2-{[Bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid
by
 , and
, and
See Mun Lee
 ,
,
Ainnul Hamidah Syahadah Azizan
, 
Kong Mun Lo
,
Sang Loon Tan
and
Edward R. T. Tiekink
* Research Centre for Crystalline Materials, School of Science and Technology, Sunway University, No. 5 Jalan Universiti, Bandar Sunway 47500, Selangor Darul Ehsan, Malaysia
*
Author to whom correspondence should be addressed.
Molbank 2019, 2019(4), M1082; https://doi.org/10.3390/M1082
Submission received: 4 September 2019
/
Revised: 17 September 2019
/
Accepted: 18 September 2019
/
Published: 20 September 2019
(This article belongs to the Section Structure Determination)
Abstract
:The title compound, (iPr)2NC(=S)SCH2C(=O)OH (1), was synthesized by conventional methods and its X-ray crystal structure was determined by X-ray crystallography. The compound was further characterized by analytical, IR, UV, 1D NMR (1H and 13C{1H}), and 2D NMR (DEPT-135) spectroscopy, and density functional theory (DFT) methods. X-ray crystallography on 1 confirms the formulation and reveals a nearly orthogonal relationship between the planar NCS2 and C2O2 residues. In the crystal, hydroxyl-O–H⋯O(carbonyl) hydrogen bonds lead dimers via an eight-membered {⋯OCOH}2 ring.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The description of the synthesis of compound 1, its characterization by spectroscopy as well its X-ray crystal structure determination (Figure 1) are reported. Recent [1,2] interest in organotin compounds containing this dithiocarbamate ester of a carboxylic acid arises from the known cytotoxicity or potential anticancer activity of organotin carboxylates [3] and organotin dithiocarbamates [4]. Compound 1 has been known for over 50 years being first described in 1963 [5,6,7,8]. Original interest in this compound and related species revolved around potential fungicidal activity [5], antibacterial investigations [6], and herbicidal activity [8]. Despite these investigations, detailed spectroscopic characterization of 1 and studies to determine its crystal structure are lacking.
2. Results and Discussion
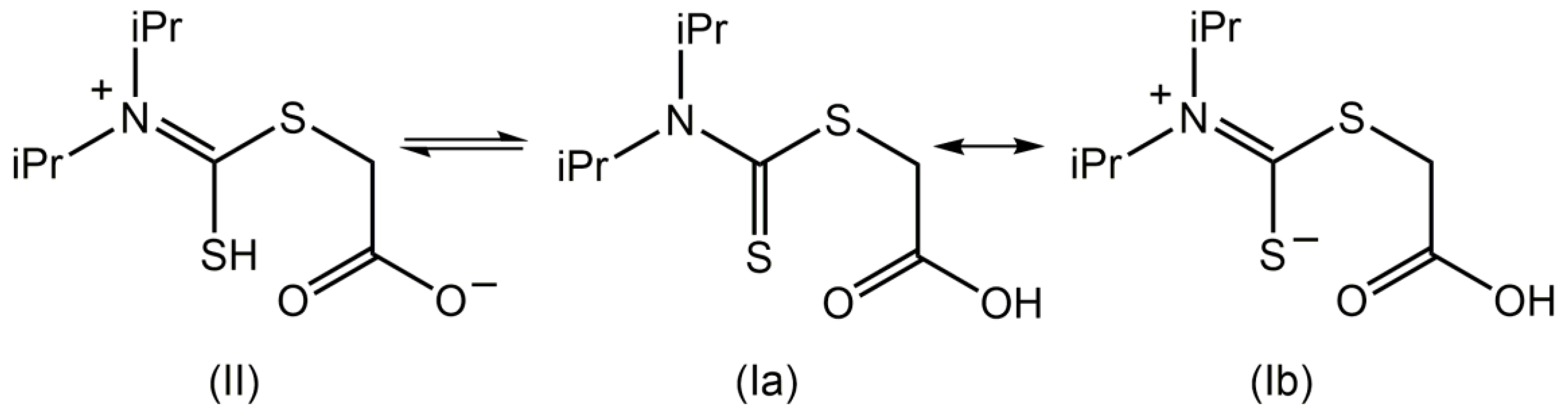
Compound 1 was prepared in good yield (60%) from a two-step reaction, that is, the in situ reaction of N,N-diisopropylamine, carbon disulfide, and sodium hydroxide to generate the dithiocarbamate, which was then reacted with sodium chloroacetate; this procedure follows several literature precedents [1,5,6,7,8]. The final product was characterized by spectroscopy (1H and 13C{1H} NMR, IR, and UV); see Supplementary Materials for original spectra. The IR spectrum showed prominent absorption bands around 1701–1660 cm−1 due to carbonyl stretching bands [1]. In the UV spectrum, a band at 280 nm is assigned to a π→π* transition, while the other band at 223 nm is assigned to a combination of π→π* and σ→σ* transitions [9]. Data for the 1H and 13C{1H} NMR of 1 are found in Section 3.2. As proposed previously for related systems [10], there are two possible tautomers for compound 1 in solution, as shown in Figure 2 (structures I and II). Structure I has two important resonance forms depicted as Ia and Ib.
