Abstract
(S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione was obtained in a three-step, one-pot synthesis, starting from optically pure (S)-2-piperazine carboxylic acid dihydrochloride. Selective acylation of the β-nitrogen atom followed by condensation with isatoic anhydride and cyclization with HATU/DIPEA to a seven-member benzodiazepine ring, led to the tricyclic benzodiazepine derivative. Crystallographic studies and initial biological screening were performed for the title compound.
1. Introduction
The tricyclic benzodiazepine analogues have attracted great attention from pharmaceutical chemists as possible scaffolds for design and discovery of new anticancer compounds (Figure 1). Indeed, the tricyclic pyrrolo[1,4]benzodiazepine 1 exhibits a potent inhibition effect on the human DNA ligase (hLig1) [1]. Anticancer activity is also exhibited by the Diazepinomycin 2, a dibenzodiazepine derivative possessing a farnesyl residue and isolated from Micromonospora strain [2]. Anthramycin 3 and its analogues, on the other hand, are isolated from Streptomyces strains, possess the tricyclic pyrrole[2,1-c][1,4]benzodiazepine scaffold and exhibit a unique feature—alkylation of the minor groove of DNA. This feature is caused by appropriate geometry and the presence of binding groups such as an imine, a carbinolamine, or a carbinolamine ether group [3].
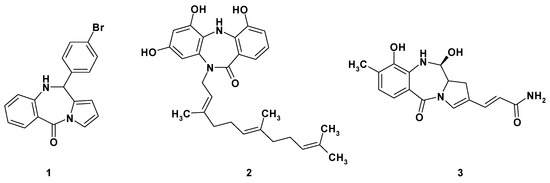
Figure 1.
Representatives of tricyclic benzodiazepine derivatives exhibiting anticancer activity.
During our continuous research on the design and synthesis of novel heterocyclic scaffolds and molecules for application in medicinal chemistry—including furo[2,3-d]pyrimidin-2(3H)-one, 3H-pyrrolo[2,3-d]pyrimidin-2(7H)-one, and 5,6-dihydropyrimido[4,5-c]pyridazin-7(8H)-one analogues [4,5], as well as oxazolo[3,2-f]pyrimidine-5,7-dione derivatives [6]—we designed and developed a novel synthesis method, leading to the fused tricyclic 1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione 4. This new, liquid-phase approach is complementary to our previously published, solid-phase synthesis of peptide mimetics, but it surpasses the old method in terms of higher yields and process upscaling capabilities [7].
2. Results and Discussion
The starting optically pure (S)-piperazine-2-carboxylic acid dihydrochloride 5, partially soluble in the 1:1 water:dioxane mixture, was treated with two equivalents of sodium hydroxide, which led to total dissolution of the amino acid in the reaction medium (Scheme 1). Next, a slight excess of 4-chlorobenzoyl chloride 6 was added and the reaction mixture was stirred at room temperature for 18 h. The reaction progress was monitored by LRMS-ESI spectrometry, which showed the disappearance of the starting material and the formation of the new product possessing an m/z matching the expected (S)-4-(4-chlorobenzoyl)piperazine-2-carboxylic acid 7. Compound 7 was not isolated from the reaction mixture, but was directly treated with three equivalents of isatoic anhydride 8, followed by addition of three equivalents of sodium carbonate and heated at 60 °C for 18 h. The isatoic anhydride could be treated