
Inflammatory Bowel Disease-Associated Colorectal Cancer: Translational and Transformational Risks Posed by Exogenous Free Hemoglobin Alpha Chain, a By-Product of Extravasated Erythrocyte Macrophage Erythrophagocytosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Core Message
2. Introduction
3. People with Inflammatory Bowel Disease Are at Escalated Risk of Colitis-Associated Colorectal Cancer with a Subsequent Poor Prognosis
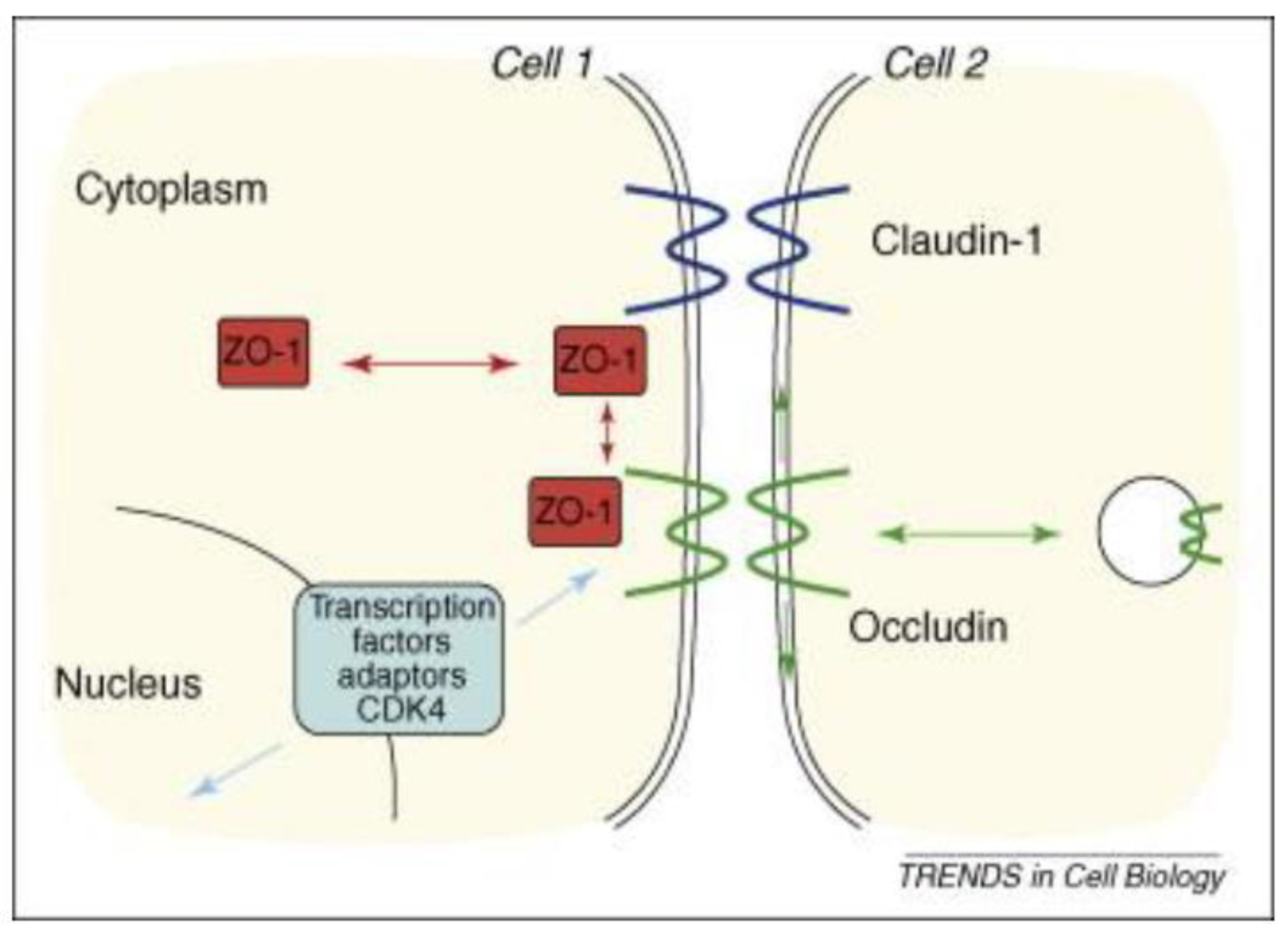
4. Malfunctioning Tight Junction Protein CALUDIN-1 Is a Source Point of Colitis-Associated Colorectal Cancer Carcinogenesis
5. Pharmacological Mitigation of Fenton Reaction to Prevent Colitis-Associated Colorectal Cancer Oncogenesis
6. Pharmaceutical Approach to Preventing Colitis-Associated Colorectal Cancer
6.1. Haptoglobin (Hp)
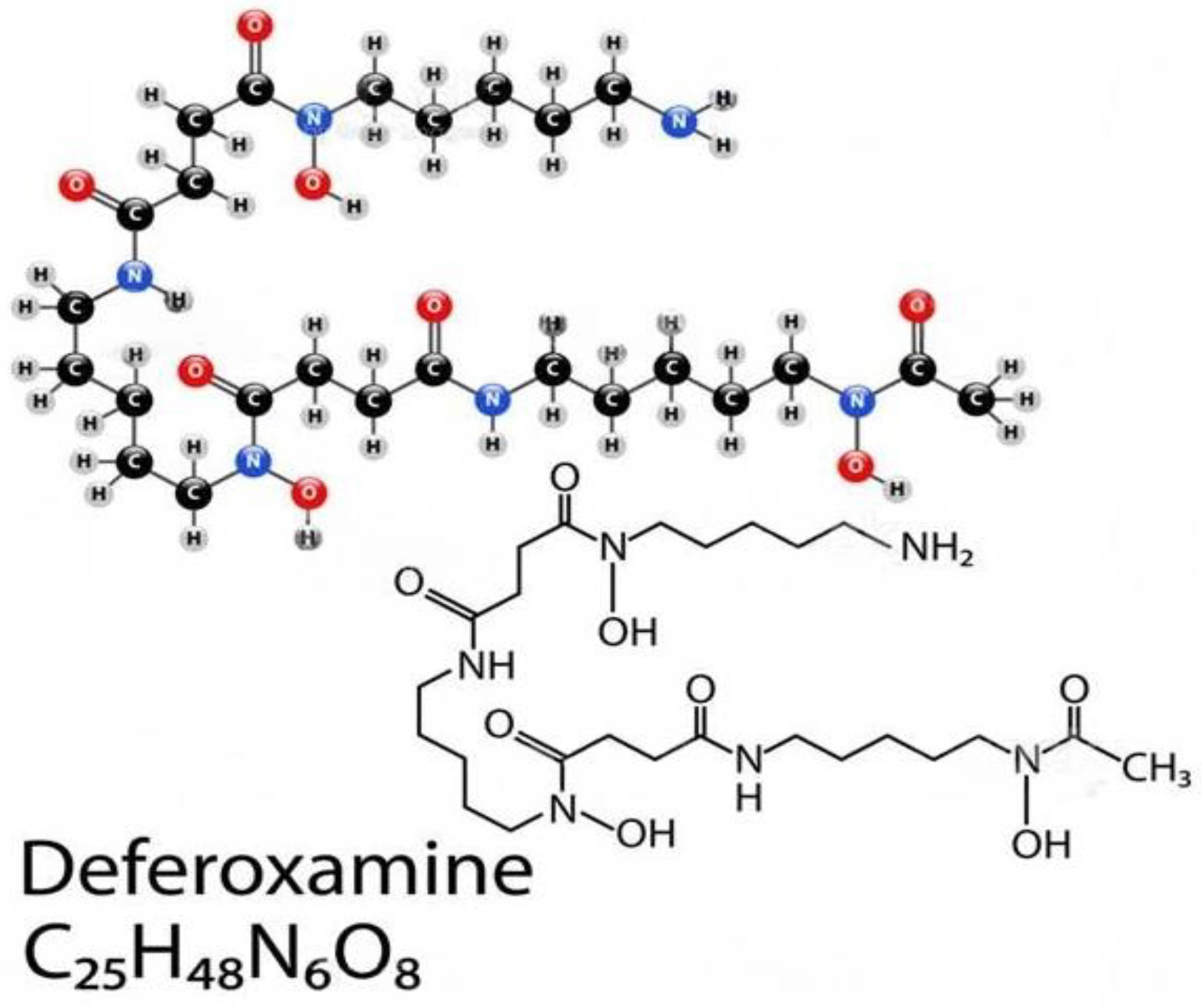
6.2. Deferoxamine (DFO)
6.3. Flavonoids
7. Closing Remarks
8. Discussion
9. Significance
10. Limitations
11. Ethical Considerations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
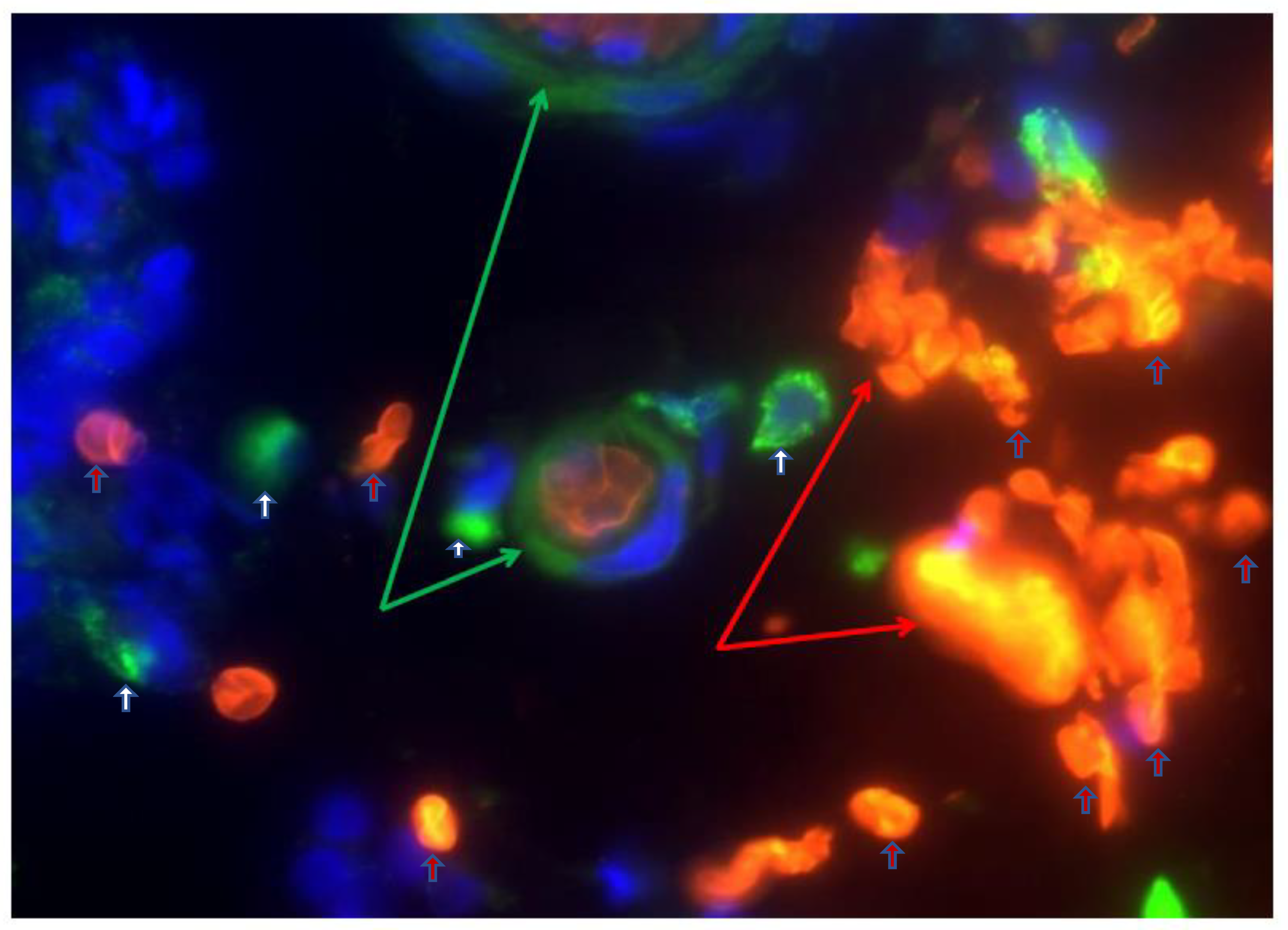
- Myers, J.N.; Schaffer, M.W.; Korolkova, O.Y.; Williams, A.D.; Gangula, P.R.; M’Koma, A.E. Implications of the Colonic Deposition of Free Hemoglobin-alpha Chain: A Previously Unknown Tissue By-product in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2014, 20, 1530–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruidenier, L.; Verspaget, H.W. Review article: Oxidative stress as a pathogenic factor in inflammatory bowel disease--radicals or ridiculous? Aliment. Pharm. Ther. 2002, 16, 1997–2015. [Google Scholar] [CrossRef] [PubMed]
- D’Odorico, A.; Bortolan, S.; Cardin, R.; D’Inca, R.; Martines, D.; Ferronato, A.; Sturniolo, G.C. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease. Scand. J. Gastroenterol. 2001, 36, 1289–1294. [Google Scholar] [PubMed]
- Krzystek-Korpacka, M.; Neubauer, K.; Berdowska, I.; Boehm, D.; Zielinski, B.; Petryszyn, P.; Terlecki, G.; Paradowski, L.; Gamian, A. Enhanced formation of advanced oxidation protein products in IBD. Inflamm. Bowel Dis. 2008, 14, 794–802. [Google Scholar] [CrossRef]
- Boehm, D.; Krzystek-Korpacka, M.; Neubauer, K.; Matusiewicz, M.; Berdowska, I.; Zielinski, B.; Paradowski, L.; Gamian, A. Paraoxonase-1 status in Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2009, 15, 93–99. [Google Scholar] [CrossRef]
- Koutroubakis, I.E.; Malliaraki, N.; Dimoulios, P.D.; Karmiris, K.; Castanas, E.; Kouroumalis, E.A. Decreased total and corrected antioxidant capacity in patients with inflammatory bowel disease. Dig. Dis. Sci. 2004, 49, 1433–1437. [Google Scholar] [CrossRef]
- Babbs, C.F. Oxygen radicals in ulcerative colitis. Free. Radic. Biol. Med. 1992, 13, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Caillet, S.; Yu, H.; Lessard, S.; Lamoureux, G.; Ajdukovic, D.; Lacroix, M. Fenton reaction applied for screening natural antioxidants. Food Chem. 2007, 100, 542–552. [Google Scholar] [CrossRef]
- de Lacerda, T.C.; Costa-Silva, B.; Giudice, F.S.; Dias, M.V.; de Oliveira, G.P.; Teixeira, B.L.; dos Santos, T.G.; Martins, V.R. Prion protein binding to HOP modulates the migration and invasion of colorectal cancer cells. Clin. Exp. Metastasis 2016, 33, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Nopel-Dunnebacke, S.; Conradi, L.C.; Reinacher-Schick, A.; Ghadimi, M. Influence of molecular markers on oncological surgery of colorectal cancer. Chirurg 2021, 92, 986–995. [Google Scholar]
- Ewing, I.; Hurley, J.J.; Josephides, E.; Millar, A. The molecular genetics of colorectal cancer. Frontline Gastroenterol. 2014, 5, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Raj, J.P. Role of biologics and biosimilars in inflammatory bowel disease: Current trends and future perspectives. J. Inflamm. Res. 2018, 11, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- M’Koma, A.E.; Moses, H.L.; Adunyah, S.E. Inflammatory bowel disease-associated colorectal cancer: Proctocolectomy andmucosectomy does not necessarily eliminate pouch related cancer incidences. Int. J. Color. Dis. 2011, 26, 533–552. [Google Scholar] [CrossRef] [Green Version]