The present NMR study confirmed the presence of two species in CDCl3 solution, assigned as II and Ib, which are tautomers. Variable temperature 1H NMR measurements indicated that the OH group exhibits temperature-dependent spectra, but not SH. Thus, at room temperature (25 °C), the resonance for OH in Ib is observed at 7.66 ppm, while that for SH in II is observed at 4.90 ppm [11,12]. When the temperature was increased from 25 to 50 °C, the chemical shift due to OH shifted upfield to 6.28 ppm, but that for SH remained relatively unchanged (4.76 ppm) consistent with reduced hydrogen bonding of the carboxylic acid residue at the higher temperature. In addition, two resonances are observed for NCH(CH3)2, that is, a singlet (major component) at 3.61 ppm and a septet (minor) at 3.34 ppm. The common feature of tautomeric structures II and Ib is the presence of a formal C=N double bond which implies restricted rotation which in turn indicates that the isopropyl groups are nonequivalent [13,14], in this case owing to the different relative orientations of the isopropyl groups to the S-/=S atoms. The isopropyl group directed away from the sulfur atom exhibits free rotation, so a singlet is observed. By contrast, the isopropyl group proximate to the sulfur allows for the formation of an intramolecular S⋯H(methine) interaction, which locks the conformation of the isopropyl group, leading to the observation of a septet [13,14]. Owing to the presence of two tautomers in solution, the integration of the septet is not equal to one proton. In the 13C{1H} NMR spectra, two resonances are observed for each of SCH2, NCH(CH3)2, and NC=S, an observation which is consistent with the above. The presence of tautomers II and Ib are also evidenced in the DEPT-135 experiments at temperatures 25, 45, and 50 °C. The presence of two distinct resonances due to SCH2, which are temperature-dependent, clearly indicates there is a difference in shielding depending on the proximity to C(=O)O- or C(=O)OH.
In the absence of X-ray crystallographic data determination for compound 1, a crystallographic analysis was performed. The molecular structure of compound 1 is shown in Figure 3 and selected interatomic parameters are included in the figure caption. Evidence for localization of the proton on the O2 atom is readily seen in the disparity of the C3–O1 and C3–O2 bond lengths of 1.214(2) and 1.314(2) Å, respectively. The pattern in C–S bond lengths follow the expected trends in that the formal C1–S1 thione bond length of 1.6611(18) Å is significantly shorter than the C1–S2 (1.7834(18) Å) and C2–S2 (1.7793(19) Å) bonds, which are equal within experimental error. The molecule comprises two planar chromophores. The central dithiocarbamate moiety, i.e., S1, S2, N1, and C1, exhibits a root mean square (r.m.s.) deviation of 0.0060 Å with the maximum deviation from the plane being 0.0104 Å for the C1 atom. The r.m.s. deviation through the O1, O2, C2, and C3 atoms is 0.0060 Å with the maximum deviation being 0.0172 Å for the C3 atom. The dihedral angle formed the aforementioned planes is 89.40(6)°, indicating an almost orthogonal relationship. Within the dithiocarbamate residue, the C1–N1 bond length of 1.334(2) Å is indicative of a double bond character in this bond, consistent with the presence of structures II and Ib in solution, Figure 2, with Ib dominant in the solid state. The carboxylic acid group is oriented to be parallel with the C2–S2 bond as seen in the S2–C2–C3–O1 torsion angle of −12.9(3)°, with the carbonyl-O1 atom folded over toward the dithiocarbamate moiety. This arrangement enables the formation of a S2⋯O1 contact with the separation of 3.0430(14) Å consistent with a stabilizing intramolecular interaction [15].
