Sarcophine-Diol Inhibits Expression of COX-2, Inhibits Activity of cPLA2, Enhances Degradation of PLA2 and PLCγ1 and Inhibits Cell Membrane Permeability in Mouse Melanoma B16F10 Cells

Abstract
:1. Introduction
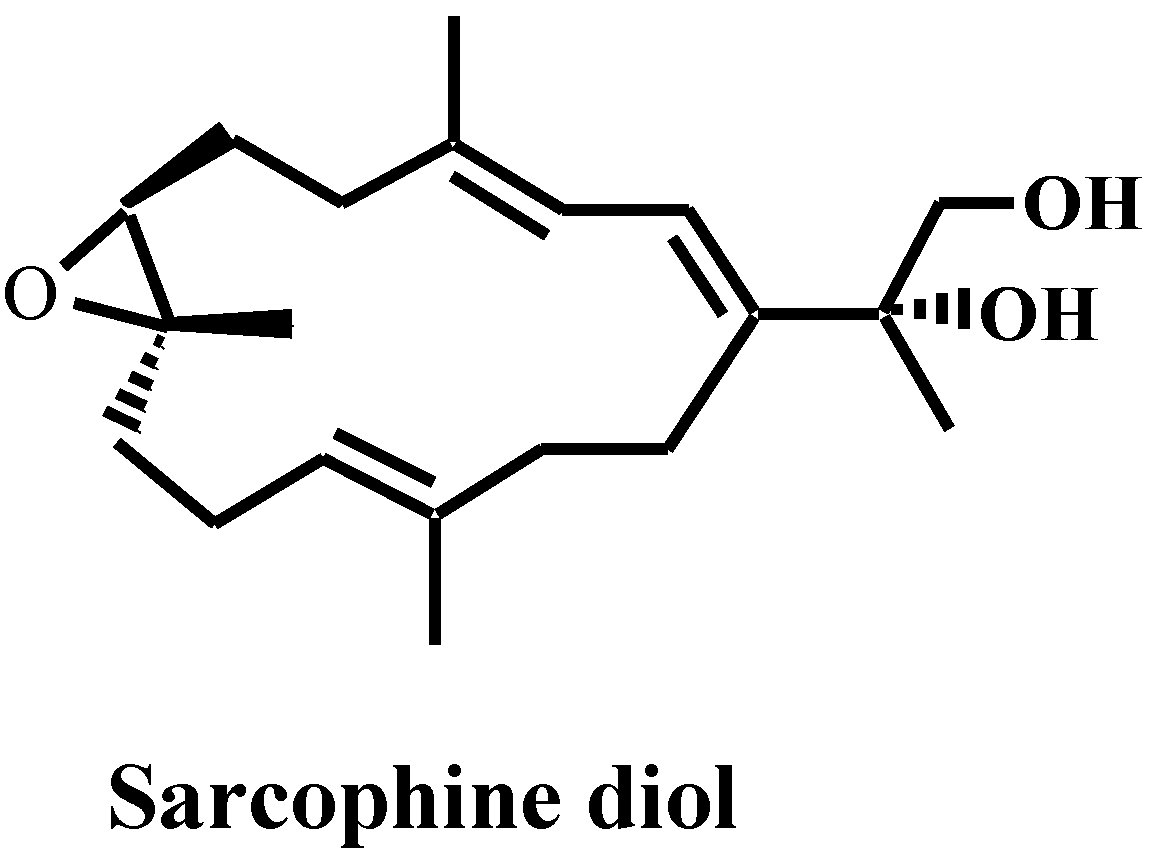
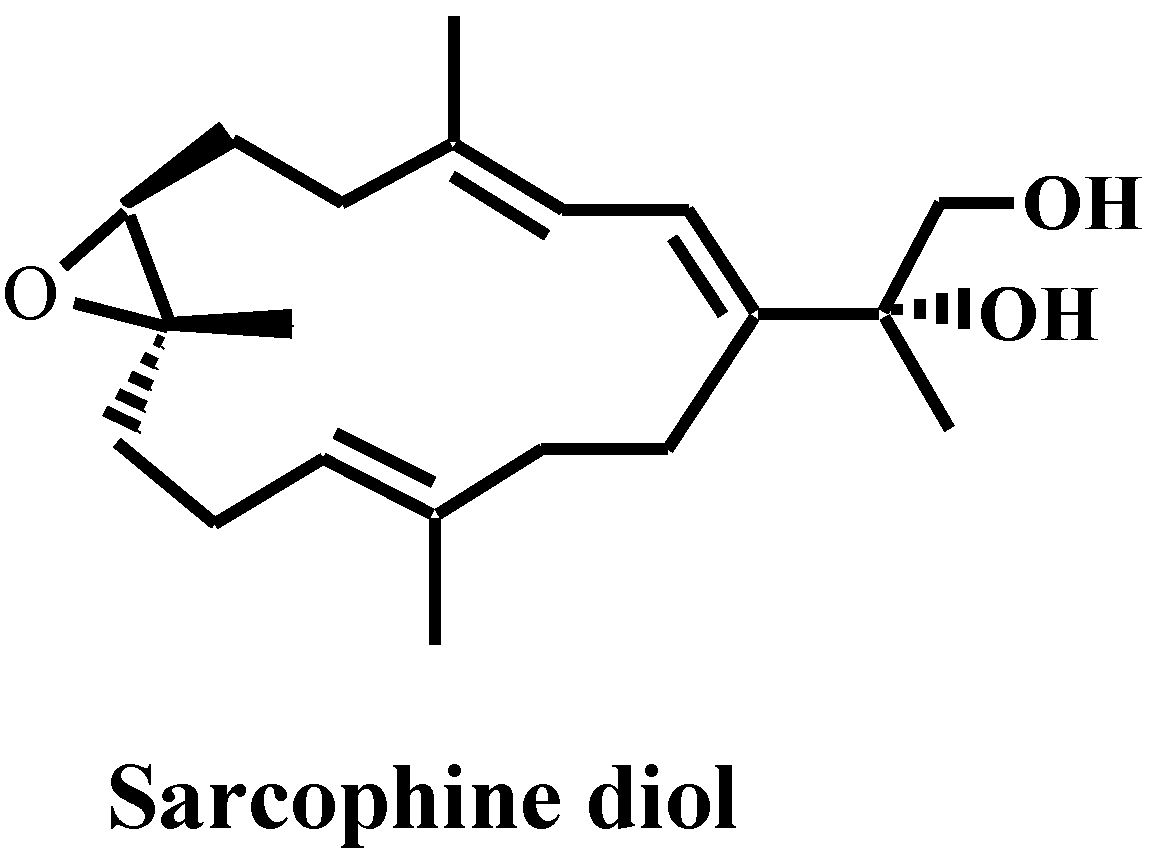
2. Results and Discussion
2.1. Wound Healing Assay

2.2. SD Inhibits Cell Multiplication

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time 0 | 0 µM | 62.5 µM | 125 µM | 250 µM | |
|---|---|---|---|---|---|
| Total protein content (µg) | 26.62 ± 5.49, n = 5 | 309.41 ± 43.93, n = 5 | 201.67 ± 35.35, n = 5 * | 165.83 ± 19.57, n = 5 * | 38.57 ± 7.32, n = 5 * |
| Total living cells | 1.86 × 105 ± 4.7 × 104, n = 5 | 2.4 × 106 ± 4.5 ×105, n = 5 | 1.68 × 106 ± 2.7 × 105, n = 5 * | 1.11 × 106 ± 1.9 × 105, n = 5 * | 2.3 × 105 ± 5.5 × 104, n = 5 * |
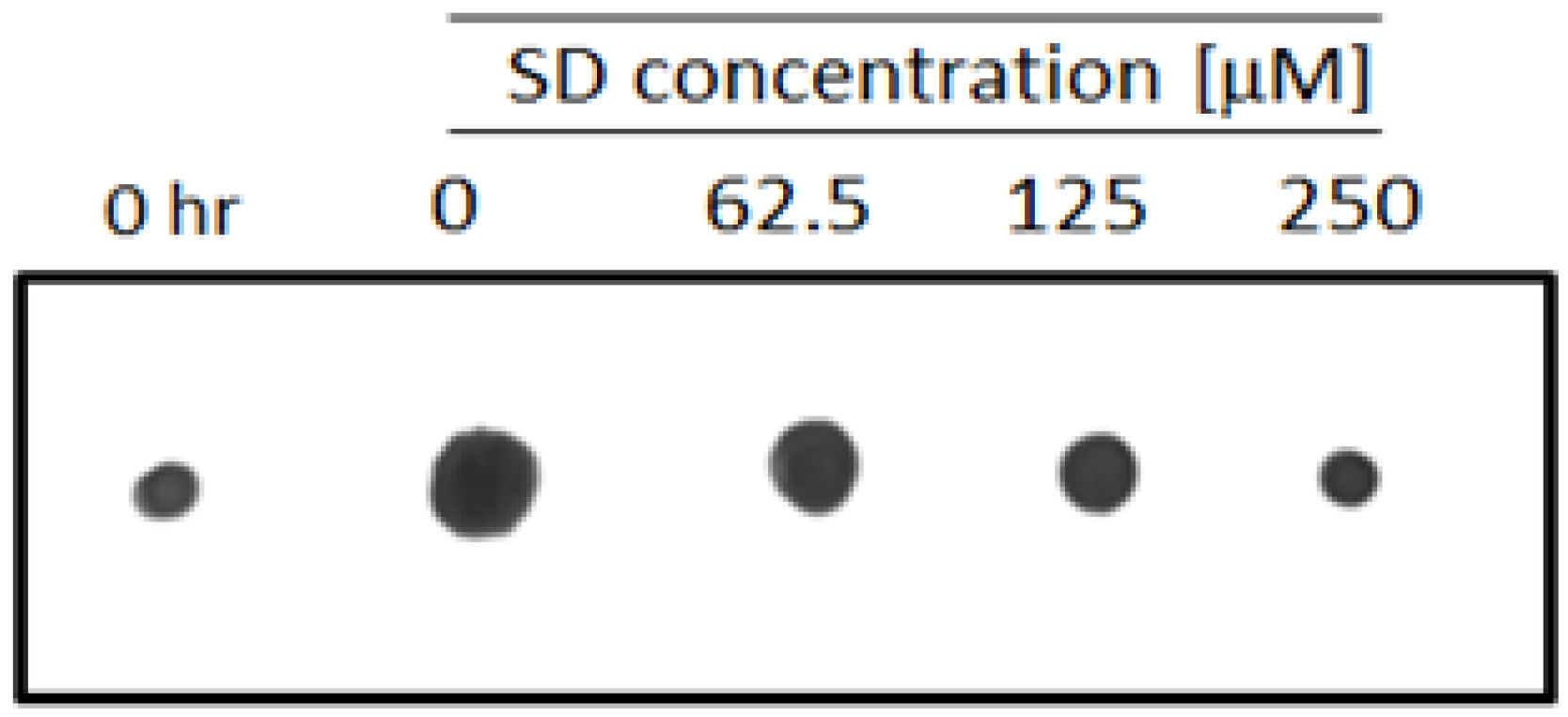
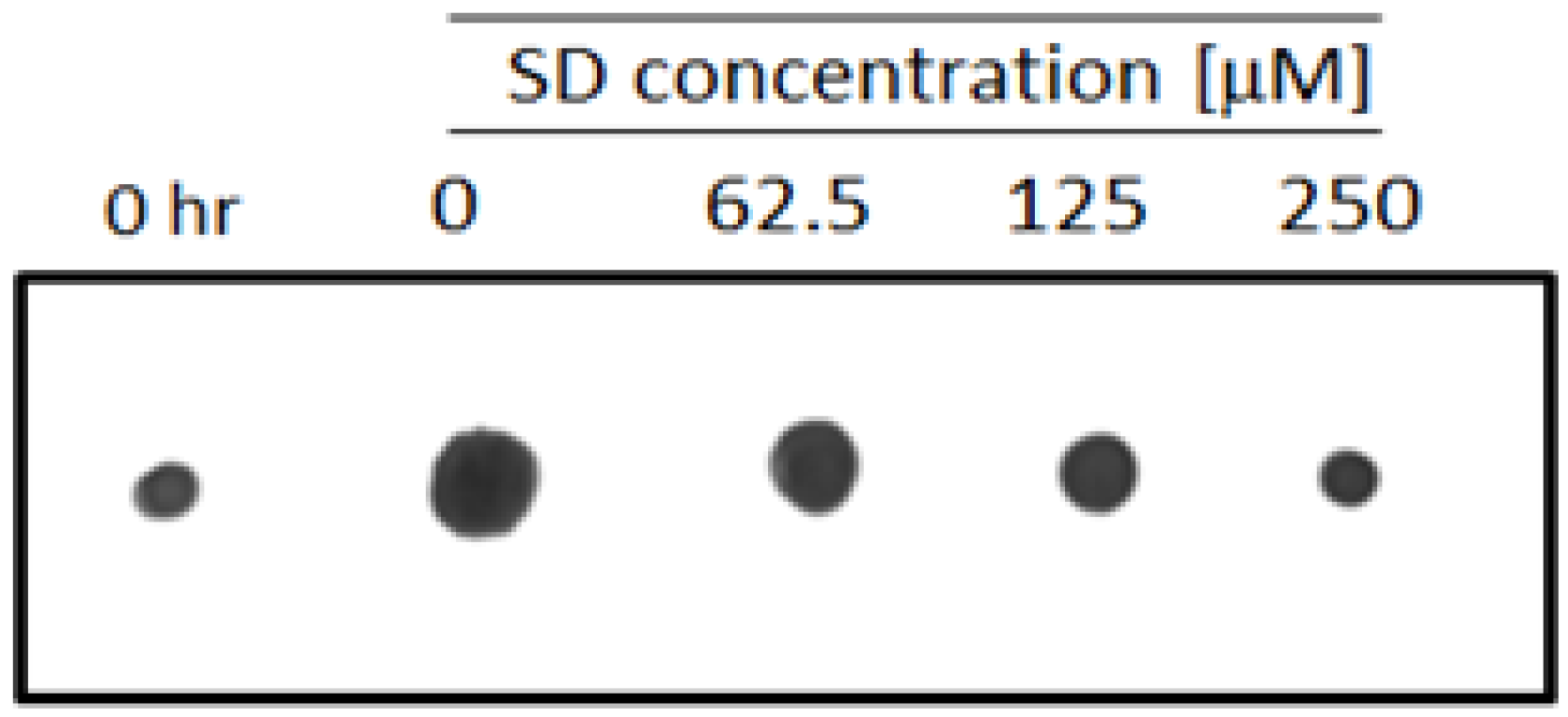
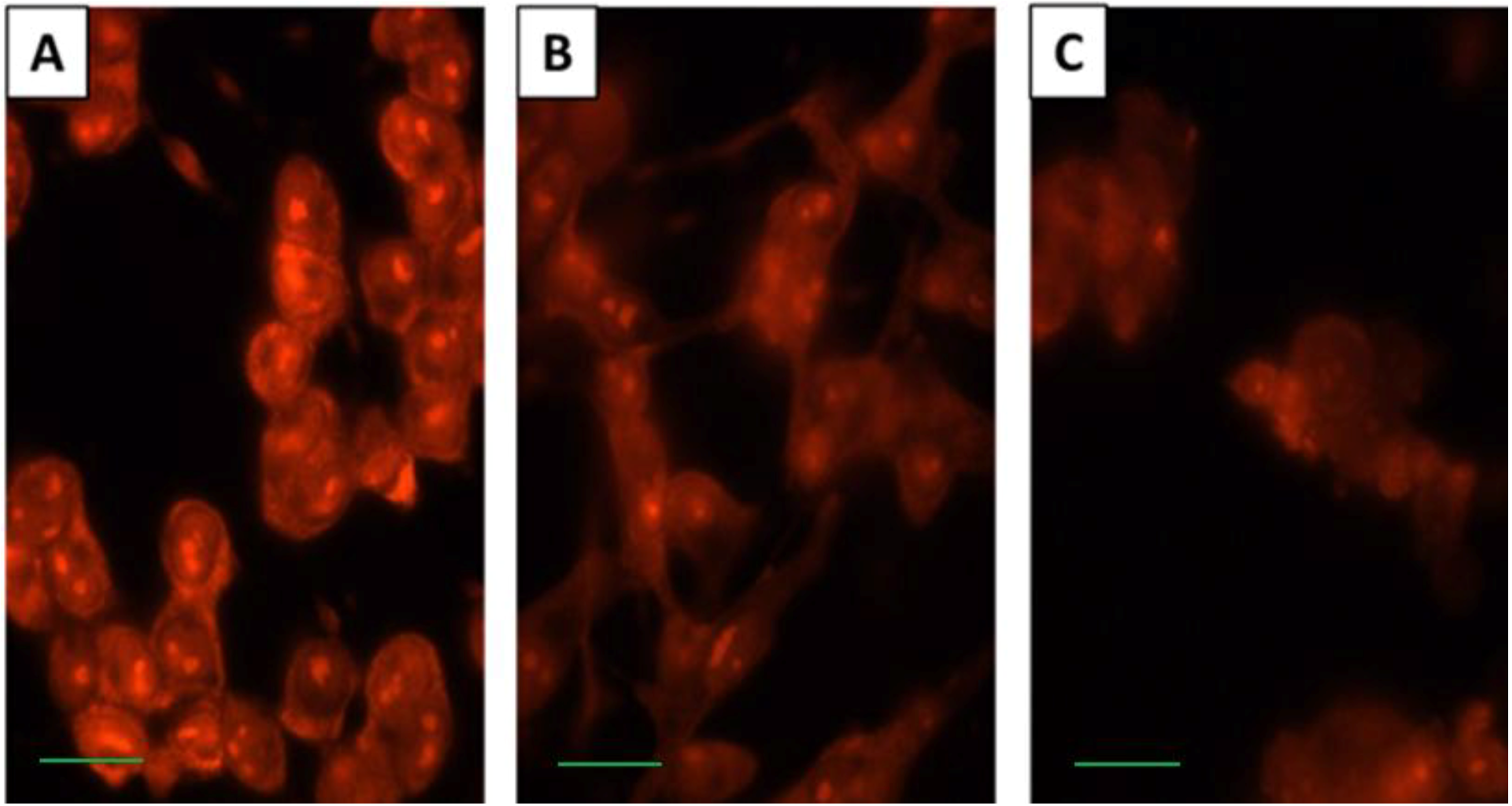
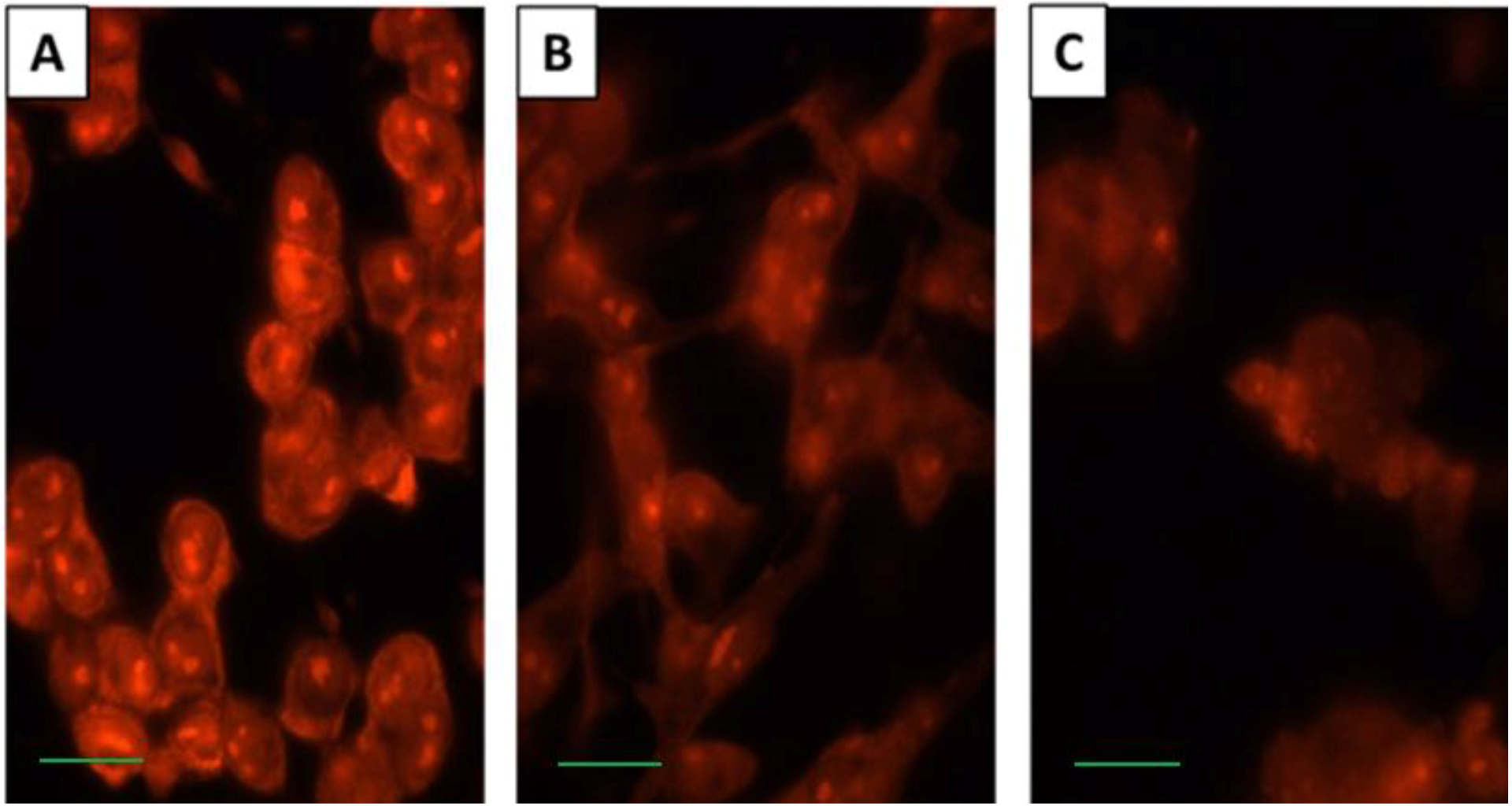
2.3. SD Inhibits Cell Membrane Permeability
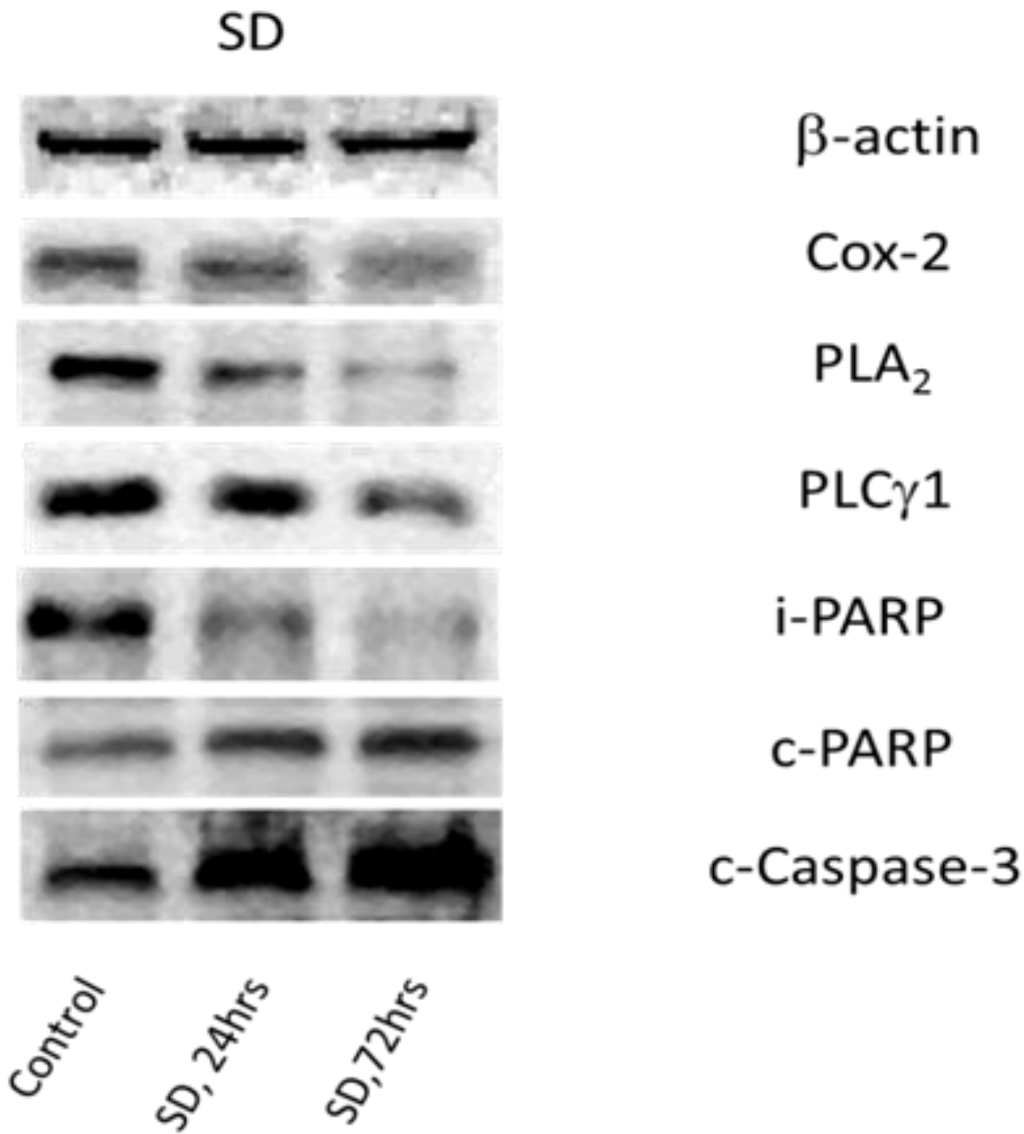
2.4. SD Inhibits Expression Levels of Cox-2 and Enhances Degradation of PLA2 and PLCγ1
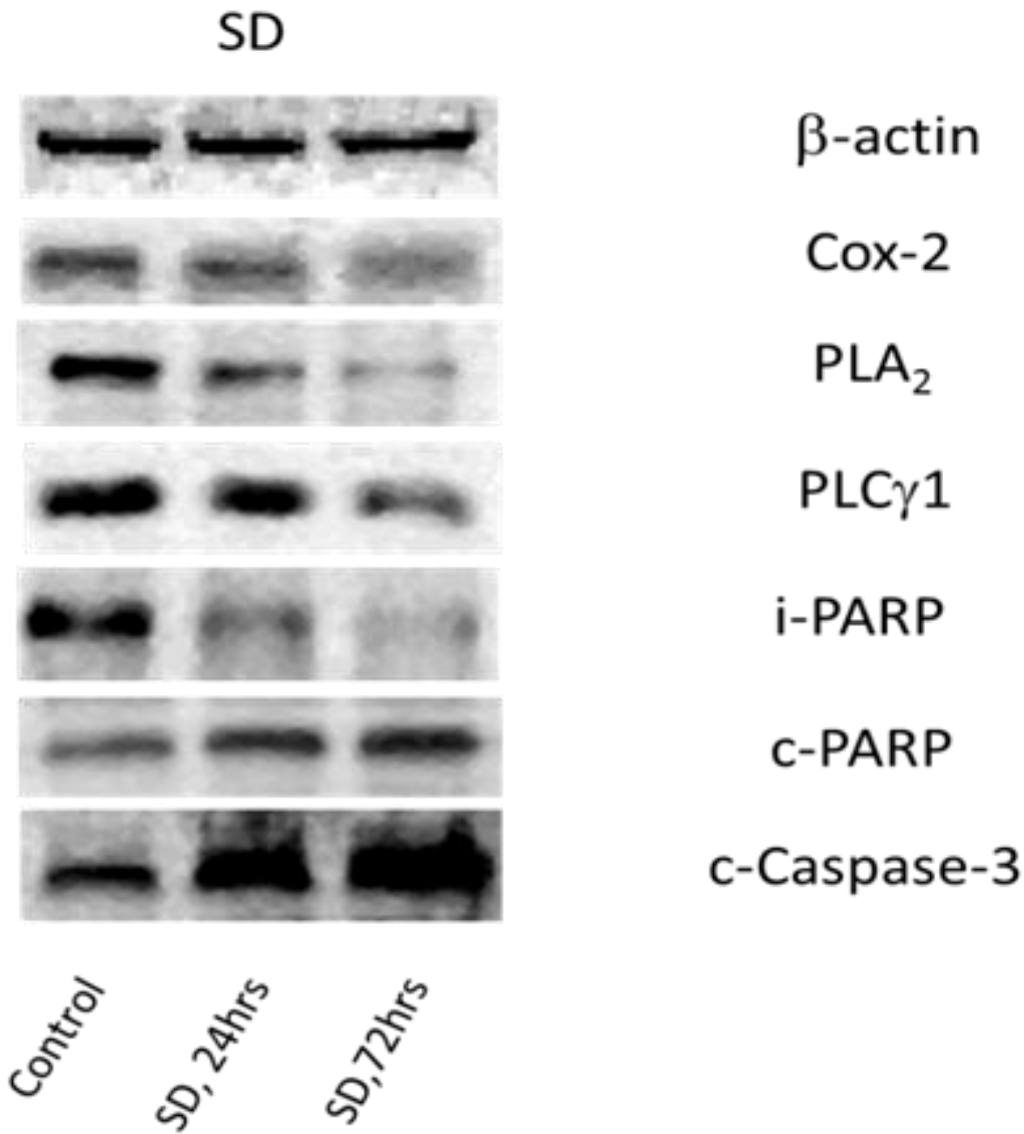
| Control | 24 h, SD | 72 h, SD | |
|---|---|---|---|
| Cox-2 | 100 | 56.81 ± 10.22, n = 4, * | 27.62 ± 6.85, n = 4, * |
| PLA2 | 100 | 57.41 ± 14.63, n = 7, * | 30.58 ± 9.67, n = 7, * |
| PLCγ1 | 100 | 59.20 ± 6.53, n = 8, * | 34.44 ± 10.05, n = 8, * |
| i-PARP | 100 | 68.51 ± 12.06, n = 4, * | 50.16 ± 13.47, n = 4, * |
| c-PARP | 100 | 150.41 ± 26.31, n = 4, * | 226.32 ± 35.27, n = 4, * |