- Marynczak, K.; Wlodarczyk, J.; Sabatowska, Z.; Dziki, A.; Dziki, L.; Wlodarczyk, M. Colitis-Associated Colorectal Cancer in Patients with Inflammatory Bowel Diseases in a Tertiary Referral Center: A Propensity Score Matching Analysis. J. Clin. Med. 2022, 11, 866. [Google Scholar] [CrossRef] [PubMed]
- Lucafo, M.; Curci, D.; Franzin, M.; Decorti, G.; Stocco, G. Inflammatory Bowel Disease and Risk of Colorectal Cancer: An Overview From Pathophysiology to Pharmacological Prevention. Front. Pharmacol. 2021, 12, 772101. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Greten, F.R. The inflammatory pathogenesis of colorectal cancer. Nat. Rev. Immunol. 2021, 21, 653–667. [Google Scholar] [CrossRef]
- M’Koma, A.E. The Multifactorial Etiopathogeneses Interplay of Inflammatory Bowel Disease: An Overview. Gastrointest. Disord. 2018, 1, 75–105. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Satsangi, J.; Silverberg, M.S.; Vermeire, S.; Colombel, J.F. The Montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 2006, 55, 749–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spekhorst, L.M.; Visschedijk, M.C.; Alberts, R.; Festen, E.A.; van der Wouden, E.J.; Dijkstra, G.; Dutch Initiative on Crohn and Colitis. Performance of the Montreal classification for inflammatory bowel diseases. World J. Gastroenterol. 2014, 20, 15374–15381. [Google Scholar] [CrossRef] [PubMed]
- Benchimol, E.I.; Fortinsky, K.J.; Gozdyra, P.; Van den Heuvel, M.; Van Limbergen, J.; Griffiths, A.M. Epidemiology of pediatric inflammatory bowel disease: A systematic review of international trends. Inflamm. Bowel Dis. 2011, 17, 423–439. [Google Scholar] [CrossRef]
- Kofla-Dlubacz, A.; Pytrus, T.; Akutko, K.; Sputa-Grzegrzolka, P.; Piotrowska, A.; Dziegiel, P. Etiology of IBD-Is It Still a Mystery? Int. J. Mol. Sci. 2022, 23, 12445. [Google Scholar] [CrossRef] [PubMed]
- Pena-Sanchez, J.N.; Osei, J.A.; Marques Santos, J.D.; Jennings, D.; Andkhoie, M.; Brass, C.; Bukassa-Kazadi, G.; Lu, X.; Johnson-Jennings, M.; Porter, L.; et al. Increasing Prevalence and Stable Incidence Rates of Inflammatory Bowel Disease Among First Nations: Population-Based Evidence From a Western Canadian Province. Inflamm. Bowel Dis. 2022, 28, 514–522. [Google Scholar] [CrossRef]
- Jones, G.R.; Lyons, M.; Plevris, N.; Jenkinson, P.W.; Bisset, C.; Burgess, C.; Din, S.; Fulforth, J.; Henderson, P.; Ho, G.-T.; et al. IBD prevalence in Lothian, Scotland, derived by capture-recapture methodology. Gut 2019, 68, 1953–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- M’Koma, A.E. Inflammatory Bowel Disease: An Expanding Global Health Problem. Clin. Med. Insights Gastroenterol. 2013, 6, 33–47. [Google Scholar] [CrossRef]
- Benchimol, E.I.; Manuel, D.G.; Guttmann, A.; Nguyen, G.C.; Mojaverian, N.; Quach, P.; Mack, D.R. Changing age demographics of inflammatory bowel disease in Ontario, Canada: A population-based cohort study of epidemiology trends. Inflamm. Bowel Dis. 2014, 20, 1761–1769. [Google Scholar] [CrossRef]
- Krzesiek, E.; Kofla-Dlubacz, A.; Akutko, K.; Stawarski, A. The Incidence of Inflammatory Bowel Disease in the Paediatric Population in the District of Lower Silesia, Poland. J. Clin. Med. 2021, 10, 3994. [Google Scholar] [CrossRef]
- Cosnes, J.; Gower-Rousseau, C.; Seksik, P.; Cortot, A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 2011, 140, 1785–1794. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Kaplan, G.G.; Ng, S.C. Changing Global Epidemiology of Inflammatory Bowel Diseases: Sustaining Health Care Delivery Into the 21st Century. Clin. Gastroenterol. Hepatol. 2020, 18, 1252–1260. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Coward, S.; Clement, F.; Benchimol, E.I.; Bernstein, C.N.; Avina-Zubieta, J.A.; Bitton, A.; Carroll, M.W.; Hazlewood, G.; Jacobson, K.; Jelinski, S.; et al. Past and Future Burden of Inflammatory Bowel Diseases Based on Modeling of Population-Based Data. Gastroenterology 2019, 156, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.C.; Tang, W.; Ching, J.Y.; Wong, M.; Chow, C.M.; Hui, A.J.; Wong, T.C.; Leung, V.K.; Tsang, S.W.; Yu, H.H.; et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology 2013, 145, 158–165.e2. [Google Scholar] [CrossRef]
