1-[N-Methyl-N-(Phenyl)amino]-3-(4-Methylphenyl)Thiourea


Research Centre for Crystalline Materials, School of Science and Technology, Sunway University, No. 5 Jalan Universiti, Bandar Sunway 47500, Selangor Darul Ehsan, Malaysia
*
Author to whom correspondence should be addressed.
Molbank 2019, 2019(1), M1052; https://doi.org/10.3390/M1052
Submission received: 20 February 2019
/
Revised: 5 March 2019
/
Accepted: 8 March 2019
/
Published: 11 March 2019
(This article belongs to the Section Structure Determination)
Abstract
:The title compound, 1-[N-methyl-N-(phenyl)amino]-3-(4-methylphenyl)thiourea (1), was synthesized by the reaction of 1-methyl-1-phenyl hydrazine and 4-tolyl isothiocyanate, and was characterized by spectroscopy (1H and 13C{1H} NMR, IR, and UV), elemental analysis as well as by single crystal X-ray crystallography. In the solid state, the molecule exists as the thioamide tautomer and features an anti-disposition of the thioamide–N–H atoms; an intramolecular N–H⋯N hydrogen bond is noted. The molecular conformation resembles that of the letter L. In the molecular packing, thioamide-N1–H⋯S1(thione) hydrogen bonds lead to centrosymmetric eight-membered {⋯HNCS}2 synthons. The dimers are assembled into a supramolecular layer in the bc-plane by phenyl- and methyl-C–H⋯π(phenyl) interactions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Thiourea, H2NC(=S)NH2, is susceptible for to up to four substitutions to give a wide range of compounds, R1(R2)NC(=S)N(R3)R4 for R1–4 = alkyl/aryl [1]. These molecules can possess interesting properties and are well known for their ability to complex metal ions [1]. Recent interest in thiourea derivatives revolves around their applications in dual hydrogen bonding catalysis for asymmetric synthesis [2,3] and for their potential biological potency [4], including when complexing metals [5]. Indeed, the original interest in the title compound 1-[N-methyl-N-(phenyl)amino]-3-(4-methylphenyl)thiourea (1), Scheme 1, related to the investigation of antiviral activity [6]; since then there have been no other reports of (1) in the literature. Herein, the synthesis, spectroscopic characterization, and X-ray crystal structure determination of the title compound, (1), are described.
2. Results and Discussion
The reaction of 1-methyl-1-phenylhydrazine and 4-tolyl isothiocyanate yielded the title compound (1) in good yield (71%). The compound was characterized by spectroscopy; see the Supplementary Materials for original spectra. The 1H spectrum showed the expected resonances and integration. The downfield shift for the NH proton proximate to the C=S and N(Me)Ph groups cf. NH(4-tolyl) is ascribed to the proximity to the nitrogen bound lone pair of electrons. The most prominent feature of the 13C{1H} spectrum was the downfield resonance at δ 180.2 ascribed to N2C(=S). In the IR spectrum, characteristic bands of ν(N–H) [3266 (m)], ν(C–N) [1485 (vs)], and ν(C=S) [1256 (vs)] were observed. In the UV spectrum, two bands were observed at λabs 242 nm and 267 nm and are ascribed to π‒π* and n‒π* transitions, respectively.