| c-Casp-3 | 100 | 143.1 ± 20.26, n = 6, * | 197.22 ± 21.75, n = 6, * |
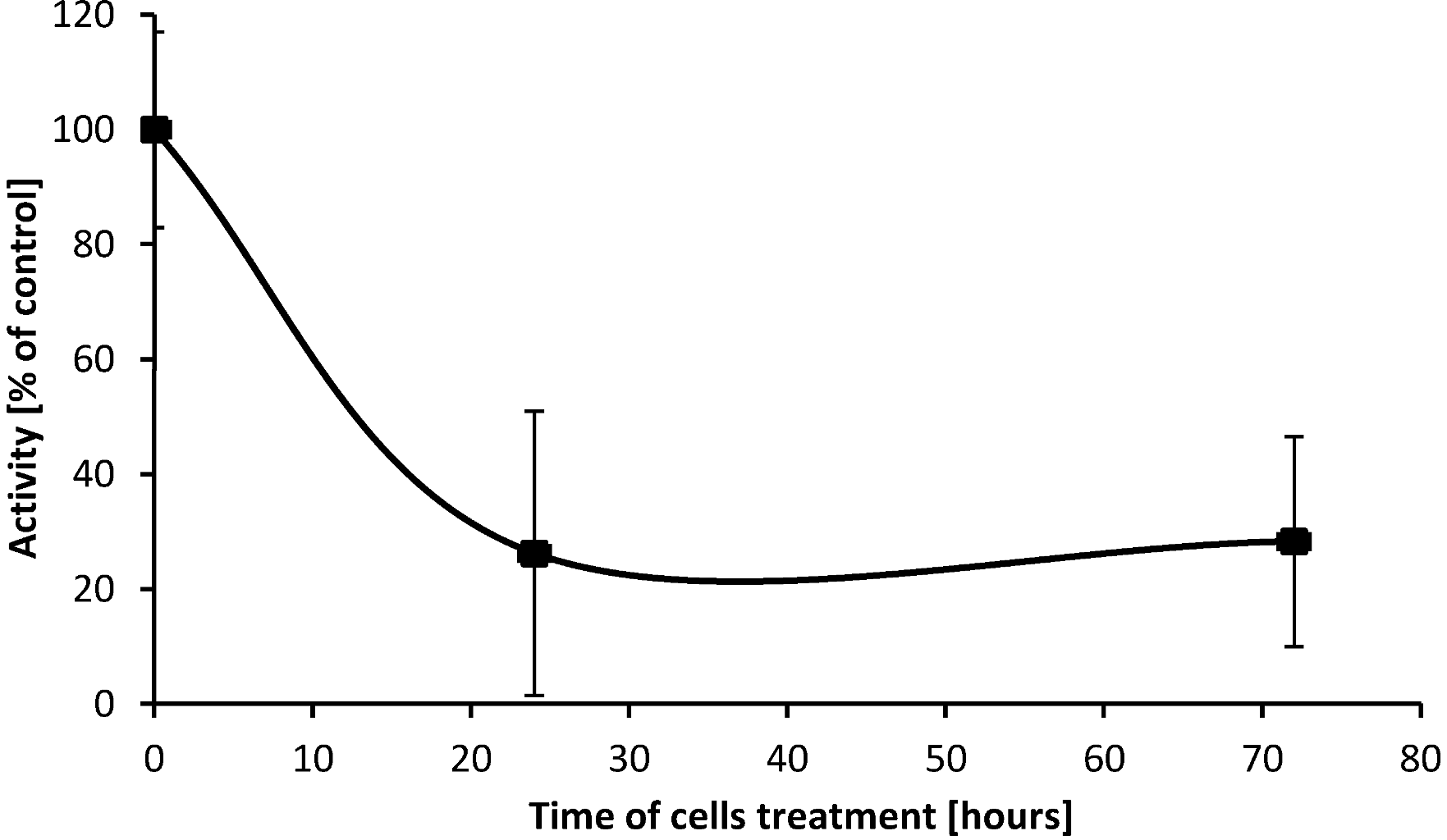
2.5. SD Inhibits Enzymatic Activity of cPLA2
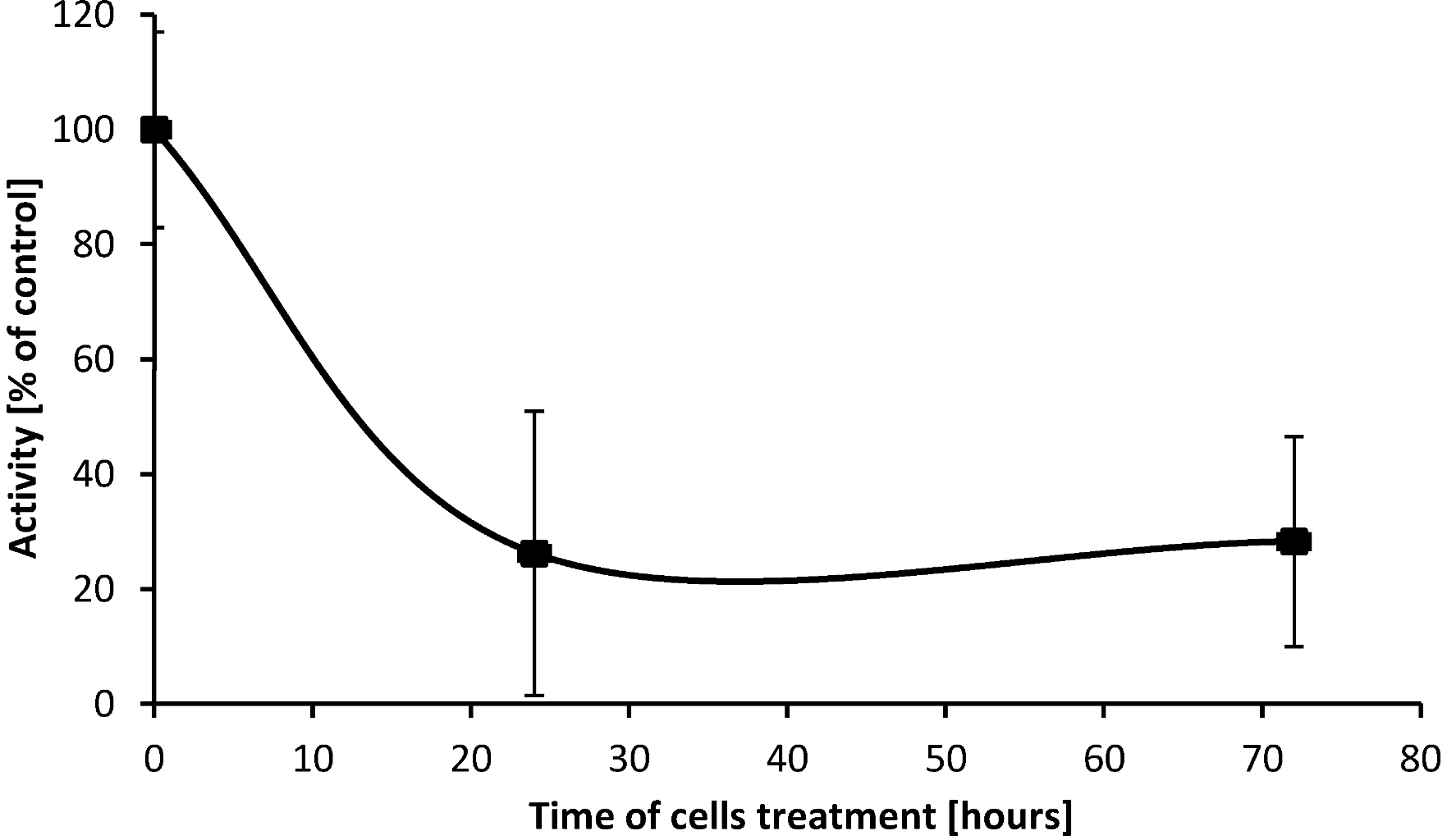
2.6. Discussion
3. Experimental Section
3.1. Chemical and Reagents
3.2. Semi-Synthesis of Sarcophine-Diol (SD)
3.3. Cell Culture
3.4. Wound Healing Assay
3.5. Cell Membrane Permeability
3.6. Determination of Cell Multiplication and Cellular Protein Content
3.7. Western Blot Analysis
3.8. Determination of Enzymatic Activity of cPLA2
3.9. Staining of the Cells with Trypan-Blue
3.10. Determination of Protein Content
3.11. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Zhang, X.; Kundoor, V.; Khalifa, S.; Zeman, D.; Fahmy, H.; Dwivedi, C. Chemopreventive effects of sarcophine-diol on skin tumor development in CD-1 mice. Cancer Lett. 2007, 253, 53–59. [Google Scholar] [CrossRef]
- Haefner, B. Chemoprevention of cancer. Cancer Res. 2003, 45, 1–8. [Google Scholar]
- Fahmy, H.; Khalifa, S.; Konoshima, T.; Zjawiony, J.K. An improved synthesis of 7,8-epoxy-1,3,11-cembratriene-15R (a), 16-diol, a cembranoid of marine origin with a potent cancer chemopreventive activity. Mar. Drugs 2004, 2, 1–7. [Google Scholar]
- Zhang, X.; Bommareddy, A.; Chen, W.; Hildreth, M.; Kaushik, R.; Zeman, D.; Khalifa, S.; Fahmy, H.; Dwivedi, C. Chemopreventive effects of sarcophine-diol on ultraviolet B-induced skin tumor development in SKH-1 hairless mice. Mar. Drugs 2009, 7, 153–165. [Google Scholar] [CrossRef]