- Archampong, T.N.; Nkrumah, K.N. Inflammatory bowel disease in Accra: What new trends. West. Afr. J. Med. 2013, 32, 40–44. [Google Scholar] [PubMed]
- Ukwenya, A.Y.; Ahmed, A.; Odigie, V.I.; Mohammed, A. Inflammatory bowel disease in Nigerians: Still a rare diagnosis? Ann. Afr. Med. 2011, 10, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agoda-Koussema, L.K.; Anoukoum, T.; Djibril, A.M.; Balaka, A.; Folligan, K.; Adjenou, V.; Amouzou, K.; N’dakéna, K.; Redah, R. Ulcerative colitis: A case in Togo. Med. St. Trop. 2012, 22, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Mebazaa, A.; Aounallah, A.; Naija, N.; Cheikh Rouhou, R.; Kallel, L.; El Euch, D.; Boubaker, J.; Mokni, M.; Filali, A.; Ben Osman, A. Dermatologic manifestations in inflammatory bowel disease in Tunisia. Tunis. Med. 2012, 90, 252–257. [Google Scholar]
- Senbanjo, I.O.; Oshikoya, K.A.; Onyekwere, C.A.; Abdulkareem, F.B.; Njokanma, O.F. Ulcerative colitis in a Nigerian girl: A case report. BMC Res. Notes 2012, 5, 564. [Google Scholar] [CrossRef] [Green Version]
- Bouzid, D.; Fourati, H.; Amouri, A.; Marques, I.; Abida, O.; Haddouk, S.; Ben Ayed, M.; Tahri, N.; Penha-Gonçalves, C.; Masmoudi, H. The CREM gene is involved in genetic predisposition to inflammatory bowel disease in the Tunisian population. Hum. Immunol. 2011, 72, 1204–1209. [Google Scholar] [CrossRef]
- O’Keefe, E.A.; Wright, J.P.; Froggatt, J.; Cuming, L.; Elliot, M. Medium-term follow-up of ulcerative colitis in Cape Town. S. Afr. Med. J. 1989, 76, 142–145. [Google Scholar] [PubMed]
- O’Keefe, E.A.; Wright, J.P.; Froggatt, J.; Zabow, D. Medium-term follow-up of Crohn’s disease in Cape Town. S. Afr. Med. J. 1989, 76, 139–141. [Google Scholar]
- Segal, I. Ulcerative colitis in a developing country of Africa: The Baragwanath experience of the first 46 patients. Int. J. Color. Dis. 1988, 3, 222–225. [Google Scholar] [CrossRef]
- Segal, I.; Tim, L.O.; Hamilton, D.G.; Walker, A.R. The rarity of ulcerative colitis in South African blacks. Am. J. Gastroenterol. 1980, 74, 332–336. [Google Scholar] [PubMed]
- Wright, J.P.; Marks, I.N.; Jameson, C.; Garisch, J.A.; Burns, D.G.; Kottler, R.E. Inflammatory bowel disease in Cape Town, 1975-1980. Part II. Crohn’s disease. S. Afr. Med. J. 1983, 63, 226–229. [Google Scholar] [PubMed]
- Wright, J.P.; Marks, I.N.; Jameson, C.; Garisch, J.A.; Burns, D.G.; Kottler, R.E. Inflammatory bowel disease in Cape Town, 1975-1980. Part I. Ulcerative colitis. S. Afr. Med. J. 1983, 63, 223–226. [Google Scholar] [PubMed]
- Brom, B.; Bank, S.; Marks, I.N.; Barbezat, G.O.; Raynham, B. Crohn’s disease in the Cape: A follow-up study of 24 cases and a review of the diagnosis and management. S. Afr. Med. J. 1968, 42, 1099–1107. [Google Scholar]
- Novis, B.H.; Marks, I.N.; Bank, S.; Louw, J.H. Incidence of Crohn’s disease at Groote Schuur Hospital during 1970–1974. S. Afr. Med. J. 1975, 49, 693–697. [Google Scholar]
- Sobel, J.D.; Schamroth, L. Ulcerative colitis in the South African Bantu. Gut 1970, 11, 760–763. [Google Scholar] [CrossRef] [Green Version]
- Giraud, R.M.; Luke, I.; Schmaman, A. Crohn’s disease in the Transvaal Bantu: A report of 5 cases. S. Afr. Med. J. 1969, 43, 610–613. [Google Scholar]
- Ananthakrishnan, A.N.; Kwon, J.; Raffals, L.; Sands, B.; Stenson, W.F.; McGovern, D.; Kwon, J.H.; Rheaume, R.L.; Sandler, R.S. Variation in treatment of patients with inflammatory bowel diseases at major referral centers in the United States. Clin. Gastroenterol. Hepatol. 2015, 13, 1197–1200. [Google Scholar] [CrossRef] [Green Version]