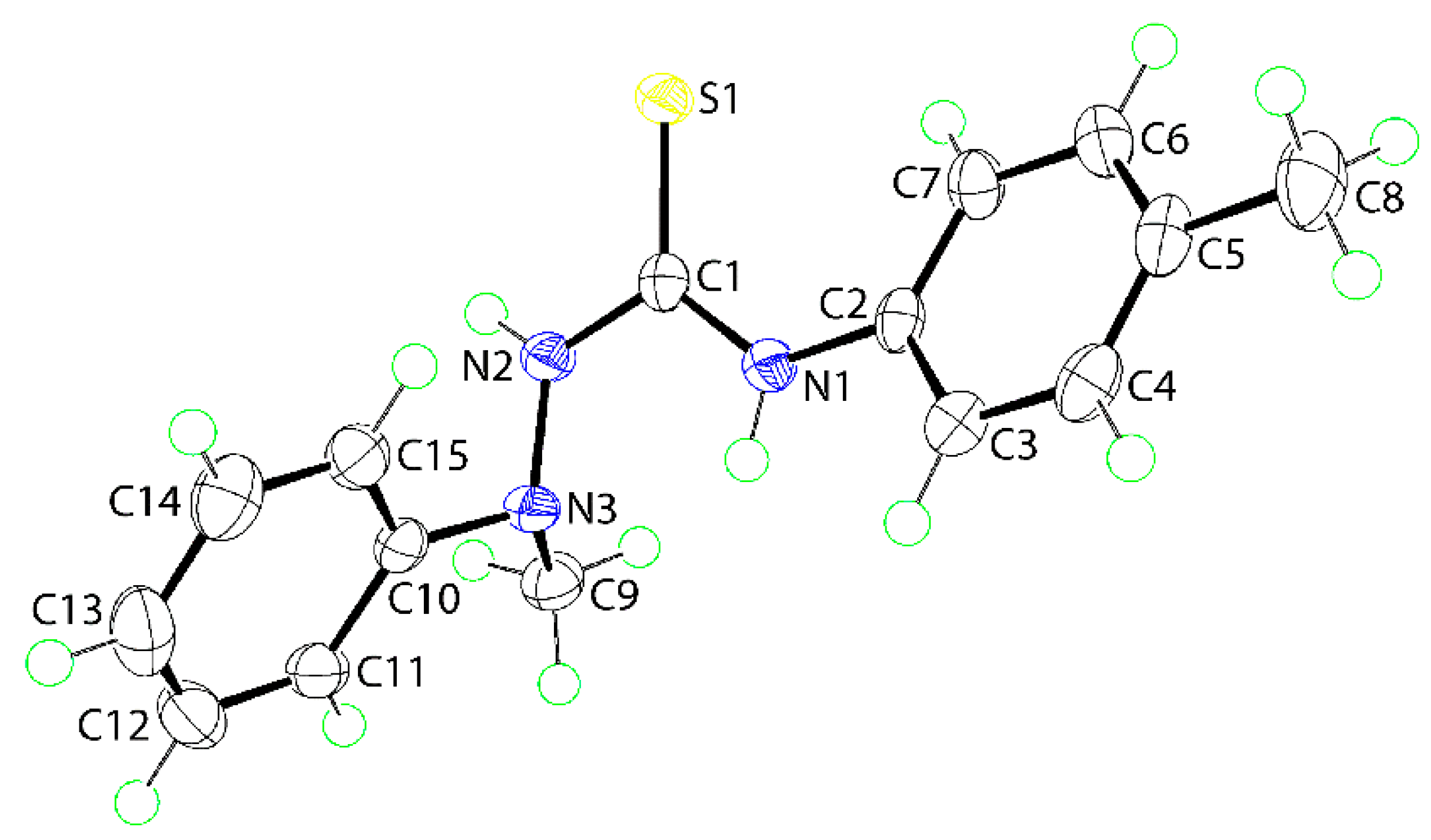
The molecular structure of (1) was also determined by X-ray crystallographic methods and is shown in Figure 1. The five atoms of the central chromophore, that is, S1, C1, and N1–N3, are effectively co-planar as evidenced in the root mean square deviation of 0.0223 Å; the maximum deviation from the least-squares plane was 0.0337(8) Å for the N2 atom. The appended C2–tolyl and C10–phenyl rings form dihedral angles of 25.42(6) and 81.18(3)° with the central residue, respectively, indicating skewed and orthogonal relationships. The dihedral angle between the terminal rings is 74.05(4)° which suggests that, to a first approximation, the molecule has the shape of the letter L. The C1–N1, N2 bond lengths are similar [1.3491(16) cf. 1.3458(16) Å] but, the S1–C1–N1 angle [127.50(10)°] is significantly wider than the S1–C1–N2 angle [117.36(9)°], presumably to minimize steric repulsion. The thioamide–NH atoms adopt an anti-disposition. An intramolecular thioamide-N1-H⋯N3 hydrogen bond is noted [N1–H1n⋯N3: H1n⋯N3 = 2.200(14) Å, N1⋯N3 = 2.6684(15) Å with angle at H1n = 113.5(11)°] that closes an S(5) loop.