- Fahmy, H.; Zjawiony, J.K.; Konoshima, T.; Tokuda, H.; Khan, S.; Khalifa, S. Potent skin cancer chemopreventiing activity of some novel semi-synthesis cembranoids from marine sources. Mar. Drugs 2006, 4, 1–9. [Google Scholar] [CrossRef]
- Kundor, V.; Zhang, X.; Khalifa, S.; Fahmy, H.; Dwivedi, C. A possible mechanism of action of the chemopreventive effects of sarcotriol on skin tumor development in CD-1 mice. Mar. Drugs 2006, 4, 274–285. [Google Scholar]
- Sawant, S.; Toussef, D.; Mayer, A.; Sylvester, P.; Wali, V.; Arnat, M.; Sayed, K.E. Anticancer and anti-inflamatory sulfur-containing semisynthetic derivatives of sarcophine. Chem. Pharm. Bull. 2006, 54, 1119–1123. [Google Scholar] [CrossRef]
- Zhang, X.; Bommareddy, A.; Chen, W.; Khalifa, S.; Kaushik, R.S.; Fahmy, H.; Dwivedi, C. Sarcophine-diol, a chemopreventive agent of skin cancer, inhibits cell growth and induces apoptosis through extrinsic pathway in human epidermoid carcinoma A431 cells. Trans. Onc. 2009, 2, 21–30. [Google Scholar]
- Skin Cancer Facts. Available online: http://www.skincancer.org (accessed on 10 May 2012).
- Szymanski, P.T.; Kuppast, B.; Ahmed, S.A.; Khalifa, S.; Fahmy, H. Sarcophine-diol inhibits proliferation and stimulates apoptosis in mouse melanoma B16F10 cell line. Mar. Drugs 2012, 10, 1–19. [Google Scholar]
- Adam-Klages, S.; Schwandner, R.; Luschen, S.; Ussat, S.; Kreder, D.; Kronke, M. Caspase-mediated inhibition of human cytosolic phospholipase A2 during apoptosis. J. Immunol. 1998, 161, 5687–5694. [Google Scholar]