- Kappelman, M.D.; Rifas-Shiman, S.L.; Porter, C.Q.; Ollendorf, D.A.; Sandler, R.S.; Galanko, J.A.; Finkelstein, J.A. Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology 2008, 135, 1907–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegmund, B.; Zeitz, M. Inflammatory bowel disease and pregnancy. Z. Gastroenterol. 2009, 47, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouraki, V.; Savoye, G.; Dauchet, L.; Vernier-Massouille, G.; Dupas, J.L.; Merle, V.; Laberenne, J.-E.; Salomez, J.-L.; Lerebours, E.; Turck, D.; et al. The changing pattern of Crohn’s disease incidence in northern France: A continuing increase in the 10- to 19-year-old age bracket (1988–2007). Aliment. Pharm. Ther. 2011, 33, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Shmidt, E.; Dubinsky, M.C. Inflammatory Bowel Disease and Pregnancy. Am. J. Gastroenterol. 2022, 117, 60–68. [Google Scholar] [CrossRef]
- Baiocco, P.J.; Korelitz, B.I. The influence of inflammatory bowel disease and its treatment on pregnancy and fetal outcome. J. Clin. Gastroenterol. 1984, 6, 211–216. [Google Scholar]
- Heetun, Z.S.; Byrnes, C.; Neary, P.; O’Morain, C. Review article: Reproduction in the patient with inflammatory bowel disease. Aliment. Pharm. Ther. 2007, 26, 513–533. [Google Scholar] [CrossRef]
- Vermeire, S.; Carbonnel, F.; Coulie, P.G.; Geenen, V.; Hazes, J.M.; Masson, P.L.; De Keyser, F.; Louis, E. Management of inflammatory bowel disease in pregnancy. J. Crohns Colitis 2012, 6, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, C.; Paerregaard, A.; Munkholm, P.; Faerk, J.; Lange, A.; Andersen, J.; Jakobsen, M.; Kramer, I.; Czernia-Mazurkiewicz, J.; Wewer, V. Pediatric inflammatory bowel disease: Increasing incidence, decreasing surgery rate, and compromised nutritional status: A prospective population-based cohort study 2007–2009. Inflamm. Bowel Dis. 2011, 17, 2541–2550. [Google Scholar] [CrossRef]
- North American Society for Pediatric Gastroenterology; Hepatology, and Nutrition; Colitis Foundation of America; Bousvaros, A.; Antonioli, D.A.; Colletti, R.B.; Dubinsky, M.C.; Glickman, J.N.; Gold, B.D.; Griffiths, A.M. Differentiating ulcerative colitis from Crohn disease in children and young adults: Report of a working group of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the Crohn’s and Colitis Foundation of America. J. Pediatr. Gastroenterol. Nutr. 2007, 44, 653–674. [Google Scholar]
- Griffiths, A.M. Specificities of inflammatory bowel disease in childhood. Best Pract. Res. Clin. Gastroenterol. 2004, 18, 509–523. [Google Scholar] [CrossRef]
- Wang, Y.R.; Loftus, E.V.; Jr Cangemi, J.R.; Picco, M.F. Racial/Ethnic and regional differences in the prevalence of inflammatory bowel disease in the United States. Digestion 2013, 88, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Afzali, A.; Cross, R.K. Racial and Ethnic Minorities with Inflammatory Bowel Disease in the United States: A Systematic Review of Disease Characteristics and Differences. Inflamm. Bowel Dis. 2016, 22, 2023–2040. [Google Scholar] [CrossRef]
- Li, D.; Collins, B.; Velayos, F.S.; Liu, L.; Lewis, J.D.; Allison, J.E.; Flowers, N.T.; Hutfless, S.; Abramson, O.; Herrinton, L.J. Racial and ethnic differences in health care utilization and outcomes among ulcerative colitis patients in an integrated health-care organization. Dig. Dis. Sci. 2014, 59, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, G.; Liu, B.; Torres, S.; Bhuket, T.; Wong, R.J. Race/Ethnicity-Specific Disparities in the Severity of Disease at Presentation in Adults with Ulcerative Colitis: A Cross-Sectional Study. Dig. Dis. Sci. 2017, 62, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Avalos, D.J.; Mendoza-Ladd, A.; Zuckerman, M.J.; Bashashati, M.; Alvarado, A.; Dwivedi, A.; Damas, O.M. Hispanic Americans and Non-Hispanic White Americans Have a Similar Inflammatory Bowel Disease Phenotype: A Systematic Review with Meta-Analysis. Dig. Dis. Sci. 2018, 63, 1558–1571. [Google Scholar] [CrossRef]