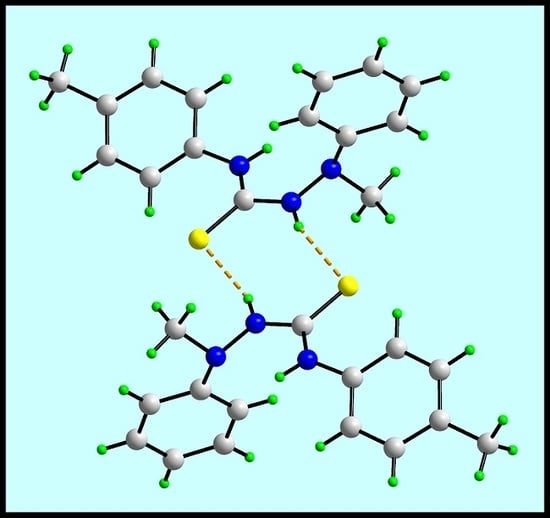
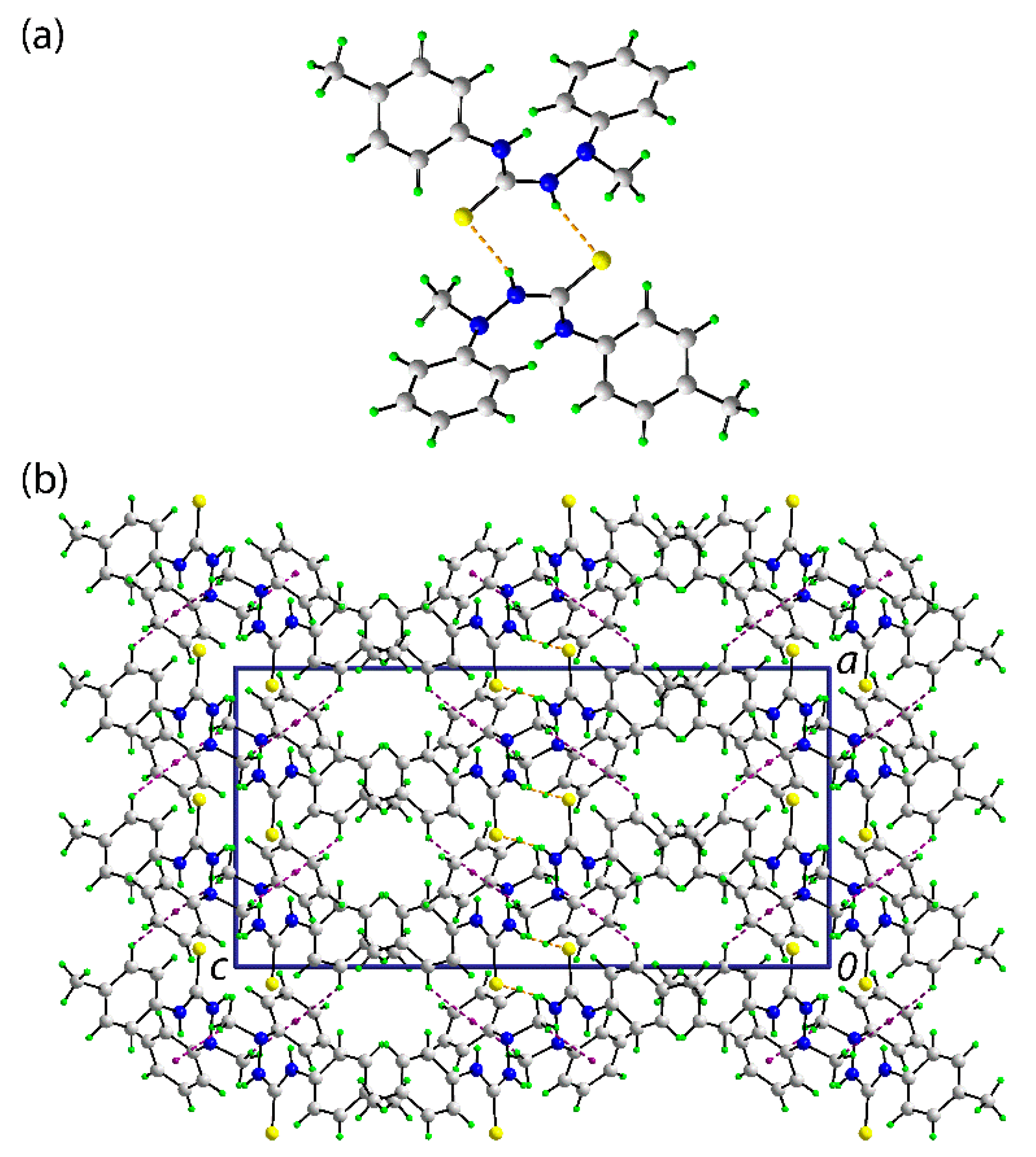
In the crystal of (1), the most prominent feature of the molecular packing is the formation of thioamide-N1–H⋯S1(thione) hydrogen bonds. These lead to the formation of a supramolecular dimer via a centrosymmetric, eight-membered {⋯HNCS}2 synthon, as shown in Figure 2a; geometric details characterizing intermolecular interactions discussed herein are given in the figure caption. The connections between the dimers are of the type phenyl- and methyl–C–H⋯π(phenyl) whereby the C10–C15 phenyl ring effectively functions as a bridge between the dimers. The result is a supramolecular layer in the bc-plane. Layers stack along the a-axis with no directional interactions between them. A view of the unit cell contents is shown in Figure 2b.