- Bae, S.S.; Perry, D.K.; Oh, Y.S.; Choi, J.H.; Galardi, S.H.; Ghayur, T.; Ryu, S.H.; Hannun, Y.A.; Suh, P.-G. Proteolytic cleavage of phospholipase C-γ1 during apoptosis in Molt-4 cells. FASEB J. 2000, 14, 1083–1092. [Google Scholar]
- Scholey, J.M.; Taylor, K.A.; Kenrick-Jones, J. Regulation of non-muscle myosin assembly by calmodulin-dependent light chain kinase. Nature 1980, 287, 233–235. [Google Scholar] [CrossRef]
- Adelstein, R.S. Calmodulin and the regulation of the actin-myosin interaction in smooth muscle and nonmuscle cells. Cell 1982, 30, 349–350. [Google Scholar] [CrossRef]
- Daly, C.J.; Gordon, J.F.; McGrath, J.C. The use of fluorescent nuclear dyes for the study of blood vessel structure and function: Novel applications of existing techniques. J. Vasc. Res. 1992, 29, 41–56. [Google Scholar]
- Murakami, M.Y.; Nakatani, G.I.; Atsumi, K.; Inoue, L.; Kudo, I. Regulatory functions of phospholipase A2. Crit. Rev. Immunol. 1997, 17, 225–283. [Google Scholar] [CrossRef]
- Leslie, C.C. Properties and regulation of cytosolic phospholipase A2. J. Biol. Chem. 1997, 272, 16707–16712. [Google Scholar] [CrossRef]
- Porter, N.A. Mechanisms for the autooxidation of polyunsaturated lipids. Acc. Chem. Res. 1986, 19, 262–268. [Google Scholar] [CrossRef]
- Hecker, M.; Volker, U.; Fischer, C.; Messe, C.O. Identification of novel arachidonic acid metabolites formed by prostaglandin H synthase. Eur. J. Biochem. 1987, 169, 113–123. [Google Scholar] [CrossRef]
- Cao, Y.; Pearman, A.T.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Intracellular unestrified arachidonic acid signals apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 11280–11285. [Google Scholar]