- Hou, J.K.; El-Serag, H.; Thirumurthi, S. Distribution and manifestations of inflammatory bowel disease in Asians, Hispanics, and African Americans: A systematic review. Am. J. Gastroenterol. 2009, 104, 2100–2109. [Google Scholar] [CrossRef]
- Ventham, N.T.; Kennedy, N.A.; Nimmo, E.R.; Satsangi, J. Beyond gene discovery in inflammatory bowel disease: The emerging role of epigenetics. Gastroenterology 2013, 145, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Price, A.B. Overlap in the spectrum of non-specific inflammatory bowel disease--’colitis indeterminate’. J Clin Pathol. 1978, 31, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Farmer, M.; Petras, R.E.; Hunt, L.E.; Janosky, J.E.; Galandiuk, S. The importance of diagnostic accuracy in colonic inflammatory bowel disease. Am. J. Gastroenterol. 2000, 95, 3184–3188. [Google Scholar] [CrossRef]
- Kader, H.A.; Tchernev, V.T.; Satyaraj, E.; Lejnine, S.; Kotler, G.; Kingsmore, S.F.; Patel, D.D. Protein microarray analysis of disease activity in pediatric inflammatory bowel disease demonstrates elevated serum PLGF, IL-7, TGF-beta1, and IL-12p40 levels in Crohn’s disease and ulcerative colitis patients in remission versus active disease. Am. J. Gastroenterol. 2005, 100, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Burczynski, M.E.; Peterson, R.L.; Twine, N.C.; Zuberek, K.A.; Brodeur, B.J.; Casciotti, L.; Maganti, V.; Reddy, P.S.; Strahs, A.; Immermann, F.; et al. Molecular classification of Crohn’s disease and ulcerative colitis patients using transcriptional profiles in peripheral blood mononuclear cells. J. Mol. Diagn. 2006, 8, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, K.; Yonezawa, H.; Fiocchi, C. Inflammatory bowel disease-associated gene expression in intestinal epithelial cells by differential cDNA screening and mRNA display. Inflamm. Bowel Dis. 2003, 9, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Shkoda, A.; Werner, T.; Daniel, H.; Gunckel, M.; Rogler, G.; Haller, D. Differential protein expression profile in the intestinal epithelium from patients with inflammatory bowel disease. J. Proteome Res. 2007, 6, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Horio, Y.; Uchino, M.; Babdo, T.; Sasaki, H.; Goto, Y.; Kuwahara, R.; Minagawa, T.; Takesue, Y.; Ikeuchi, H. Incidence, Risk Factors and Outcomes of Cancer of the Anal Transit Zone in Patients with Ulcerative Colitis. J. Crohns Colitis 2020, 14, 1165–1571. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.G.; Meltzer, S.J.; Jankowski, J.A. ABC of colorectal cancer. Molecular basis for risk factors. BMJ 2000, 321, 886–889. [Google Scholar] [CrossRef]
- Rubin, D.T.; Parekh, N. Colorectal cancer in inflammatory bowel disease: Molecular and clinical considerations. Curr. Treat. Options Gastroenterol. 2006, 9, 211–220. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Harpaz, N.; Talbot, I.C. Colorectal cancer in idiopathic inflammatory bowel disease. Semin. Diagn. Pathol. 1996, 13, 339–357. [Google Scholar]
- Rubio, C.A.; Befrits, R.; Ljung, T.; Jaramillo, E.; Slezak, P. Colorectal carcinoma in ulcerative colitis is decreasing in Scandinavian countries. Anticancer. Res. 2001, 21, 2921–2924. [Google Scholar]
- Loftus, E.V., Jr. Epidemiology and risk factors for colorectal dysplasia and cancer in ulcerative colitis. Gastroenterol. Clin. N. Am. 2006, 35, 517–531. [Google Scholar] [CrossRef]