There is a closely related structure in the literature where the N-bound phenyl group of (1) is substituted by a methyl group and the original 4-tolyl ring is now a phenyl ring, that is, Me2NN(H)C(=S)N(H)Ph (2) [7]. Molecule (2) adopts essentially the same conformation as just described for (1) but, some systematic changes in the key bond lengths are evident. Thus, while the C1=S1 [1.6813(18) Å] bond length in (2) is experimentally equivalent to that in (1), the C1–N1 [1.330(2) Å] and C1–N2 [1.330(3) Å] bond lengths in (2) are shorter. The reduction in the specified bond lengths is consistent with there being reduced delocalization of π-electron density over the ring as a result of replacing phenyl in (1) with electron donating methyl in (2), and 4-tolyl with electron-withdrawing phenyl. The molecular packing of (2) features the same eight-membered {⋯HNCS}2 synthon seen in the crystal of (1).
In conclusion, an X-ray crystallographic analysis on (1) confirms the molecule to exist as the thioamide tautomer, to have an anti-disposition of the thioamide–N–H atoms and an intramolecular N1–H⋯N3 hydrogen bond. Overall, the conformation of the molecule resembles that of the letter L. The presence of thioamide–N–H⋯S(thione) hydrogen bonds in the molecular packing leads to the formation of eight-membered {⋯HNCS}2 synthons.
3. Materials and Methods
3.1. General Information
All standard chemicals and solvents were sourced from Merck (Darmstadt, Germany) and used without further purification. The melting point was determined on a Biobase automatic melting point apparatus MP450 (Biobase Group, Jinan, Shandong Province, China). The IR spectrum was measured on a Bruker Vertex 70v FTIR (Billerica, MA, USA) spectrophotometer from 4000 to 80 cm−1. Elemental analyses were performed on a Leco TruSpec Micro CHN Elemental Analyzer (Saint Joseph, MI, USA). 1H and 13C{1H} NMR spectra were recorded in CDCl3 solution on a Bruker Ascend 400 MHz NMR (Billerica, MA, USA) spectrometer with chemical shifts relative to tetramethylsilane. The optical absorption spectra were obtained from an acetonitrile solution of 1 × 10−5 M in the range 200–800 nm on a Shimadzu UV-3600 plus UV/VIS/NIR (Shimadzu Corporation, Kyoto, Japan) spectrophotometer.