as a β-amino acid derivative, a reaction product between 2-aminobenzoic acid (anthranilic acid) and phosgene. The isatoic anhydride could react as an ‘activated’ amino acid, capable of peptide coupling and amide bond formation with the amine group of another amino acid residue. Indeed, mass spectra showed appearance of a new product possessing an m/z matching the expected (S)-1-(2-aminobenzoyl)-4-(4-chlorobenzoyl)piperazine-2-carboxylic acid 9—resulting from the coupling of isatoic anhydride with the secondary amine group in the α position of (S)-piperazine-2-carboxylic acid. Finally, we focused on the intramolecular cyclization of intermediate 9 to a (S)-2-(4-chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione 4. We observed that heating of 9 in boiling acetic acid for several hours resulted in the formation of desired tricyclic derivative 4. Unfortunately, we observed concurrent partial or total racemization of the final product, leading to the mixture of two enantiomers. This inconvenience forced us to investigate other approaches for the intramolecular formation of the benzodiazepine ring. Finally, we found that treatment of 9 with the excess HATU, in the presence of DIPEA, resulted in formation of the desired tricyclic derivative 4, with a moderate yield. The 1H- and 13C-NMR spectra revealed that the synthesized compound in solution exists in two conformations which undergo fast interconversion in a deuterated dimethyl sulfoxide solution. Additionally, the presence of magnetically non-equivalent axial and equatorial hydrogens in the three methylene groups of piperazine ring resulted in a complex multiplet, in both aliphatic and aromatic parts of the spectra. The identity of 4, crystallized from ethyl acetate solution, was proven by a single-crystal X-ray diffraction analysis. It showed that the investigated compound crystallizes in a triclinic P1 space group, with two molecules of compound in the asymmetric unit of the crystal lattice (Figure 2). Details of the crystallographic data and the refinement parameters are summarized in Table S1 (Supplementary Information). The full list of bond length values, valence, and torsion angles can also be found in the Supplementary Information (Tables S2–S4). The alignment of molecules labelled as A and B reveals clearly that the overall geometry of the 1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione moiety is essentially the same (Figure 3). In the case of both molecules of 4, the (S)-configuration of the chiral centre is observed and heterocyclic seven- and six-membered rings adopt the boat and chair conformations, respectively. On the other hand, a noticeable difference is observed comparing the spatial orientation of the 4-chlorobenzoyl substituent. Values of the dihedral angle between the mean-quadratic planes delineated by the carbon atoms of the phenyl rings of the 4-chlorobenzoyl and the 1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione moieties are 64.5(2)° and 85.7(2)° in A and B, respectively.
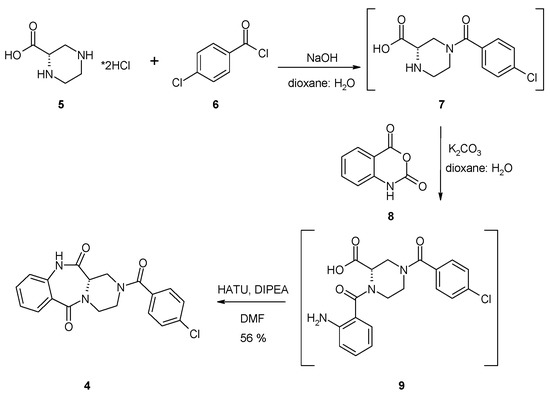
Scheme 1.
Yield was added to the scheme.
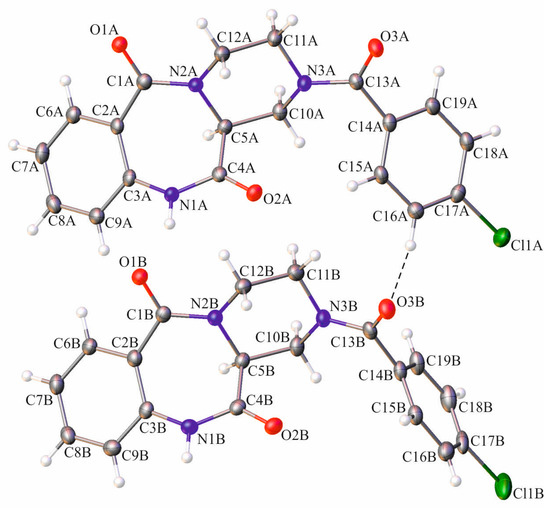
Figure 2.