- Delaunoit, T.; Limburg, P.J.; Goldberg, R.M.; Lymp, J.F.; Loftus, E.V., Jr. Colorectal cancer prognosis among patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2006, 4, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Keller, D.S.; Windsor, A.; Cohen, R.; Chand, M. Colorectal cancer in inflammatory bowel disease: Review of the evidence. Tech. Coloproctol. 2019, 23, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Hakeem, A.R.; Scott, N.; Saunders, R.N. Significance of R1 resection margin in colon cancer resections in the modern era. Color. Dis. 2015, 17, 943–953. [Google Scholar] [CrossRef]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment. Pharm. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Stidham, R.W.; Higgins, P.D.R. Colorectal Cancer in Inflammatory Bowel Disease. Clin. Colon Rectal Surg. 2018, 31, 168–178. [Google Scholar]
- Munkholm, P. Review article: The incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment. Pharm. Ther. 2003, 18 (Suppl. S2), 1–5. [Google Scholar] [CrossRef]
- Axelrad, J.E.; Lichtiger, S.; Yajnik, V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J. Gastroenterol. 2016, 22, 4794–4801. [Google Scholar] [CrossRef]
- Baker, A.M.; Cross, W.; Curtius, K.; Al Bakir, I.; Choi, C.R.; Davis, H.L.; Temko, D.; Biswas, S.; Martinez, P.; Williams, M.; et al. Evolutionary history of human colitis-associated colorectal cancer. Gut 2019, 68, 985–995. [Google Scholar] [CrossRef]
- Um, J.W.; M’Koma, A.E. Pouch-related dysplasia and adenocarcinoma following restorative proctocolectomy for ulcerative colitis. Tech. Coloproctol. 2011, 15, 7–16. [Google Scholar] [CrossRef]
- Gallo, G.; Kotze, P.G.; Spinelli, A. Surgery in ulcerative colitis: When? How? Best Pract. Res. Clin. Gastroenterol. 2018, 32–33, 71–78. [Google Scholar] [CrossRef]
- Yashiro, M. Ulcerative colitis-associated colorectal cancer. World J. Gastroenterol. 2014, 20, 16389–16397. [Google Scholar] [CrossRef] [PubMed]
- Stjarngrim, J.; Widman, L.; Schmidt, P.T.; Ekbom, A.; Forsberg, A. Post-endoscopy colorectal cancer after colectomy in inflammatory bowel disease patients: A population-based register study. Eur. J. Gastroenterol. Hepatol. 2023, 35, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Vleggaar, F.P.; Lutgens, M.W.; Oldenburg, B.; Schipper, M.E.; Samsom, M. British and American screening guidelines inadequate for prevention of colorectal carcinoma in patients with inflammatory bowel disease. Ned. Tijdschr. Geneeskd. 2007, 151, 2787–2791. [Google Scholar]
- M’Koma, A.E.; Seeley, E.H.; Wise, P.E.; Washingtoin, M.K.; Schwartz, D.A.; Herline, A.J.; Muldon, R.L.; Caprioli, R.M. Proteomic analysis of colonic submucosa differentiates Crohn’s and ulcerative colitis. Gastroenterology 2009, 136 (Suppl. S1), A-349. [Google Scholar] [CrossRef]
- M’Koma, A.E.; Seeley, E.H.; Washington, M.K.; Schwartz, D.A.; Muldoon, R.L.; Herline, A.J.; Wise, P.E.; Caprioli, R.M. Proteomic profiling of mucosal and submucosal colonic tissues yields protein signatures that differentiate the inflammatory colitides. Inflamm. Bowel Dis. 2011, 17, 875–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeley, E.H.; Washington, M.K.; Caprioli, R.M.; M’Koma, A.E. Proteomic patterns of colonic mucosal tissues delineate Crohn’s colitis and ulcerative colitis. Proteom. Clin. Appl. 2013, 7, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Steed, E.; Balda, M.S.; Matter, K. Dynamics and functions of tight junctions. Trends Cell Biol. 2010, 20, 142–149. [Google Scholar] [CrossRef]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. 2008, 88, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Steed, E.; Rodrigues, N.T.; Balda, M.S.; Matter, K. Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 2009, 10, 95. [Google Scholar] [CrossRef] [Green Version]
- Porter, R.J.; Arends, M.J.; Churchhouse, A.M.D.; Din, S. Inflammatory Bowel Disease-Associated Colorectal Cancer: Translational Risks from Mechanisms to Medicines. J. Crohns Colitis 2021, 15, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
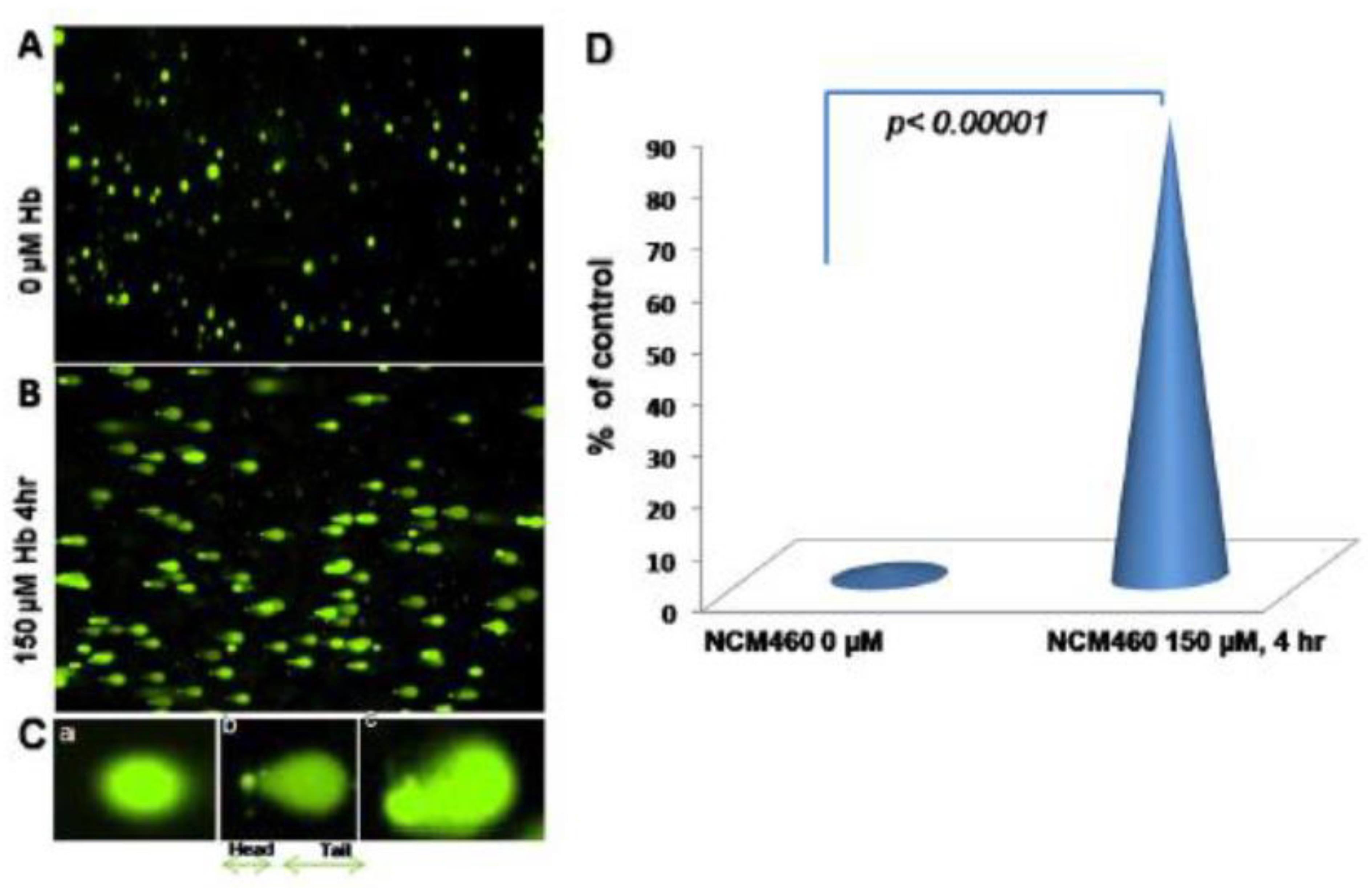
- Bystrom, L.M.; Guzman, M.L.; Rivella, S. Iron and reactive oxygen species: Friends or foes of cancer cells? Antioxid. Redox Signal. 2014, 20, 1917–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielinska, A.K.; Salaga, M.; Siwinski, P.; Wlodarczyk, M.; Dziki, A.; Fichna, J. Oxidative Stress Does Not Influence Subjective Pain Sensation in Inflammatory Bowel Disease Patients. Antioxidants 2021, 10, 1237. [Google Scholar] [CrossRef] [PubMed]
