0.00 ± 0.00 | 0.67 ± 0.27 |
| 200 | - | - | 0.36 ± 0.10 |
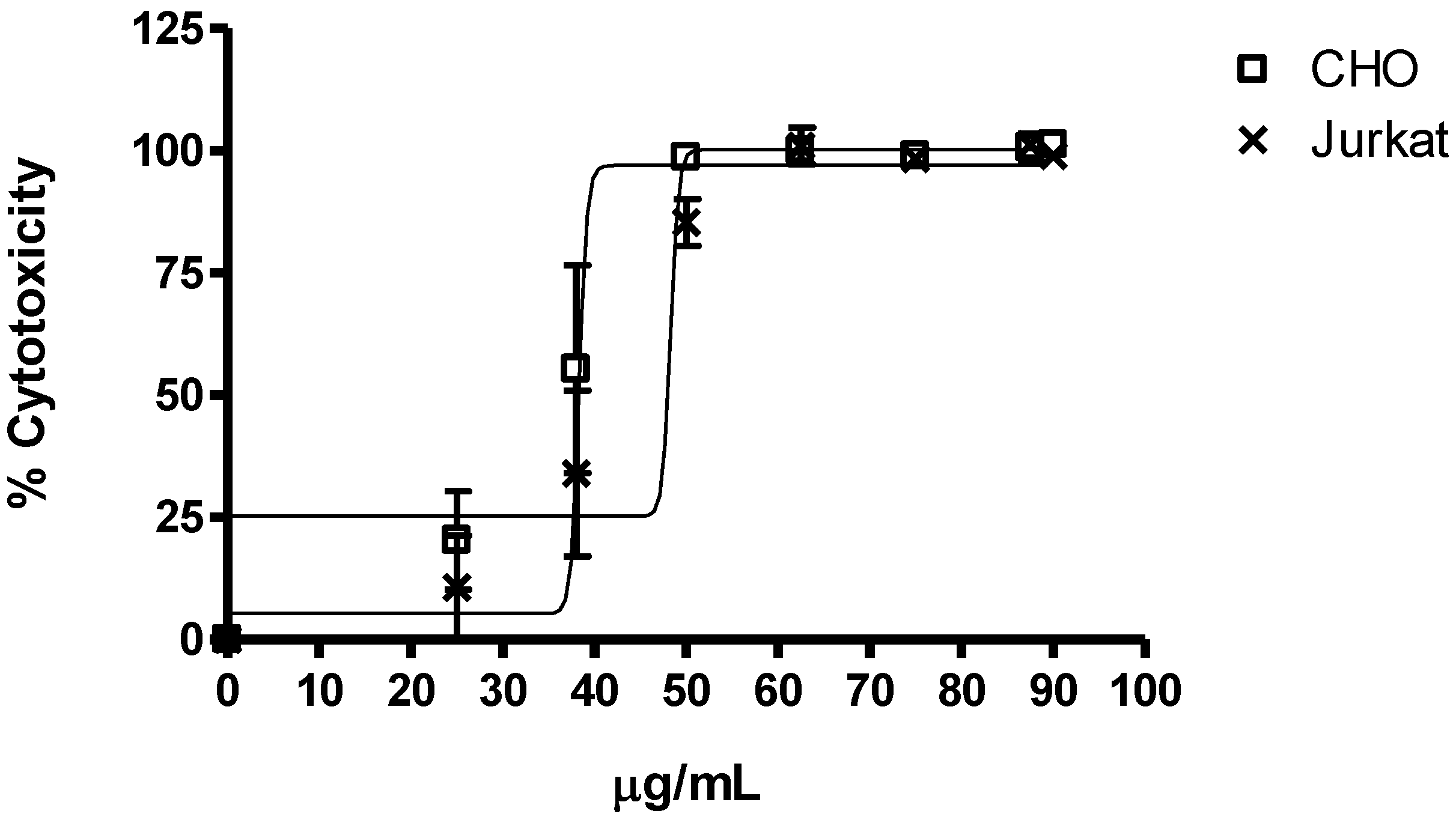
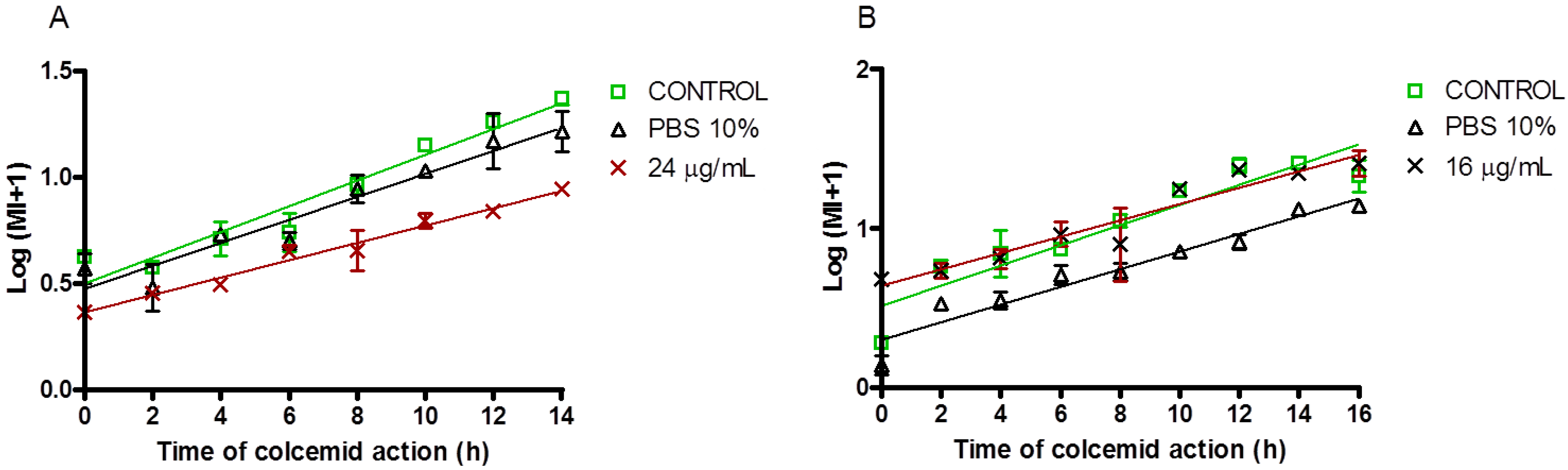
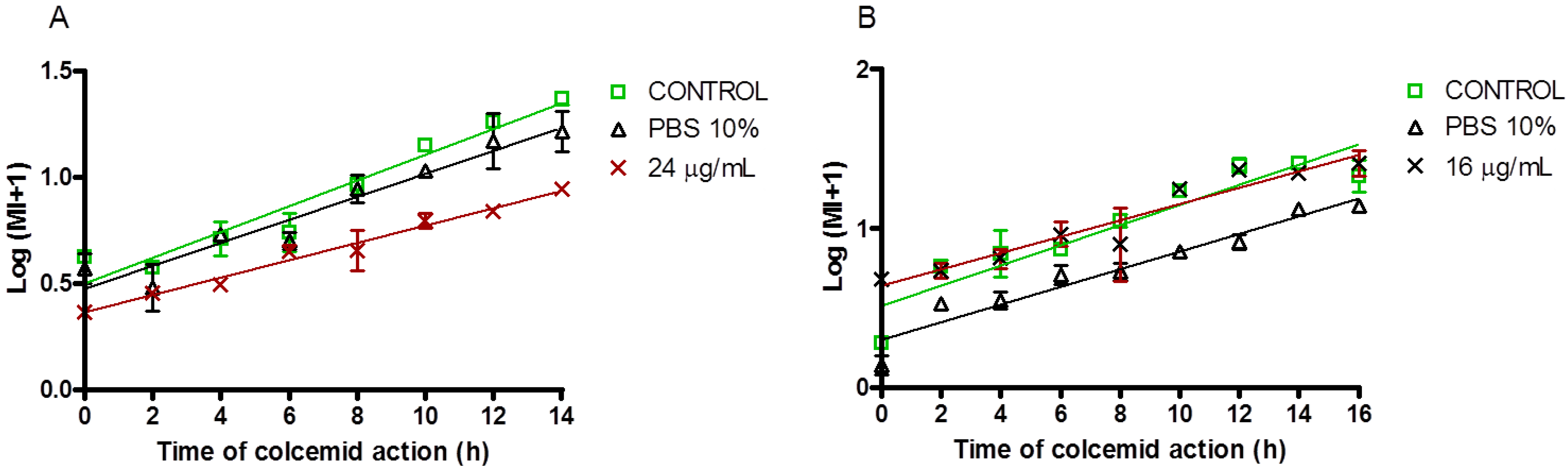
2.2.2. Antiproliferative and Cell Cycle Progression
| Treatment µg/mL | Absolute cloning efficiency | Relative cloning efficiency | ||||
|---|---|---|---|---|---|---|
| Mean ± SEM × 100 | Low limit | High limit | Mean ± SEM × 100 | Low limit | High limit | |
| CHO cell line | ||||||
| 0 | 1.13 ± 0.15 | 0.96 | 1.31 | |||
| Solvent (PBS 10%) | 0.84 ± 0.08 | 0.67 | 1.02 | 0.77 ± 0.17 | 0.57 | 0.96 |
| 7.5 | 0.98 ± 0.01 | 0.81 | 1.15 | 0.87 ± 0.07 | 0.68 | 1.06 |
| 15 | 0.68 ± 0.09 | 0.51 | 0.86 | 0.62 ± 0.02 | 0.42 | 0.81 |
| 30 | 0.55 ± 0.09 | 0.38 | 0.73 | 0.48 ± 0.03 | 0.29 | 0.68 |
| Jurkat cell line | ||||||
| 0 | 2,46 ± 0.2 | 1.43 | 3.48 | |||
| Solvent (PBS 10%) | 1.53 ± 1.35 | 0.51 | 2.55 | 0.67 ± 0.6 | 0.07 | 1.27 |
| 8.6 | 2.87 ± 0.44 | 1.84 | 3.89 | 1.16 ± 0.09 | 0.56 | 1.76 |
| 17 | 1.56 ± 0.17 | 0.54 | 2.58 | 0.63 ± 0.02 | 0.03 | 1.23 |
| 26 | 0.19 ± 0.01 | −0.84 | 1.21 | 0.08 ± 0.0 | −0.52 | 0.67 |
3. Experimental Section
3.1. Biological Material
3.2. Extraction of Saponins
3.3. Liquid Chromatography/Electrospray Ionization Mass Spectrometry
3.4. Cell Cultures
3.5. MTT Test
3.6. Cell Cloning Efficiency Assay
3.7. Mitotic Index
3.8. Hemolysis Assay
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar]
- Belarbi, E.H.; Contreras, A.; Chisti, Y.; García, F.; Emilio, M. Producing drugs from marine sponges. Biotechnol. Adv. 2003, 21, 585–598. [Google Scholar] [CrossRef]
- Mollica, A.; Locatelli, M.; Stefanucci, A.; Pinnen, F. Synthesis and bioactivity of secondary metabolites from marine sponges containing dibrominated indolic systems. Molecules 2012, 17, 6083–6099. [Google Scholar] [CrossRef]
- Sabdono, A.; Karna, O. Microbial symbionts in marine sponges: Marine natural product factory. J. Coast. Dev. 2012, 29, 144–222. [Google Scholar]