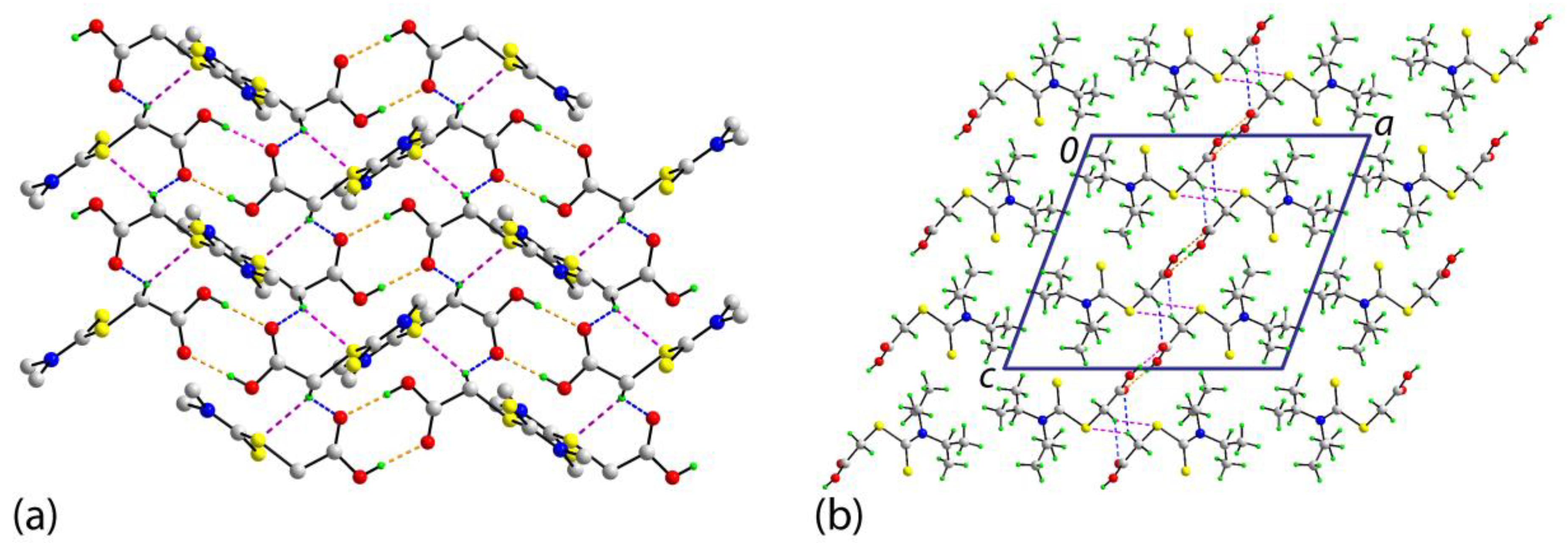
In the crystal of compound 1, hydroxyl-O2-H⋯O1(carbonyl) hydrogen bonds feature leading to the formation of a centrosymmetric, eight-membered {⋯HOCO}2 ring; geometric details characterizing these interactions are given in the caption for Figure 3. The dimers are connected into a supramolecular layer parallel to [1 0 1] via methylene-C-H⋯O(carbonyl) and methylene-C-H⋯S(ester) interactions, Figure 4a. The S2CN(iPr)2 residues project to either side of the plane and stack along the a-axis direction and no directional interactions are apparent between the layers, Figure 4b.
The molecular packing in compound 1 was also investigated via an analysis of the Hirshfeld surfaces along with the overall and decomposed two-dimensional fingerprint plots by employing the CrystalExplorer [16] and following established literature procedures [17]; calculations were performed on the major disorder component only. This analysis proves the importance of hydrophobic contacts in the crystal with 55.7% of the surface contacts being of the type H⋯H. The next most dominant surface contacts are O⋯H/H⋯O and S⋯H/H⋯S of 21.8 and 17.2%, respectively. These three contacts account for 94.7% of all contacts on the surface with the next most prominent being 3.8% for C⋯H/H⋯C.