- Mrowicka, M.; Mrowicki, J.; Mik, M.; Dziki, L.; Dziki, A.; Majsterek, I. Assessment of DNA damage profile and oxidative /antioxidative biomarker level in patients with inflammatory bowel disease. Pol. Prz. Chir. 2020, 92, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J.; Pattichi, K.; Hadjigavriel, M.; Kolnagou, A. Transfusional iron overload and chelation therapy with deferoxamine and deferiprone (L1). Transfus. Sci. 2000, 23, 211–223. [Google Scholar] [CrossRef]
- Szymonik, J.; Wala, K.; Gornicki, T.; Saczko, J.; Pencakowski, B.; Kulbacka, J. The Impact of Iron Chelators on the Biology of Cancer Stem Cells. Int. J. Mol. Sci. 2021, 23, 89. [Google Scholar] [CrossRef]
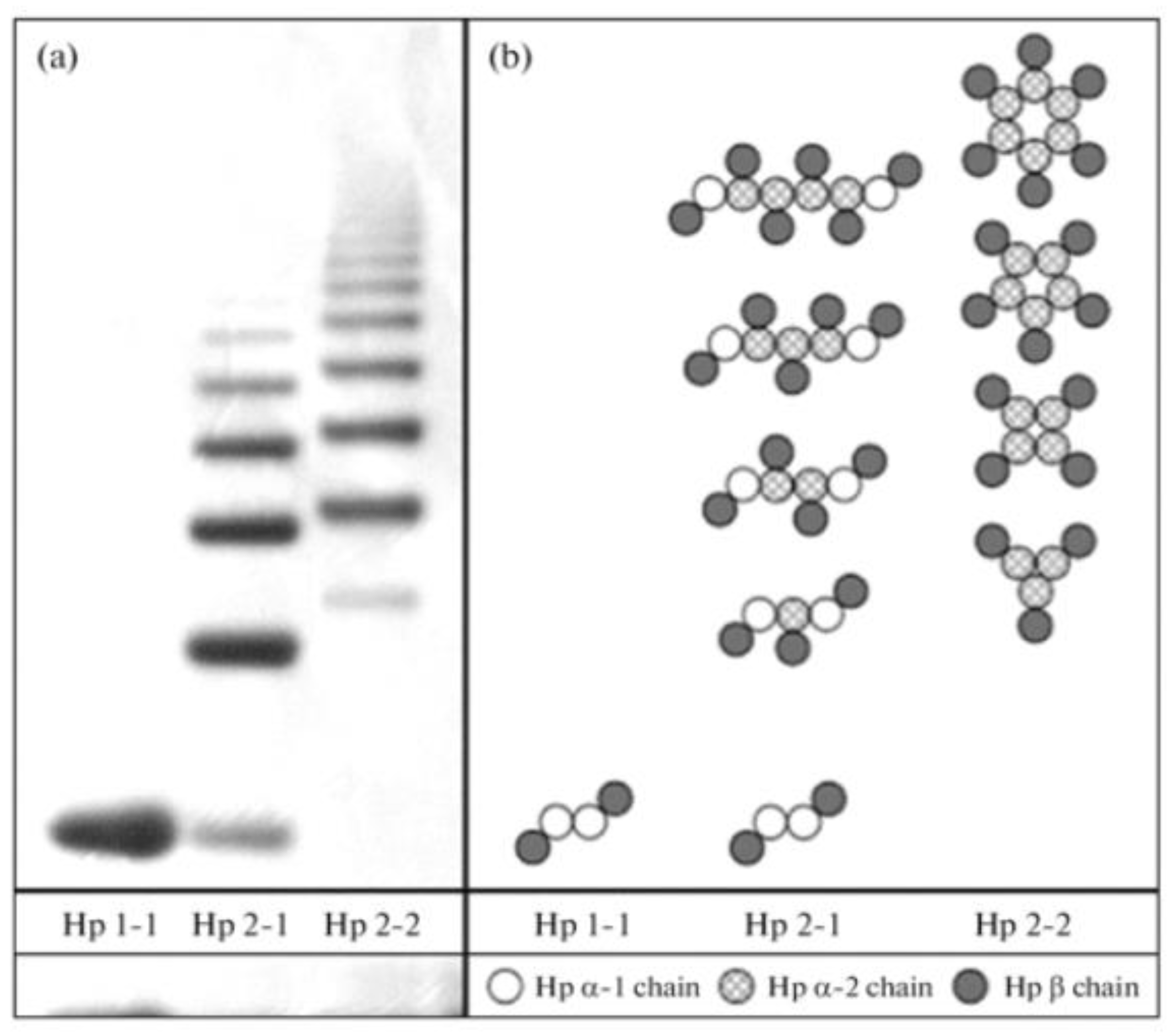
- Naryzny, S.N.; Legina, O.K. Haptoglobin as a Biomarker. Biochem. Suppl. Ser. B Biomed. Chem. 2021, 15, 184–198. [Google Scholar] [CrossRef]
- Wobeto, V.P.; Zaccariotto, T.R.; Sonati, M.D. Polymorphism of human haptoglobin and its clinical importance. Genet. Mol. Biol. 2008, 31, 602–620. [Google Scholar] [CrossRef] [Green Version]
- Marquez, L.; Cleynen, I.; Machiels, K.; Organe, S.; Ferrante, M.; Henckaerts, L.; Ballet, V.; Rutgeerts, P.J.; Vermeire, S. Haptoglobin Poymorphisms are Associated With Ulcerative Colitis and Crohn’s Disease. Gastroenterology 2010, 138, S-680. [Google Scholar] [CrossRef]
- Tayari, M.; Afsharzadeh, D. Amplification of antioxidant activity of haptoglobin(2-2)-hemoglobin at pathologic temperature and presence of antibiotics. Indian J. Clin. Biochem. 2012, 27, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Marquez, L.; Shen, C.; Machiels, K.; Perrier, C.; Ballet, V.; Organe, S. Role of Haptoglobin in Susceptibility of IBD and in Triggering Murine Colitis. Gastroenterology 2011, 140, S28. [Google Scholar] [CrossRef]
- Meczynska, S.; Lewandowska, H.; Sochanowicz, B.; Sadlo, J.; Kruszewski, M. Variable inhibitory effects on the formation of dinitrosyl iron complexes by deferoxamine and salicylaldehyde isonicotinoyl hydrazone in K562 cells. Hemoglobin 2008, 32, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Martino, E.A.; Mendicino, F.; Lucia, E.; Olivito, V.; Bova, C.; Filippelli, G.; Capodanno, I.; Neri, A.; Morabito, F.; Gentile, M.; et al. Iron chelation therapy. Eur. J. Haematol. 2023, 110, 490–497. [Google Scholar]
- Cao, Y.; Liu, M.; Cheng, J.; Yin, J.; Huang, C.; Cui, H.; Zhang, X.; Zhao, G. Acidity-Triggered Tumor-Targeted Nanosystem for Synergistic Therapy via a Cascade of ROS Generation and NO Release. ACS Appl. Mater. Interfaces. 2020, 12, 28975–28984. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, Y.; Wang, X.; Zhu, Y.; Jiao, Y.; Bao, Y.; Shi, W. Cuscuta chinensis flavonoids reducing oxidative stress of the improve sperm damage in 35 bisphenol A exposed mice offspring. Ecotoxicol. Environ. Saf. 2023, 255, 114831. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, W.; Li, Y.; Cong, Y. Enteroendocrine Cells: Sensing Gut Microbiota and Regulating Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2020, 26, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, D.; Proenca, C.; Rocha, S.; Lima, J.; Carvalho, F.; Fernandes, E.; Freitas, M. Immunomodulatory Effects of Flavonoids in the Prophylaxis and Treatment of Inflammatory Bowel Diseases: A Comprehensive Review. Curr. Med. Chem. 2018, 25, 3374–3412. [Google Scholar] [CrossRef]
- Li, M.; Weigmann, B. A Novel Pathway of Flavonoids Protecting against Inflammatory Bowel Disease: Modulating Enteroendocrine System. Metabolites 2022, 12, 31. [Google Scholar] [CrossRef]
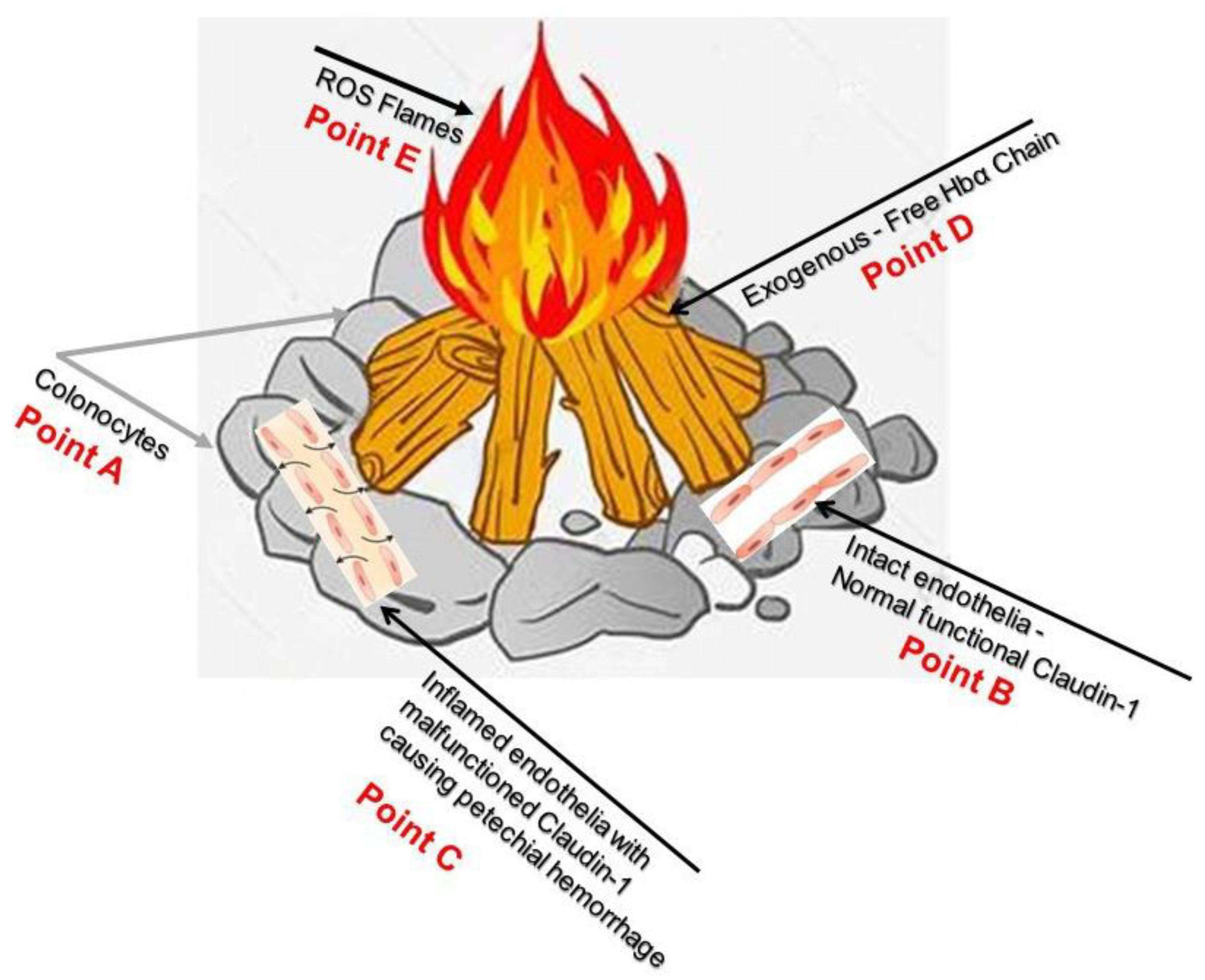
- Available online: https://pikbest.com/png-images/qianku-drawing-burning-wood-illustration_1963420.html (accessed on 27 May 2023).