3.2. Synthesis and Characterization of (1)
1-Methyl-1-phenylhydrazine (0.01 mol, 1.18 mL) was added dropwise to 4-tolyl isothiocyanate (0.01 mol, 1.49 g) in acetone (15 mL). The resulting mixture was stirred for 2 h and then left for slow evaporation under ambient conditions. The solid obtained was recrystallized in the solvent mixture 1:1 v/v chloroform/acetonitrile. Colorless crystals were formed after two weeks. Yield: 1.93 g, 71%. M. pt: 159.5–161.7 °C; Lit. 165 °C [6]. Anal. Calc. for C15H17N3S: C, 66.39; H, 6.31; N, 15.48%. Found: C, 66.34; H, 6.26; N, 15.35%. IR (cm−1): 3266 (m) ν(N-H), 3138 (m) ν(C–H), 1485 (vs) ν(C–N), 1256 (vs) ν(C=S). 1H NMR (CDCl3): δ 8.78 (s, br, 1H, Ph–NH), 7.69 (s, br, 1H, N–NH), 7.41 (d, 2H, C6H4-3, 3JHH = 8.26 Hz), 7.36–7.32 (m, 2H, C6H5-3), 7.15 (d, 2H, C6H4-2, 3JHH = 8.13 Hz), 7.05–7.00 (m, 3H, C6H5-2,4), 3.21 (s, 3H, NCH3), 2.32 (s, 3H, aryl–CH3). 13C{1H} NMR (CDCl3): δ 180.2 (Cq), 148.9 (C6H5, C1), 136.1 (C6H4, C1), 135.0 (C6H4, C4), 129.5 (C6H4, C3, C5), 129.3 (C6H4, C2, C6), 124.7 (C6H5, C3, C5), 122.4 (C6H5, C4), 114.8 (C6H5, C2, C6), 41.8 (NCH3), 21.0 (C6H4–CH3). UV (acetonitrile; nm, L·cm−1·mol−1): λabs = 267, ε = 26,700; λabs = 242, ε = 22,500.
3.3. Crystallography
Intensity data for (1) were measured at T = 100(2) K on a XtaLAB Synergy Dual AtlasS2 (Rigaku Polska SP. Z O O, Wrocław, Poland) diffractometer fitted with Cu Kα radiation (λ = 1.54184 Å) using ω-scans in the θmax range 3.8°–67.1°. Data reduction, including absorption correction, was accomplished with CrysAlis Pro [8]. Of the 18,012 reflections measured, 2550 were unique (Rint = 0.030), and of these, 2356 data satisfied the I ≥ 2σ(I) criterion of observability. The structure was solved by direct methods [9] and refined (anisotropic displacement parameters and C-bound H atoms in the riding model approximation) on F2 [10]. The thioamide–N–H atoms were located from a difference map and refined with a N–H restraint of 0.88 ± 0.01 Å. A weighting scheme of the form w = 1/[σ2(Fo2) + (0.039P)2 + 1.297P] was introduced, where P = (Fo2 + 2Fc2)/3). Based on the refinement of 180 parameters, the final values of R and wR (all data) were 0.028 and 0.077, respectively. The molecular structure diagram was generated with ORTEP for Windows [11] and the packing diagram using DIAMOND [12].
Crystal data for C15H17N3S (1): M = 271.38, orthorhombic, Pbca, a = 10.50030(10) Å, b = 11.70230(10) Å, c = 23.2486(3) Å, V = 2856.73(5) Å3, Z = 8, Dx = 1.262 g cm−3, F(000) = 1152 and μ = 1.918 mm−1. CCDC deposition number: 1898244.
Supplementary Materials
The following are available online: 1H and 13C{1H} NMR, IR, and UV spectra, and crystallographic data for (1) in crystallographic information file (CIF) format. CCDC 1898244 also contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Author Contributions
Synthesis and spectroscopic characterization was carried out by C.I.Y. X-ray crystallography was performed by E.R.T.T. The manuscript was written by C.I.Y. and E.R.T.T.
Funding
This research received no external funding. The APC was funded by Sunway University.
Acknowledgments
Sunway University Sdn Bhd is thanked for financial support of this work. The X-ray crystallography laboratory at Sunway University is thanked for the X-ray intensity data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Saeed, A.; Flörke, U.; Erben, M.F. A review on the chemistry, coordination, structure and biological properties of 1-(acyl/aroyl)-3-(substituted) thioureas. J. Sulfur Chem. 2014, 35, 318–355. [Google Scholar] [CrossRef]
- Zhang, Z.; Schreiner, P.R. (Thio)urea organocatalysis—What can be learnt from anion recognition? Chem. Soc. Rev. 2009, 38, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, Y. Recent topics in dual hydrogen bonding catalysis. Tetrahedron Lett. 2018, 59, 216–223. [Google Scholar] [CrossRef]
- Kožurková, M.; Sabolová, D.; Kristian, P. A review on acridinylthioureas and its derivatives: Biological and cytotoxic activity. J. Appl. Toxicol. 2017, 37, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Larik, F.A.; Jabeen, F.; Mehfooz, H.; Ghumro, S.A.; El-Seedi, H.R.; Ali, M.; Channar, P.A.; Ashraf, H. Synthesis, antibacterial and antileishmanial activity, cytotoxicity, and molecular docking of new heteroleptic copper(I) complexes with thiourea ligands and triphenylphosphine. Russ. J. Gen. Chem. 2018, 88, 541–550. [Google Scholar] [CrossRef]
- Buu-Hoï, N.P.; Xuong, N.D.; Nam, N.H. Potential antiviral thiourea derivatives. J. Chem. Soc. 1956, 2160–2165. [Google Scholar] [CrossRef]
- López-Torres, E.; Dilworth, J.R. Reactivity of thiosemicarbazides with redox active metal ions: Controlled formation of coordination complexes versus heterocyclic compounds. Chem. Eur. J. 2009, 15, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Oxford Diffraction. CrysAlis PRO; Rigaku Corporation: Oxford, UK, 2017. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]
Sample Availability: Not available. |
Scheme 1.