There is a sole example of a related species to compound 1 in the crystallographic literature, namely 2-(4-morpholinecarbothioylsulfanyl)acetic acid (2) [18], where the N(iPr)2 residue in compound 1 has been substituted for a morpholine group. In compound 2, there are two independent molecules in the asymmetric unit. As anticipated, the same general trends in the C–O and C–S bond lengths are evident as for compound 1. The molecules in compound 2 are also twisted, as for compound 1, as reflected in the dihedral angles between the CNS2 and C2O2 chromophores of 88.70(14) and 89.55(15)°. The two independent molecules of compound 2 are connected into a dimeric aggregate via hydroxyl-O2-H⋯O1(carbonyl) hydrogen bonds and an eight-membered {⋯HOCO}2 ring as described for 1. Reflecting the reduced hydrogen atom content in compound 2 cf. compound 1, the contribution of H⋯H contacts to the Hirshfeld surface reduces to 37.5%; the given values are the average for the independent molecules. Other prominent surface contacts are O⋯H/H⋯O (31.2%), S⋯H/H⋯S (22.1%), and C⋯H/H⋯C (5.4%), all more important than the comparable contacts found in the crystal of 1.
The experimental structure of compound 1 was subjected to geometry optimization calculations to ascertain any influence of the crystalline manifold upon the solid-state molecular geometry. As seen in Figure 5, there is a high degree of agreement between the experimental (crystallographic) and theoretical (gas phase) structures. Overall, the r.m.s. deviation between the two molecules is 0.0191 Å.
In summary, a combined X-ray crystallographic and DFT investigation on compound 1 indicates a highly twisted molecule in the solid state, which persists in the gas phase. In the molecular packing, supramolecular dimers are formed mediated by hydroxyl-O-H⋯O(carbonyl) hydrogen bonds.
3. Materials and Methods
3.1. General Information
All chemicals were purchased from commercial sources: Sodium chloroacetate, sodium hydroxide, N,N-diisopropylamine, carbon disulfide, and hydrochloric acid fuming 37% (Merck, Darmstadt, Germany) and used as received without further purification. Solvents used were of EMSURE grade (Merck, Darmstadt, Germany) and used without further purification. The melting point was determined using a Melt-Temp automatic melting point apparatus (Cole Palmer, Staffordshire, UK) and was uncorrected. Elemental analyses were performed on a Leco TruSpec Micro CHN elemental analyzer (Leco, Saint Joseph, MI, USA). The IR spectrum was measured on a Bruker Vertex 70v FTIR (Bruker, Billerica, MA, USA) spectrophotometer from 4000 to 80 cm−1. The optical absorption spectrum was obtained from an acetonitrile solution (1 × 10−5 M) in the range 185–800 nm on a Shimadzu UV-3600 plus UV/VIS/NIR (Shimadzu Corporation, Kyoto Prefecture, Japan) spectrophotometer. 1H and 13C{1H} NMR, and DEPT-135 spectra were recorded on a Bruker Ascend 400 MHz NMR (Bruker, Billerica, MA, USA) spectrometer. Compound 1 was dissolved in deuterated chloroform and the chemical shifts were recorded in ppm relative to tetramethylsilane.
3.2. Synthesis and Characterization
N,N-diisopropylamine (1.4 mL, 0.01 mol) was slowly added dropwise into a solution of carbon disulfide (0.6 mL, 0.01 mol) and sodium hydroxide (0.4 g, 0.01 mol) in water (2.5 mL) at 10 °C. Then, the mixture was kept at 30 °C and stirred for 2 h. Next, sodium chloroacetate (1.16 g, 0.01 mol) in water (2.5 mL) was added to the mixture followed by stirring for 1.5 h. The obtained mixture was cooled in an ice bath and was neutralized with 5 M hydrochloric acid to afford white solids. These were recrystallized from a mixture of chloroform–ethanol (1:2) to give 1. Yield: 1.40 g, 60%. M. p.: 120–122 °C cf. 125–126 °C [5], 123 °C [6], 124–125 °C [7], and 127–128 °C [8]. Anal. Calc. for C9H17NO2S2: C, 45.93; H, 7.28; N, 5.95%. Found: C, 45.67; H, 7.46; N, 6.03%. IR (ATR, cm−1): 2970(m), 2930(m), 1701(s), 1652(s), 1370(m), 1316(m), 1283(s), 1138(s), 1033(s), 820(m), 667(m), and 622(m). 1H NMR (CDCl3, 25 °C) (Ib): 7.66 (1H, br, COOH), 4.17 (2H, s, SCH2), 3.61 (1H, s, N{CH(CH3)2}2), and 1.17–1.39 (12H, m, N{CH(CH3)2}2). 1H NMR (CDCl3, 25 °C) (II): 4.90 (1H, br, SH), 4.17 (2H, s, SCH2), 3.34* (1H, septet, N{CH(CH3)2}2), and 1.17–1.39 (12H, m, N{CH(CH3)2}2). (*approximate integration value of NCH not equal to one; see text and Figure S3). 13C{1H} NMR (CDCl3, 25 °C) (Ib): 167.39(NC=S), 162.08 (COOH), 42.39 (NCH(CH3)2), 28.00 (SCH2), 15.71 and 14.46 (NCH(CH3)2). 13C{1H} NMR (CDCl3, 25 °C) (II): 167.02 (NC=S), 162.08 (COO-), 42.39 (NCH(CH3)2), 33.80 (SCH2), 14.94 (NCH(CH3)2). UV (acetonitrile; nm, L·cm−1·mol−1): λabs = 280, ε = 13,100; λabs = 223, and ε = 26,160.