- Hillier, T.A.; Vesco, K.K.; Pedula, K.L.; Beil, T.L.; Whitlock, E.P.; Pettitt, D.J. Screening for gestational diabetes mellitus: A systematic review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2008, 148, 766–775. [Google Scholar] [CrossRef]
- Lin, J.S.; Piper, M.A.; Perdue, L.A.; Rutter, C.M.; Webber, E.M.; O’Connor, E.; Smith, N.; Whitlock, E.P. Screening for Colorectal Cancer: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2016, 315, 2576–2594. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.S.; Perdue, L.A.; Henrikson, N.B.; Bean, S.I.; Blasi, P.R. Screening for Colorectal Cancer: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2021, 325, 1978–1998. [Google Scholar] [CrossRef] [PubMed]
- Miller-Wilson, L.A.; Limburg, P.; Helmueller, L.; Joao Janeiro, M.; Hartlaub, P. The impact of multi-target stool DNA testing in clinical practice in the United States: A real-world evidence retrospective study. Prev. Med. Rep. 2022, 30, 102045. [Google Scholar] [CrossRef]
- Kim, A.; Gitlin, M.; Fadli, E.; McGarvey, N.; Cong, Z.; Chung, K.C. Breast, Colorectal, Lung, Prostate, and Cervical Cancer Screening Prevalence in a Large Commercial and Medicare Advantage Plan, 2008–2020. Prev. Med. Rep. 2022, 30, 102046. [Google Scholar] [CrossRef]
- Poulson, M.R.; Geary, A.; Papageorge, M.; Laraja, A.; Sacks, O.; Hall, J.; Kenzik, K.M. The effect of medicare and screening guidelines on colorectal cancer outcomes. J. Natl. Med. Assoc. 2022, 115, 90–98. [Google Scholar] [CrossRef]
- Meester, R.G.S.; Peterse, E.F.P.; Knudsen, A.B.; de Weerdt, A.C.; Chen, J.C.; Lietz, A.P.; Dwyer, A.; Ahnen, D.J.; Siegel, R.L.; Smith, R.A.; et al. Optimizing colorectal cancer screening by race and sex: Microsimulation analysis II to inform the American Cancer Society colorectal cancer screening guideline. Cancer 2018, 124, 2974–2985. [Google Scholar] [CrossRef]
- Peterse, E.F.P.; Meester, R.G.S.; Siegel, R.L.; Chen, J.C.; Dwyer, A.; Ahnen, D.J.; Smith, R.A.; Zauber, A.G.; Lansdorp-Vogelaar, I. The impact of the rising colorectal cancer incidence in young adults on the optimal age to start screening: Microsimulation analysis I to inform the American Cancer Society colorectal cancer screening guideline. Cancer 2018, 124, 2964–2973. [Google Scholar] [CrossRef] [PubMed]
- Almario, C.V.; May, F.P.; Ponce, N.A.; Spiegel, B.M. Racial and Ethnic Disparities in Colonoscopic Examination of Individuals With a Family History of Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2015, 13, 1487–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, F.P.; Almario, C.V.; Ponce, N.; Spiegel, B.M. Racial minorities are more likely than whites to report lack of provider recommendation for colon cancer screening. Am. J. Gastroenterol. 2015, 110, 1388–1394. [Google Scholar] [CrossRef]
- Naylor, K.B. Addressing Colorectal Cancer Disparities Among African American Men Beyond Traditional Practice-Based Settings. Am. J. Public Health 2017, 107, 1356–1358. [Google Scholar] [CrossRef]
- Mehta, S.J.; Jensen, C.D.; Quinn, V.P.; Schottinger, J.E.; Zauber, A.G.; Meester, R.; Laiyemo, A.O.; Fedewa, S.; Goodman, M.; Fletcher, R.H.; et al. Race/Ethnicity and Adoption of a Population Health Management Approach to Colorectal Cancer Screening in a Community-Based Healthcare System. J. Gen. Intern. Med. 2016, 31, 1323–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naylor, K.; Ward, J.; Polite, B.N. Interventions to improve care related to colorectal cancer among racial and ethnic minorities: A systematic review. J. Gen. Intern. Med. 2012, 27, 1033–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.K.; Erdman, S.H. Double-balloon Enteroscopy: Pediatric Experience. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.; Li, H.; Fan, X.; Liu, Y.; Liu, J.H.; Rao, V.P.; Poutahidis, T.; Taylor, C.L.; Jackson, E.A.; Hewes, C.; et al. Mutations in Bone Marrow-Derived Stromal Stem Cells Unmask Latent Malignancy. Stem Cells Dev. 2010, 19. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Bielecki, C.; Felber, J.; Stallmach, A. Surveillance strategies in inflammatory bowel disease. Minerva Gastroenterol. Dietol. 2010, 56, 189–201. [Google Scholar] [PubMed]
- Burrill, B.; Crohn, B.B.R.H. The Sigmoidoscopic Picture of Chronic Ulcerative Colitis (Non-Specific). Am. J. Med. Sci. 1925, 170, 220–228. [Google Scholar]
- Warren, S.; Sommers, S.C. Cicatrizing enteritis as a pathologic entity; analysis of 120 cases. Am. J. Pathol. 1948, 24, 475–501. [Google Scholar]
- Soderlund, S.; Brandt, L.; Lapidus, A.; Karlen, P.; Brostrom, O.; Lofberg, R.; Ekbom, A.; Askling, J. Decreasing time-trends of colorectal cancer in a large cohort of patients with inflammatory bowel disease. Gastroenterology 2009, 136, 1561–1567. [Google Scholar] [CrossRef]
- Itzkowitz, S.H.; Present, D.H. Crohn’s, Colitis Foundation of America Colon Cancer in IBDSG. Consensus conference: Colorectal cancer screening and surveillance in inflammatory bowel disease. Inflamm. Bowel Dis. 2005, 11, 314–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthet, M.; Gay, G.; Sautereau, D.; Ponchon, T.; Napoleo, B.; Boyer, J.; Canard, J.M.; Dalbies, P.; Escourrou, J.; Greff, M.; et al. Endoscopic surveillance of chronic inflammatory bowel disease. Endoscopy 2005, 37, 597–599. [Google Scholar] [CrossRef]
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [Green Version]
- Averboukh, F.; Ziv, Y.; Kariv, Y.; Zmora, O.; Dotan, I.; Klausner, J.M.; Rabau, M.; Tulchinsky, H. Colorectal carcinoma in inflammatory bowel disease: A comparison between Crohn’s and ulcerative colitis. Color. Dis. 2011, 13, 1230–1235. [Google Scholar] [CrossRef]
- Sadrzadeh, S.M.; Graf, E.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin. A biologic fenton reagent. J. Biol. Chem. 1984, 259, 14354–14356. [Google Scholar] [CrossRef] [PubMed]
- Timoshnikov, V.A.; Kobzeva, T.V.; Polyakov, N.E.; Kontoghiorghes, G.J. Inhibition of Fe(2+)- and Fe(3+)- induced hydroxyl radical production by iron-chelating drug deferiprone. Free Radic. Biol. Med. 2015, 78, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Abe, C.; Miyazawa, T.; Miyazawa, T. Current Use of Fenton Reaction in Drugs and Food. Molecules 2022, 27, 5451. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Wang, M.; Zhou, M.; Lu, H.; Zhang, J.; Gao, J.; Chen, J.; Hu, Y. Doxorubicin-Metal Coordinated Micellar Nanoparticles for Intracellular Codelivery and Chemo/Chemodynamic Therapy in Vitro. ACS Appl. Bio Mater. 2019, 2, 4703–4707. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.shutterstock.com/create/editor/CiQ3YTEzMTI1YS0zOTdiLTQzNTAtOTE2ZC0yYWM5N2U0NmRlYWM (accessed on 27 May 2023).
- Woodford, F.P. Ethical experimentation and the editor. N. Engl. J. Med. 1972, 286, 892. [Google Scholar] [CrossRef]
- World Medical Association. Declaration of Helsinki: Ethical principles for Medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bragg, M.A.; Breaux, W.A.; M’Koma, A.E. Inflammatory Bowel Disease-Associated Colorectal Cancer: Translational and Transformational Risks Posed by Exogenous Free Hemoglobin Alpha Chain, a By-Product of Extravasated Erythrocyte Macrophage Erythrophagocytosis. Medicina 2023, 59, 1254. https://doi.org/10.3390/medicina59071254
Bragg MA, Breaux WA, M’Koma AE. Inflammatory Bowel Disease-Associated Colorectal Cancer: Translational and Transformational Risks Posed by Exogenous Free Hemoglobin Alpha Chain, a By-Product of Extravasated Erythrocyte Macrophage Erythrophagocytosis. Medicina. 2023; 59(7):1254. https://doi.org/10.3390/medicina59071254
Chicago/Turabian StyleBragg, Maya A., Williams A. Breaux, and Amosy E. M’Koma. 2023. "Inflammatory Bowel Disease-Associated Colorectal Cancer: Translational and Transformational Risks Posed by Exogenous Free Hemoglobin Alpha Chain, a By-Product of Extravasated Erythrocyte Macrophage Erythrophagocytosis" Medicina 59, no. 7: 1254. https://doi.org/10.3390/medicina59071254