- Figueiredo, A.; Caissie, A.L.; Callejo, S.A.; McLean, I.W.; Gold, P.; Burnier, M.N., Jr. Cyclooxygenase-2 expression in uveal melanoma: Novel classification of mixed-cell-type tumors. Can. J. Ophthalmol. 2003, 38, 352–356. [Google Scholar]
- Marshall, J.-C.; Caissie, A.L.; Cruess, S.R.; Cools-Lartique, J.; Burnier, M.N., Jr. The effects of a cyclooxygenase-2 (Cox-2) expression and inhibition on human uveal melanoma cell proliferation and macrophage nitric oxide production. J. Carcinog. 2007, 6, 17–27. [Google Scholar] [CrossRef]
- Karin, M. Inflammation and cancer: The long reach of RAs. Nat. Med. 2005, 11, 20–21. [Google Scholar] [CrossRef]
- Kokkotou, E.; Moss, A.C.; Torres, D.; Karaginnides, I.; Cheifez, A.; Liu, S.; O’Brien, M.; Maratos-Flier, E.; Pothoulakis, C. Melanin-concentrating hormone as a mediator of intestinal inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 10613–10618. [Google Scholar]
- Sala, G.; Dituri, F.; Raimondi, C.; Prevedi, S.; Maffucci, T.; Mazzoletti, M.; Rossi, C.; Iezzi, M.; Lattanzio, R.; Pianetelli, M.; et al. Phospholipase Cγ1 is required for metastasis development and progression. Cancer Res. 2008, 68, 10187–10196. [Google Scholar] [CrossRef]
- Clark, J.D.; Schievella, A.R.; Nalefski, E.A.; Lin, L.-L. Cytosolic phospholipase A2. J. Lipid Mediat. Cell Signal. 1995, 12, 83–117. [Google Scholar] [CrossRef]
- Weltzien, H.U. Cytolytic and membrane-perturbing properties of lysophosphatidylcholine. Biochim. Biophys. Acta 1979, 559, 259–287. [Google Scholar] [CrossRef]