Molecular structure of 4 with crystallographic numbering. Displacement ellipsoids are drawn at the 50% probability level and the H-atoms are shown as small spheres of arbitrary radius. The weak intermolecular C–H···O hydrogen bond is represented by a dashed line.
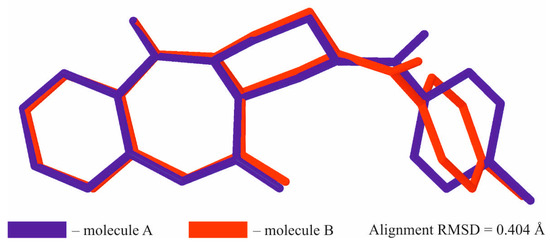
Figure 3.
Superimposed molecules A and B of 4. The H-atoms were omitted for clarity.
The packing of molecules in the crystal is dominated by the formation of hydrogen bonds (Figure 4, Table S5). Full list of molecular interactions identified using PLATON software [8] can be found in the SI (Tables S5–S7).
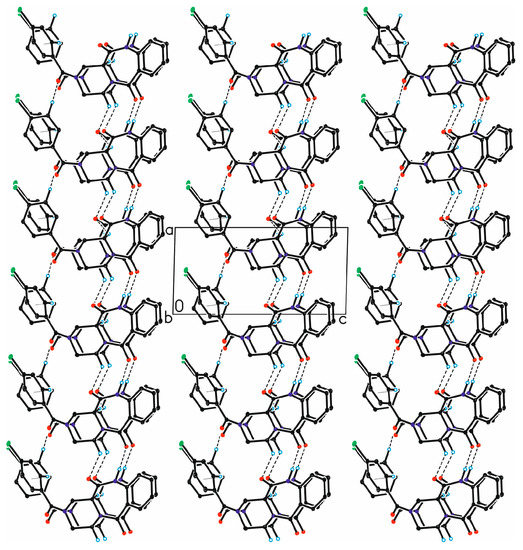
Figure 4.
The arrangement of molecules in the crystal of 4, viewed along b-direction. The H-atoms not involved in the intermolecular interactions have been omitted for clarity. Hydrogen bonds are represented by the dashed lines, while the C–H···π contacts by dotted lines.
Product 4 was tested for its antiproliferative activity on MV-4-11 (biphenotypic B myelomonocytic leukemia) using the MTT method [9] and UM-UC-3 (transitional cell carcinoma) using the SRB method [10], but no significant cytotoxic effect was observed up to 200 μM after 72 h of treatment.
3. Materials and Methods
Commercially available chemicals were of reagent grade and used as received. The reactions were monitored by thin layer chromatography (TLC) analysis, using silica gel plates (Kieselgel 60F254, E. Merck, Darmstadt, Germany). Column chromatography was performed on Silica Gel 60M (0.040–0.063 mm, E. Merck). Melting points are uncorrected and were measured on a Kofler apparatus. The 1H- and 13C-NMR spectra, in DMSO-d6, were recorded at the Department of Chemistry, Warsaw University, using the Varian Unity Plus spectrometer (500 MHz); shift values in parts per million are relative to the SiMe4 internal reference. The resonance assignments are based on peak integration, peak multiplicity, and 2D correlation experiments. Multiplets were assigned as s (singlet) and m (multiplet). High Resolution Mass spectra were performed by the Laboratory of Mass Spectrometry at the Institute of Biochemistry and Biophysics PAS, on an LTQ Orbitrap Velos instrument, Thermo Scientific (Waltham, MA, USA).