- Goulitquer, S.; Potin, P.; Tonon, T. Mass spectrometry-based metabolomics to elucidate functions in marine organisms and ecosystems. Mar. Drugs 2012, 10, 849–880. [Google Scholar] [CrossRef]
- Ivanchina, N.V.; Kicha, A.A.; Stonik, V.A. Steroid glycosides from marine organisms. Steroids 2011, 76, 425–454. [Google Scholar] [CrossRef]
- Kalinin, V.I.; Ivanchina, N.V.; Krasokhin, V.B.; Makarieva, T.N.; Stonik, V.A. Glycosides from marine sponges (Porifera, Demospongiae): Structures, taxonomical distribution, biological activities and biological roles. Mar. Drugs 2012, 10, 1671–1710. [Google Scholar] [CrossRef]
- Regalado, E.L.; Jimenez-Romero, C.; Genta-Jouve, G.; Tasdemir, D.; Amade, P.; Nogueiras, C.; Thomas, O.P. Acanthifoliosides, minor steroidal saponins from the caribbean sponge Pandaros acanthifolium. Tetrahedron 2011, 67, 1011–1018. [Google Scholar] [CrossRef]
- Regalado, E.L.; Tasdemir, D.; Kaiser, M.; Cachet, N.; Amade, P.; Thomas, O.P. Antiprotozoal steroidal saponins from the marine sponge Pandaros acanthifolium. J. Nat. Prod. 2010, 73, 1404–1410. [Google Scholar] [CrossRef]
- Colorado, J.; Muñoz, D.; Marquez, D.; Marquez, M.E.; Lopez, J.; Thomas, O.P.; Martinez, A. Ulososides and urabosides—Triterpenoid saponins from the Caribbean marine sponge Ectyoplasia ferox. Molecules 2013, 18, 2598–2610. [Google Scholar]
- Kelly, S.; Jensen, P.; Henkel, T.; Fenical, W.; Pawlik, J. Effects of Caribbean sponge extracts on bacterial attachment. Aquat. Microb. Ecol. 2003, 31, 175–182. [Google Scholar] [CrossRef]
- Podolak, I.; Galanty, A.; Sobolewska, D. Saponins as cytotoxic agents: A review. Phytochem. Rev. 2010, 9, 425–474. [Google Scholar] [CrossRef]
- Locatelli, M.; Governatori, L.; Carlucci, G.; Genovese, S.; Mollica, A.; Epifano, F. Recent application of analytical methods to phase I and phase II drugs development: A review. Biomed. Chromatogr. 2012, 26, 283–300. [Google Scholar]