- Satoh, M.S.; Lindhal, T. Role of poly (ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: structure, activation, substrates and functions during apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. [Google Scholar] [CrossRef]
- Thornberry, N.; Lazebnik, Y.A. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Rembold, C.M.; Murphy, R.A. [Ca2+]-dependent myosin phosphorylation in phorbol diester stimulated smooth muscle contraction. Am. J. Physiol. 1988, 255, C719–C723. [Google Scholar]
- Murthy, K.S.; Yee, Y.S.; Grider, J.R.; Makhlouf, G.M. Phorbol-stimulated Ca2+ mobilization and contraction in dispersed intestinal smooth muscle cells. J. Pharmacol. Exp. Ther. 2000, 294, 991–996. [Google Scholar]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Tewari, M.; Quan, L.T.; O’Rourke, K.; Desnoyers, S.; Zeng, Z.; Beider, D.R.; Poirer, G.G.; Salven, G.S.; Dixit, V.M. Yamma/CPP32 beta, a mammalian homolog of CED-3, is a Cma-inhibitable protease that cleaves the death substrate by poly(ADP-ribose) polymerase. Cell 1995, 81, 801–809. [Google Scholar] [CrossRef]
- Ihle, J.N. STAT’s signal transducers and activators of transcription. Cell 1996, 84, 331–334. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.F., Jr. STAT3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Benchimol, S. p53-dependent pathway of apoptosis. Cell Death Diff. 2001, 8, 1049–1051. [Google Scholar] [CrossRef]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the ant-apoptotic surviving gene by wild type p53. J. Biol. Chem. 2002, 277, 3247–3257. [Google Scholar]