Diffraction data were collected on the Agilent Technologies SuperNova Dual Source diffractometer with CuKα radiation (λ = 1.54184 Å), using CrysAlis RED software [11]. The multi-scan empirical absorption correction using spherical harmonics implemented in the SCALE3 ABSPACK scaling algorithm was applied [11]. The structural determination procedure was carried out using the SHELX package [12]. The structures were solved with direct methods and then successive least-square refinement was carried out, based on the full-matrix least-squares method on F2, using the SHELXL program [12]. All H-atoms linked to the N-atoms were located on a Fourier difference map and refined as riding, with Uiso(H) = 1.2Ueq(N). Other H-atoms were positioned geometrically, with C–H equal to 0.93, 0.97, and 0.98 Å for the aromatic, methylene, and methine H-atoms, respectively, and constrained to ride on their parent atoms with Uiso(H) = xUeq(C), where x = 1.2 for the aromatic, methylene, and methine H-atoms. All presented molecular interactions were found using the PLATON program [11]. All figures for this publication were prepared using the Olex2 and ORTEP-3 programs [13,14].
Synthesis of the (S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione (4)
(S)-piperazine-2-carboxylic acid dihydrochloride (5, 700 mg, 3.45 mmol, 1 equiv.) was dispersed in 50 mL of 1:1 water:dioxane mixture and treated with sodium hydroxide (276 mg, 6.89 mmol, 2 equiv.). After dissolution of the starting material, 4-chlorobenzoyl chloride (6, 0.49 mL, 3.79 mmol, 1.1 equiv.) was added and reaction mixture was stirred in room temperature for 18 h. The next day, the disappearance of starting material and formation of (S)-4-(4-chlorobenzoyl)piperazine-2-carboxylic acid (7) was confirmed by LRMS-ESI spectra. Then, isatoic anhydride (8, 1.69 g, 10.34 mmol, 3 equiv.) was added, followed by addition of sodium carbonate (1.10 g, 10.34 mmol, 3 equiv.); the reaction mixture was heated in 60 °C for 18 h. The following day, formation of the (S)-1-(2-aminobenzoyl)-4-(4-chlorobenzoyl)piperazine-2-carboxylic acid 9 was confirmed by LRMS-ESI spectra. The volatiles were evaporated under reduced pressure, then the residue was co-evaporated with toluene (3 × 50 mL) and dissolved in dry DMF. For cyclization of 9, HATU (3.93 g, 10.34 mmol, 3 equiv.) and DIPEA (1.80 mL, 10.34 mmol, 3 equiv.) were added, and reaction mixture was stirred in room temperature for 18 h. The day after, the volatiles were evaporated under reduced pressure and residue was dissolved in water:ethyl acetate biphasic system. The organic phase was washed with water (2 × 50 mL), 0.5 M HCl (3 × 50 mL), saturated sodium bicarbonate (1 × 50 mL), and dried over magnesium sulphate. The crude product dissolved in ethyl acetate was evaporated with silica gel (2 g) and purified by column chromatography using hexane:ethyl acetate 2:8 v/v mixture, followed by pure ethyl acetate. Yield: 711 mg (56%). 1H-NMR (500 MHz, DMSO-d6): 10.55, 10.45 (2 × s, 2 × NH); 7.80–7.70 (m, 1H, HAr); 7.70–7.40 (m, 5H, HAr); 7.30–7.20 (m, 1H, HAr); 7.20–7.00 (m, 1H, HAr); 4.45–3.30 (m, 7H, 3 × CH2, 1 × CH); 13C-NMR (125 MHz, DMSO-d6): 170.5, 169.5, 166.6, 136.6, 135.0, 134.2, 132.3, 130.9, 129.3, 129.0, 128.5, 128.3, 125.6, 124.0, 120.9, 51.7, 42.7, 42.2, 38.2; HRMS (ESI): m/z [M + H]+ calcd. for C19H17ClN3O3: 370.09530, 372.09235, found: 370.09517, 372.09206; m.p. 248–250 °C. = +290 (c 1.0, DMSO). IR (KBr): cm−1 3465, 3369, 3229, 3160, 3109, 3068, 2909, 2866, 1694, 1657, 1620, 1521, 1477, 1431, 1407, 1339, 1303, 1262, 1219, 1181, 1162, 1092, 1035, 1010.