Chemical diagram for 1-[N-methyl-N-(phenyl)amino]-3-(4-methylphenyl)thiourea (1).
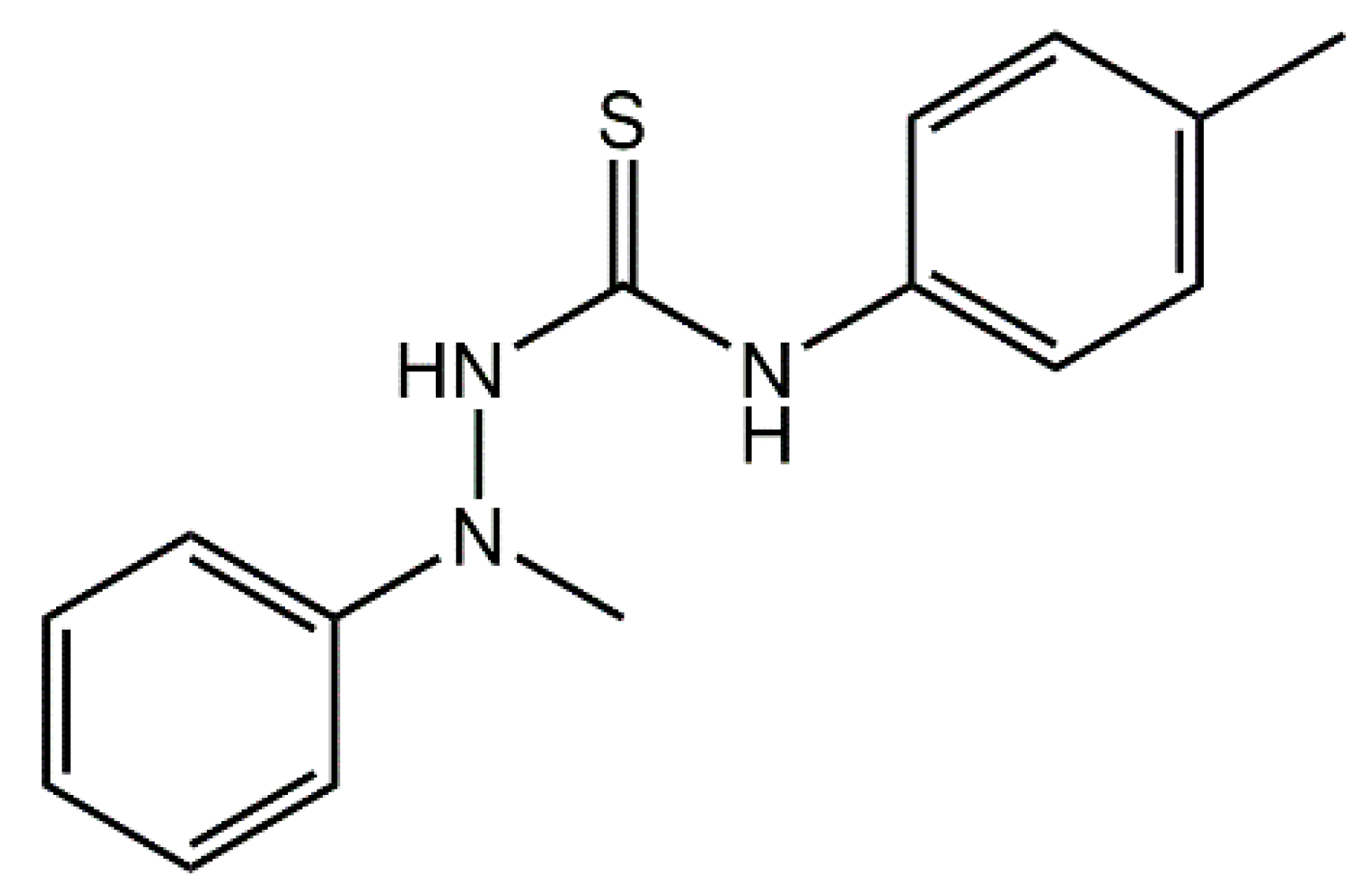
Figure 1.