3.3. Crystallography
The intensity data for compound 1 were measured on a XtaLAB Synergy Dual AtlasS2 (Rigaku Corporation, Oxford, UK) diffractometer fitted with Cu Kα radiation (λ = 1.54184 Å) at T = 100(2) K. Data were measured using ω-scans with θmax being 67.1°. The raw data were reduced and corrected for absorption with the use of CrysAlis Pro [19]. Of the 14,979 reflections measured, 2154 were unique (Rint = 0.031), and of these, 2000 data satisfied the I ≥ 2σ(I) criterion. Direct methods were employed to solve the structure [20], which was then refined on F2 [21]. Non-hydrogen atoms were refined with anisotropic displacement parameters. While the C-bound H atoms were included in the riding model approximation, the hydroxyl-H atom was located from a difference map and refined with O–H restrained to 0.84 ± 0.01 Å. At this stage, disorder in each isopropropyl group was noted. The disorder was modeled over two sites with the major component having a refined site occupancy factor of 0.646(6). The major component was refined anisotropically and the minor component, isotropically. The N–C (1.43 ± 0.01 Å) and C–C (1.50 ± 0.01 Å) bonds were restrained. Finally, the anisotropic displacement parameter for the C7 site was restrained to be approximately isotropic. A weighting scheme of the form w = 1/[σ2(Fo2) + (0.045P)2 + 0.837P] was introduced, where P = (Fo2 + 2Fc2)/3). Based on the refinement of 163 parameters, the final values of R and wR (all data) were 0.034 and 0.090, respectively. The molecular structure diagram was generated with ORTEP for Windows [22] and the packing diagrams using DIAMOND [23].
Crystal data for C9H17NO2S2 (1): M = 235.35, monoclinic, P21/c, a = 15.1909(2), b = 6.22180(10), c = 13.6404(2) Å, β = 110.589(2)°, V = 1206.87(3) Å3, Z = 4, Dx = 1.295 g cm−3, F(000) = 504, and μ = 3.825 mm−1. CCDC deposition number: 1923162.
3.4. Computational Studies
All density functional theory (DFT) calculations were performed using Gaussian16 software [24], while the input and output files were processed using GaussView6 [25]. The ground state molecule was optimized in the gas phase using the atomic coordinates generated from the X-ray crystallographic study as the starting point through the long range corrected wB97XD functional [26] with the 6-311++G(d,p) Pople basis set [27].
Supplementary Materials
The following are available online: IR, UV, 1D-NMR (1H and 13C{1H}), and 2D-NMR (DEPT-135), and crystallographic data for Compound 1 in crystallographic information file (CIF) format. CCDC 1923162 also contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Author Contributions
Synthesis and spectroscopic characterization was carried out by S.M.L. and A.H.S.A. X-ray crystallography was performed by K.M.L. and E.R.T.T. Theoretical study was performed by S.L.T. The manuscript was written by S.M.L., A.H.S.A., K.M.L., S.L.T., and E.R.T.T.