- Montoya, G.L.; Arango, G.J.; Ramírez-Pineda, J.R. Rapid differentiation of isobaric and positional isomers of structurally related glycosides from Phytolacca bogotensis. Rapid Commun. Mass Spectrom. 2009, 23, 3361–3371. [Google Scholar] [CrossRef]
- Fang, S.; Hao, C.; Sun, W.; Liu, Z.; Liu, S. Rapid analysis of steroidal saponin mixture using electrospray ionization mass spectrometry combined with sequential tandem mass spectrometry. Rapid Commun. Mass Spectrom. 1998, 12, 589–594. [Google Scholar] [CrossRef]
- Campagnuolo, C.; Fattorusso, E.; Taglialatela-Scafati, O. Feroxosides a-b, two norlanostane tetraglycosides from the caribbean sponge Ectyoplasia ferox. Tetrahedron 2001, 57, 4049–4055. [Google Scholar] [CrossRef]
- Cafieri, F.; Fattorusso, E.; Taglialatela-Scafati, O. Ectyoplasides A–B: Unique triterpene oligoglycosides from the caribbean sponge ectyoplasia ferox. Eur. J. Org. Chem. 1999, 1999, 231–238. [Google Scholar] [CrossRef]
- Domon, B.; Costello, C.E. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
- Antonov, A.S.; Kalinovskii, A.I.; Stonik, V.A.; Evtushenko, E.V.; Elyakov, G.B. Structure of ulososide a, a new triterpenoid glucoside from sponge ulosa sp. Russ. Chem. Bull. 1994, 43, 1265–1269. [Google Scholar] [CrossRef]
- Antonov, A.S.; Kalinovskii, A.I.; Dmitrenok, P.S.; Stonik, V.A. New triterpene glycosides from an ulosa sp. Sponge. Russ. J. Bioorg. Chem. 2002, 28, 183–188. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Freshney, R.I. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications, 6th ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2010; pp. 207–225. [Google Scholar]
- Regalado, E.L.; Turk, T.; Tasdemir, D.; Gorjanc, M.; Kaiser, M.; Thomas, O.P.; Fernandez, R.; Amade, P. Cytotoxic and haemolytic steroidal glycosides from the Caribbean sponge Pandaros acanthifolium. Steroids 2011, 76, 1389–1396. [Google Scholar] [CrossRef]
- Li, H.-J.; Chen, T.; Xie, Y.-L.; Chen, W.-D.; Zhu, X.-F.; Lan, W.-J. Isolation and structuralelucidation of chondrosterins F-H from the marine fungus Chondrostereum sp. Mar. Drugs 2013, 11, 551–558. [Google Scholar] [CrossRef]