The molecular structure of (1) showing atom labeling and displacement ellipsoids at the 70% probability level. Selected geometric parameters: C1–S1 = 1.6895(13) Å, N2–N3 = 1.4081(15) Å, C1–N1 = 1.3491(16) Å, and C1–N2 = 1.3458(16) Å; C1–N1–C2 = 129.77(11)°, C1–N2–N3 = 122.11(11)°, S1–C1–N1 = 127.50(10)°, S1–C1–N2 = 117.36(9)°, and N1–C1–N2 = 115.14(11)°.
Figure 1.
The molecular structure of (1) showing atom labeling and displacement ellipsoids at the 70% probability level. Selected geometric parameters: C1–S1 = 1.6895(13) Å, N2–N3 = 1.4081(15) Å, C1–N1 = 1.3491(16) Å, and C1–N2 = 1.3458(16) Å; C1–N1–C2 = 129.77(11)°, C1–N2–N3 = 122.11(11)°, S1–C1–N1 = 127.50(10)°, S1–C1–N2 = 117.36(9)°, and N1–C1–N2 = 115.14(11)°.
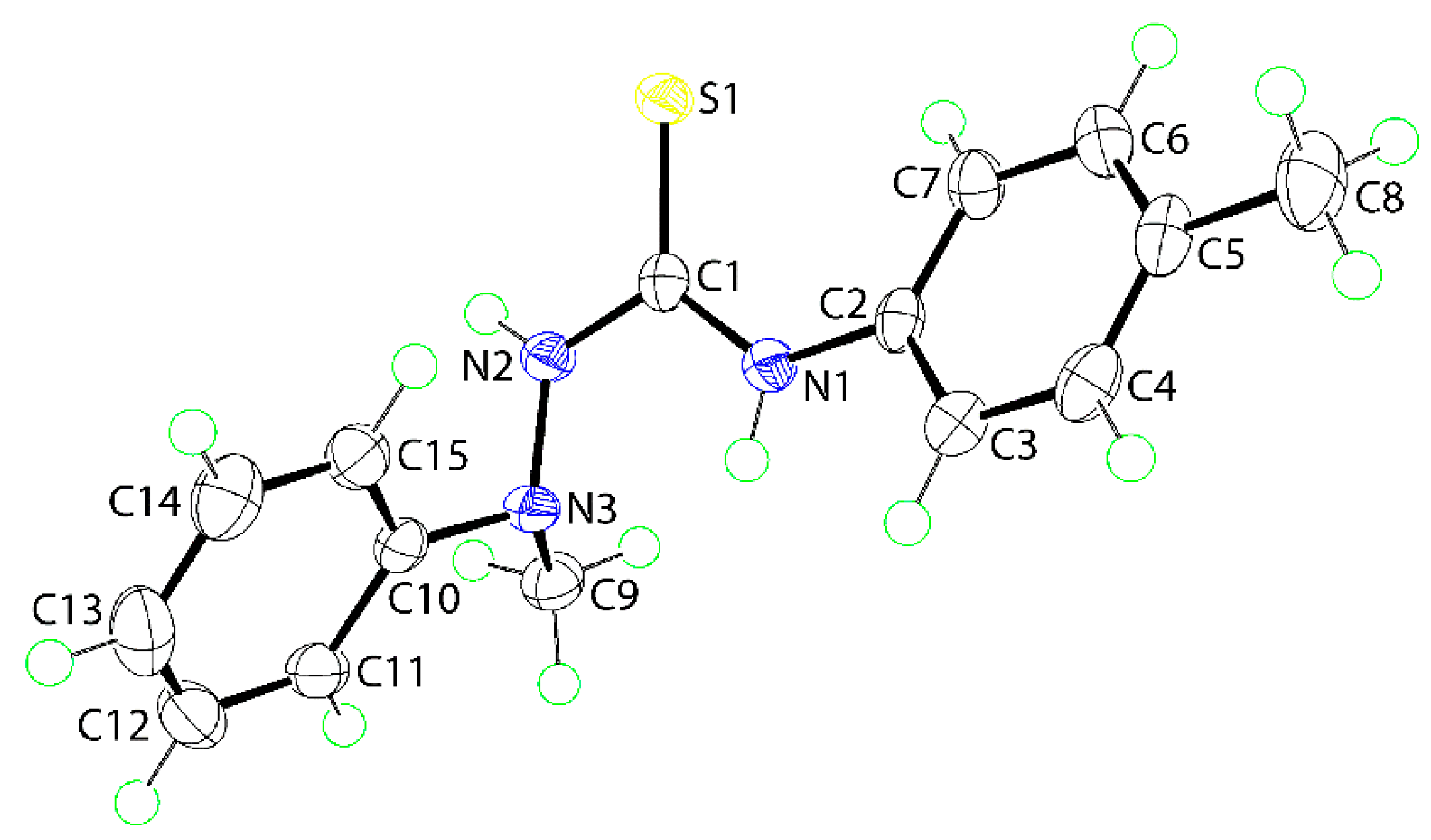
Figure 2.