Funding
The APC was funded by the Sunway University Sdn Bhd. Sunway University Sdn Bhd is thanked for financial support of this work (Grant. no. STR-RCTR-RCCM-001-2019).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anasamy, T.; Thy, C.K.; Lo, K.M.; Chee, C.F.; Yeap, S.K.; Kamalidehghan, B.; Chung, L.Y. Tribenzyltin carboxylates as anticancer drug candidates: Effect on the cytotoxicity, motility and invasivenessof breast cancer cell lines. Eur. J. Med. Chem. 2017, 125, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Lo, K.M.; Tiekink, E.R.T. Crystal structure of bis[(μ3-oxido)-(μ2-(N,N-diisopropylthiocarbamoylthio)acetato-κ2O,O’)-((N,N-diisopropylthiocarbamoylthio)acetato-κO)-bis(di-4-methylbenzyl-tin(IV))], C100H136N4O10S8Sn4. Z. Kristallogr. New Cryst. Struct. 2019, 234. in press. [Google Scholar] [CrossRef]
- Gielen, M.; Tiekink, E.R.T. 50Sn Tin compounds and their therapeutic potential. In Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine; Gielen, M., Tiekink, E.R.T., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2005; Chapter 22; pp. 421–439. [Google Scholar]
- Tiekink, E.R.T. Tin dithiocarbamates: Applications and structures. Appl. Organomet. Chem. 2008, 22, 533–550. [Google Scholar] [CrossRef]
- Carter, G.A.; Garraway, J.L.; Spencer, D.M.; Wain, R.L. Fungicides. VI. The antifungal activity of certain dithiocarbamic and hydroxydithioformic acid derivatives. Ann. Appl. Biol. 1963, 51, 135–151. [Google Scholar] [CrossRef]
- Nardi, D.; Massarani, E.; Tajana, A.; Degen, L.; Magistretti, M.J. Antibacterial nitrofuran derivatives. I. 5-Nitro-2-furaldehyde semicarbazones and thiosemicarbazones. J. Med. Chem. 1967, 10, 530–533. [Google Scholar] [CrossRef]
- Jensen, K.A.; Anthoni, U.; Kagi, B.; Larsen, C.; Pedersen, C.T. Studies of thioacids and their derivatives. IX. Thiosemicarbazides. Acta Chem. Scand. 1968, 22, 1–50. [Google Scholar] [CrossRef]
- Wakamori, S.; Yoshida, Y.; Ishii, Y. Syntheses and herbicidal activities of dithiocarbamates. I. Benzyl esters of N-substituted dithiocarbamic acids and related compounds. Agric. Biol. Chem. 1969, 33, 1367–1376. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.; Bhattacharya, B.; Majhi, K.; Majee, P.; Sarkar, U.; Seikh, M.M. Benzthiazoline-2-thione (BTT) revisited: An experimental and theoretical endeavor to understand UV-spectra. Chem. Phys. Lett. 2017, 686, 88–96. [Google Scholar] [CrossRef]
- Ng, S.W.; Das, V.G.K. Organotin esters of dithiocarbamylacetic acids. J. Organomet. Chem. 1991, 409, 143–156. [Google Scholar] [CrossRef]
- Le-Thanh, H.; Vocelle, D. 1H NMR studies of proton transfer in Schiff base and carboxylic acid systems. Can. J. Chem. 1990, 68, 1909–1916. [Google Scholar] [CrossRef]
- Couperus, P.A.; Clague, A.D.H.; van Dongen, J.P.C.M. Carbon-13 chemical shifts of some model carboxylic acids and esters. Org. Mag. Res. 1978, 11, 1590–1597. [Google Scholar] [CrossRef]
- Takeda, Y.; Tanaka, T. Three rotational isomers of se-methyl N,N-di-isopropyldiselenocarbamate and the sulphur analogue found in low temperature 1H NMR spectra. Org. Mag. Reson. 1975, 7, 107–108. [Google Scholar] [CrossRef]
- Liljefors, T.; Sandström, J. The gear effect. VII†—Conformational analysis of methyl N,N-diisopropylcarbamate, its thiol and diseleno analogues. An experimental proof for a two-step conformational interconversion mechanism. Org. Mag. Reson. 1977, 9, 276–280. [Google Scholar] [CrossRef]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalogen bonding: An overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Mckinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer v17.; The University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. E 2019, 75, 308–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, K.M.; Ng, S.W. 2-(4-Morpholine-carbothio-ylsulfan-yl)-acetic acid. Acta Crystallogr. E 2010, 66, o1078. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlis PRO; Rigaku Corporation: Oxford, UK, 2017. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND.; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculation. 1. 2nd row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
Figure 1.
Chemical diagram for 2-{[bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid (1).

Figure 2.
Possible structures for (1) in solution.

Figure 3.