- Arabski, M.; Wegierek-Ciuk, A.; Czerwonka, G.; Lankoff, A.; Kaca, W. Effects of saponins against clinical E. coli strains and eukaryotic cell line. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Van-Dyck, S.; Gerbaux, P.; Flammang, P. Qualitative and quantitative saponin contents in five sea cucumbers from the indian ocean. Mar. Drugs 2010, 8, 173–189. [Google Scholar] [CrossRef]
- Taniyama, S.; Arakawa, O.; Terada, M.; Nishio, S.; Takatani, T.; Mahmud, Y.; Noguchi, T. Ostreopsis sp., a possible origin of palytoxin (ptx) in parrotfish Scarus ovifrons. Toxicon 2003, 42, 29–33. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Colorado-Ríos, J.; Muñoz, D.; Montoya, G.; Márquez, D.; Márquez, M.-E.; López, J.; Martínez, A. HPLC-ESI-IT-MS/MS Analysis and Biological Activity of Triterpene Glycosides from the Colombian Marine Sponge Ectyoplasia ferox. Mar. Drugs 2013, 11, 4815-4833. https://doi.org/10.3390/md11124815
Colorado-Ríos J, Muñoz D, Montoya G, Márquez D, Márquez M-E, López J, Martínez A. HPLC-ESI-IT-MS/MS Analysis and Biological Activity of Triterpene Glycosides from the Colombian Marine Sponge Ectyoplasia ferox. Marine Drugs. 2013; 11(12):4815-4833. https://doi.org/10.3390/md11124815
Chicago/Turabian StyleColorado-Ríos, Jhonny, Diana Muñoz, Guillermo Montoya, Diana Márquez, Maria-Elena Márquez, Juan López, and Alejandro Martínez. 2013. "HPLC-ESI-IT-MS/MS Analysis and Biological Activity of Triterpene Glycosides from the Colombian Marine Sponge Ectyoplasia ferox" Marine Drugs 11, no. 12: 4815-4833. https://doi.org/10.3390/md11124815
APA StyleColorado-Ríos, J., Muñoz, D., Montoya, G., Márquez, D., Márquez, M.-E., López, J., & Martínez, A. (2013). HPLC-ESI-IT-MS/MS Analysis and Biological Activity of Triterpene Glycosides from the Colombian Marine Sponge Ectyoplasia ferox. Marine Drugs, 11(12), 4815-4833. https://doi.org/10.3390/md11124815