- Kim, P.; Yoshimoto, Y.; Iino, M.; Sasaki, T.; Kirino, T.; Nonomura, Y. Impaired calcium regulation of smooth muscle during chronic vasospasm following subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 1996, 16, 334–341. [Google Scholar]
- Kim, P.; Yoshimoto, Y.; Nakaguchi, H.; Mori, T.; Asai, A.; Sasaki, T.; Kirino, T.; Nonomura, Y. Increased sarcolemmal permeability in cerebral artery during chronic spasm: An assessment using DNA-binding dyes and detection of apoptosis. J. Cereb. Blood Flow Metab. 1999, 19, 889–897. [Google Scholar]
- Sidik, K.; Smerdon, M.J. Bleomycin-induced DNA damage and repair in human cells permeabilized with lysophosphatidylcholine. Cancer Res. 1990, 50, 1613–1619. [Google Scholar]
- Lorenz, J.D.; Watkins, J.F.; Smerdon, M.J. Excision repair of UV damage in human fibroblasts reversibly permeabilized with lysophosphatidylcholine. Mutat. Res. 1988, 193, 167–719. [Google Scholar] [CrossRef]
- Chen, O.; Morimoto, S.; Kitano, E.; Koh, K.; Fukuo, B.; Jiang, S.; Chen, O.; Yasuda, A.; Hirotani, A.; Ogihara, T. Lysophosphatidylcholine causes Ca2+ influx, enhanced DNA synthesis and cytotoxity in cultured vascular smooth muscle cells. Atherosclerosis 1995, 112, 69–76. [Google Scholar] [CrossRef]
- Chai, Y.C.; Howe, P.H.; DiCorleto, P.E.; Chisolm, G.M. Oxidized low density lipoprotein and lysophosphatidylcholine stimulates cell cycle entry in vascular smooth muscle cells: Evidence for release of FGF-2. J. Biol. Chem. 1996, 271, 17791–17797. [Google Scholar]
- Hong, K.H.; Bonventre, J.O.; O’Leary, E.; Bonventre, J.V.; Lander, E.S. Deletion of cytosolic phospholipase A(2) suppresses Apc(Min)-induced tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 3935–3939. [Google Scholar]