Supplementary Materials
Copies of the 1H-NMR, 13C-NMR, IR, HRMS-ESI mass spectra and detailed crystallographic data are available online www.mdpi.com/1422-8599/2017/4/M964. The CCDC 1574755 4 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
Acknowledgments
The synthetic part of this work was supported by the National Science Center (decision number: DEC-2011/01/B/NZ4/03566). The equipment used for the synthesis was sponsored, in part, by the Center for Preclinical Research and Technology (CePT), a project co-sponsored by European Regional Development Fund and Innovative Economy, The National Cohesion Strategy of Poland. We thank Jacek Olędzki for recording the ES-MS spectra. The structural study was carried out at the Biological and Chemical Research Center, University of Warsaw, established within the project co-financed by European Union from the European Regional Development Fund, under the Operational Program Innovative Economy, 2007–2013. This study was also supported by the National Science Centre, Poland, MAESTRO grant—DEC-2012/04/A/ST5/00609 (D.T. and K.W.)—which enabled the performance of X-ray structural analysis.
Author Contributions
A.M.: synthesis planning, writing of manuscript; M.B.: experimental synthetic work; B.B.: MS and NMR interpretation; M.M.: literature research, writing of manuscript; M.W.: NMR recording and interpretation; M.P.: screening of biological activity; D.T.: crystallographic studies, writing of manuscript; K.W.: writing of manuscript.
References
- Shameem, M.; Kumar, R.; Krishna, S.; Kumar, C.; Siddiqui, M.I.; Kundu, B.; Banerjee, D. Synthetic modified pyrrolo[1,4]benzodiazepine molecules demonstrate selective anticancer activity by targeting the human ligase 1 enzyme: An in silico and in vitro mechanistic study. Chem. Biol. Interact. 2015, 237, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Rambabu, V.; Vijayakumar, S. In vitro cytotoxicity perspective of deazepinomycin (ECO-4601) on human hepatoma cell line (HEPG2). Biomed. Aging Pathol. 2014, 4, 65–70. [Google Scholar] [CrossRef]
- Cipolla, L.; Araujo, A.C.; Airoldi, C.; Bini, D. Pyrrolo[2,1-c]benzodiazepine as a scaffold for the design and synthesis of antitumor drugs. Anti-Cancer Med. Chem. 2009, 9, 1–31. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Makowska, M.; Sekuła, J.; Tomczyk, E.; Zalewska, E.; Nasulewicz-Goldeman, A.; Wietrzyk, J. Bicyclic cytarabine analogues: Synthesis and investigation of antitumor properties of novel, 6-aryl- and 6-alkyl-3H-pyrrolo[2,3-d]pyrimidin-2(7H)-one arabinosides. Tetrahedron 2015, 71, 8454–8461. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Tomczyk, E.; Makowska, M.; Nasulewicz-Goldeman, A.; Gajda, R.; Woźniak, K.; Wietrzyk, J. The synthesis and investigation of anti-tumor properties of novel, bicyclic furopyrimidine, pyrrolopyrimidine and pyrimidopyridazine nucleoside analogues. Synthesis 2016, 48, 566–572. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Bazlekova, M.; Bagiński, M.; Wójcik, J.; Winczura, A.; Miazga, A.; Gajda, R.; Woźniak, K.; Tudek, B. A mild and efficient approach to 6H-oxazolo[3,2-f]pyrimidine-5,7-dione scaffold via unexpected rearrangement of 2,3-dihydropyrimido[6,1-b][1,5,3]dioxazepine-7,9(5H,8H)-diones: A synthesis, crystallographic studies and cytotoxic activity screening. Tetrahedron Lett. 2016, 57, 743–746. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Jurczak, J. A traceless solid-supported synthesis of novel pyrazinediazepinedione derivatives. Tetrahedron 2010, 66, 2514–2519. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis CCD and CrysAlis RED, Version 1.171.32.15; Oxford Diffraction Ltd.: Yarnton, UK, 2008.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).