The molecular packing in the crystal of (1): (a) a view of the supramolecular dimer sustained by thioamide–N–H⋯S(thione) hydrogen bonding [N2–H2n⋯S1i: H2n⋯S1i = 2.474(14) Å, N2⋯S1i = 3.3155(12) Å, angle at H1n = 161.2(12)° for symmetry operation i: 1 − x, 1 − y, 1 − z] shown as orange dashed lines and (b) a view of the unit cell contents shown in projection down the b-axis. The phenyl– and methyl–C–H⋯π(phenyl) interactions [C6–H6⋯Cg(C10–C15)ii: H6⋯Cg(C10–C15)ii = 2.77 Å, C9⋯Cg(C10–C15)ii = 3.6338(15) Å with angle at H6 = 137°, and C9–H9c⋯Cg(C10–C15)ii: H9c⋯Cg(C10–C15)ii = 2.72 Å, C9⋯Cg(C10–C15)ii = 3.5286(15) Å, angle at H9c = 156° for symmetry operations ii: ½ − x, ½ + y, z and iii: –½ + x, ½ − y, 1 − z] are shown as purple dashed lines.
Figure 2.
The molecular packing in the crystal of (1): (a) a view of the supramolecular dimer sustained by thioamide–N–H⋯S(thione) hydrogen bonding [N2–H2n⋯S1i: H2n⋯S1i = 2.474(14) Å, N2⋯S1i = 3.3155(12) Å, angle at H1n = 161.2(12)° for symmetry operation i: 1 − x, 1 − y, 1 − z] shown as orange dashed lines and (b) a view of the unit cell contents shown in projection down the b-axis. The phenyl– and methyl–C–H⋯π(phenyl) interactions [C6–H6⋯Cg(C10–C15)ii: H6⋯Cg(C10–C15)ii = 2.77 Å, C9⋯Cg(C10–C15)ii = 3.6338(15) Å with angle at H6 = 137°, and C9–H9c⋯Cg(C10–C15)ii: H9c⋯Cg(C10–C15)ii = 2.72 Å, C9⋯Cg(C10–C15)ii = 3.5286(15) Å, angle at H9c = 156° for symmetry operations ii: ½ − x, ½ + y, z and iii: –½ + x, ½ − y, 1 − z] are shown as purple dashed lines.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yeo, C.I.; Tiekink, E.R.T. 1-[N-Methyl-N-(Phenyl)amino]-3-(4-Methylphenyl)Thiourea. Molbank 2019, 2019, M1052. https://doi.org/10.3390/M1052
AMA Style
Yeo CI, Tiekink ERT. 1-[N-Methyl-N-(Phenyl)amino]-3-(4-Methylphenyl)Thiourea. Molbank. 2019; 2019(1):M1052. https://doi.org/10.3390/M1052
Chicago/Turabian StyleYeo, Chien Ing, and Edward R. T. Tiekink. 2019. "1-[N-Methyl-N-(Phenyl)amino]-3-(4-Methylphenyl)Thiourea" Molbank 2019, no. 1: M1052. https://doi.org/10.3390/M1052
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.