- Segal, A.; van Duuren, B.L.; Mate, U. The identification of phorbol myristate acetate as a new metabolite of phorbol myristate acetate in mouse skin. Cancer Res. 1975, 35, 2154–2159. [Google Scholar]
- Dissanayake, S.K.; Weeraratna, A.T. Detecting PKC phosphorylation as part of the Wnt/calcium pathway in cutaneous melanoma. Methods Mol. Biol. 2008, 468, 157–172. [Google Scholar] [CrossRef]
- Foskett, K.J.; White, C.; Cheung, K.-H.; Mak, D.D.-O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Szymanski, P.T.; Muley, P.; Ahmed, S.A.; Khalifa, S.; Fahmy, H. Sarcophine-Diol Inhibits Expression of COX-2, Inhibits Activity of cPLA2, Enhances Degradation of PLA2 and PLCγ1 and Inhibits Cell Membrane Permeability in Mouse Melanoma B16F10 Cells. Mar. Drugs 2012, 10, 2166-2180. https://doi.org/10.3390/md10102166
Szymanski PT, Muley P, Ahmed SA, Khalifa S, Fahmy H. Sarcophine-Diol Inhibits Expression of COX-2, Inhibits Activity of cPLA2, Enhances Degradation of PLA2 and PLCγ1 and Inhibits Cell Membrane Permeability in Mouse Melanoma B16F10 Cells. Marine Drugs. 2012; 10(10):2166-2180. https://doi.org/10.3390/md10102166
Chicago/Turabian StyleSzymanski, Pawel T., Pratik Muley, Safwat A. Ahmed, Sherief Khalifa, and Hesham Fahmy. 2012. "Sarcophine-Diol Inhibits Expression of COX-2, Inhibits Activity of cPLA2, Enhances Degradation of PLA2 and PLCγ1 and Inhibits Cell Membrane Permeability in Mouse Melanoma B16F10 Cells" Marine Drugs 10, no. 10: 2166-2180. https://doi.org/10.3390/md10102166