The molecular structure of compound 1 showing atom labeling and displacement ellipsoids at the 50% probability level. Selected geometric parameters: C1–S1 = 1.6611(18) Å, C1–S2 = 1.7834(18) Å, C2–S2 = 1.7793(19) Å, C3–O1 = 1.214(3) Å, C3–O2 = 1.314(3) Å, C1–N1 = 1.334(2) Å; S1–C1–S2 = 119.62(10)°, S1–C1–N1 = 126.49(14)°, S2–C1–N1 = 113.87(14)°, C1–N1–C4 = 117.92(18)°, O1–C3–O2 = 124.59(16)°, O1–C3–C2 = 124.32(17)°, O2–C3–C2 = 111.00(17)°.
Figure 3.
The molecular structure of compound 1 showing atom labeling and displacement ellipsoids at the 50% probability level. Selected geometric parameters: C1–S1 = 1.6611(18) Å, C1–S2 = 1.7834(18) Å, C2–S2 = 1.7793(19) Å, C3–O1 = 1.214(3) Å, C3–O2 = 1.314(3) Å, C1–N1 = 1.334(2) Å; S1–C1–S2 = 119.62(10)°, S1–C1–N1 = 126.49(14)°, S2–C1–N1 = 113.87(14)°, C1–N1–C4 = 117.92(18)°, O1–C3–O2 = 124.59(16)°, O1–C3–C2 = 124.32(17)°, O2–C3–C2 = 111.00(17)°.

Figure 4.
The molecular packing in the crystal of compound 1: (a) A view of the supramolecular layer parallel to [1 0 1] sustained by hydroxyl-O-H⋯O(carbonyl) hydrogen bonding [O2-H2o...O1i: H2o...O1i = 1.85(3) Å, O2⋯O1i = 2.685(2) Å, and angle at H2o = 177(2)° for symmetry operation i: 1 − x, 1 − y, 1 − z], methylene-C-H⋯O(carbonyl) [C2-H2b⋯O1ii: H2b⋯O1ii = 2.49 Å, C2⋯O1ii = 3.320(2) Å, and angle at H2b = 141° for ii: 1 − x, ½ + y, ½ − z], and methylene-C-H⋯S(ester) [C2-H2b⋯S2ii: H2b⋯S2ii = 2.77 Å, C2⋯S2ii = 3.652(2) Å, and angle at H2b = 148°] interactions shown as orange, blue and pink dashed lines, respectively; non-participating hydrogen atoms and methyl groups have been omitted for reasons of clarity, and (b) a view of the unit cell contents shown in projection down the b-axis highlighting the inter-digitation of the layers shown in (a).
Figure 4.
The molecular packing in the crystal of compound 1: (a) A view of the supramolecular layer parallel to [1 0 1] sustained by hydroxyl-O-H⋯O(carbonyl) hydrogen bonding [O2-H2o...O1i: H2o...O1i = 1.85(3) Å, O2⋯O1i = 2.685(2) Å, and angle at H2o = 177(2)° for symmetry operation i: 1 − x, 1 − y, 1 − z], methylene-C-H⋯O(carbonyl) [C2-H2b⋯O1ii: H2b⋯O1ii = 2.49 Å, C2⋯O1ii = 3.320(2) Å, and angle at H2b = 141° for ii: 1 − x, ½ + y, ½ − z], and methylene-C-H⋯S(ester) [C2-H2b⋯S2ii: H2b⋯S2ii = 2.77 Å, C2⋯S2ii = 3.652(2) Å, and angle at H2b = 148°] interactions shown as orange, blue and pink dashed lines, respectively; non-participating hydrogen atoms and methyl groups have been omitted for reasons of clarity, and (b) a view of the unit cell contents shown in projection down the b-axis highlighting the inter-digitation of the layers shown in (a).

Figure 5.
An overlay diagram of the experimental (green image) and optimized molecules (red).

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, S.M.; Azizan, A.H.S.; Lo, K.M.; Tan, S.L.; Tiekink, E.R.T. 2-{[Bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid. Molbank 2019, 2019, M1082. https://doi.org/10.3390/M1082
AMA Style
Lee SM, Azizan AHS, Lo KM, Tan SL, Tiekink ERT. 2-{[Bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid. Molbank. 2019; 2019(4):M1082. https://doi.org/10.3390/M1082
Chicago/Turabian StyleLee, See Mun, Ainnul Hamidah Syahadah Azizan, Kong Mun Lo, Sang Loon Tan, and Edward R. T. Tiekink. 2019. "2-{[Bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid" Molbank 2019, no. 4: M1082. https://doi.org/10.3390/M1082
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.