Threading Cyclodextrins in Chloroform: A [2]Pseudorotaxane

Dipartimento di Chimica Organica e Biologica, Università di Messina, Salita Sperone 31, I-98166 Messina, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2007, 8(10), 1052-1063; https://doi.org/10.3390/i8101052
Submission received: 13 September 2007
/
Revised: 27 September 2007
/
Accepted: 30 September 2007
/
Published: 22 October 2007
(This article belongs to the Section Molecular Recognition)
Abstract
:The unusual self-assembly in chloroform of a novel cyclodextrin-based [2]pseudorotaxane, composed of heptakis-2,6-di-O-methyl-3-O-acetyl-β-cyclodextrin and a viologen-containing linear component, is reported.
1. Introduction
Cyclodextrins [1–3] (CDs) are a well-known family of cyclic oligosaccharides – composed of 1→4-linked α-d-(+)-glucopyranose units – that have found over the past few years an ever-increasing number of commercial applications, in various sectors ranging from pharmaceuticals and cosmetics to the food industry [4–6]. CDs owe their attractiveness mainly to two different features. Firstly, they have been found to be perfectly biocompatible, showing an absolutely negligible toxicity [7]. The second key to their success is the fact that they possess rigid and highly preorganized cavities of different dimensions, which make them among the most efficient molecular receptors presently available to the researcher. They come in three different sizes, α-CD (composed of six glucose units, and possessing a cavity with an internal diameter of 4.7–5.3 Å), β-CD (seven units, 6.0–6.5 Å) and γ−CD (eight units, 7.5–8.3 Å), all of them displaying remarkable complexing abilities [3].
Supramolecular chemists showed an early interest in these molecular receptors [8,9]. CDs and their chemically modified derivatives have been used over the years as building blocks for the construction of the most diverse supramolecular architectures. Interlocked supermolecules [10–12], such as rotaxanes [13,14] and catenanes [15], supramolecular polymers [16], and artificial enzymes [17,18], have all been self-assembled or synthesized using CDs as the macrocyclic component, harnessing their innate tendency to form, with suitable guests, inclusion compounds with a pseudorotaxane-like geometry. Cyclodextrin-based rotaxanes and pseudorotaxanes have also been employed for the construction of molecular switches [11,19,20], activated by electrochemical [21], photochemical [22–24], thermal [22] or chemical stimuli [25].
Most of the work involving cyclodextrins has been carried out in aqueous media, as a consequence of their mode of binding, that mostly relies on hydrophobic interactions taking place between the interior of the cavity of the torus-shaped CD and insoluble or poorly-soluble aliphatic or aromatic guest molecules [26]. In order to take advantage of the complexing abilities of cyclodextrins in less polar media, chemical modifications of the hydroxyl groups of the CDs are necessary, both to ensure sufficient solubility and to provide additional functionalities to increase their affinity towards guests with different structural features. This would greatly expand the potential applications of cyclodextrins, and would enable them to compete in non-aqueous media with known fully-synthetic receptors for the construction of supramolecular architectures.
In this respect, taking advantage of the development of efficient methods for exhaustive and selective functionalization [27–29], a number of papers have appeared in recent years on the construction of supramolecular structures composed of chemically-modified CDs and viologen derivatives [9,30–33]. In particular, cyclodextrins bearing acyl groups on the secondary face can efficiently yield inclusion complexes in polar organic media (acetonitrile, acetone) where a dicationic bipyridinium moiety is tightly bound owing to carbonyl oxygen-cation interactions. Following this lead, we report here on the self-assembly in chloroform of a novel cyclodextrin pseudorotaxane.
2. Results and Discussion
2.1. Design and Synthesis of the Linear and Macrocyclic Components
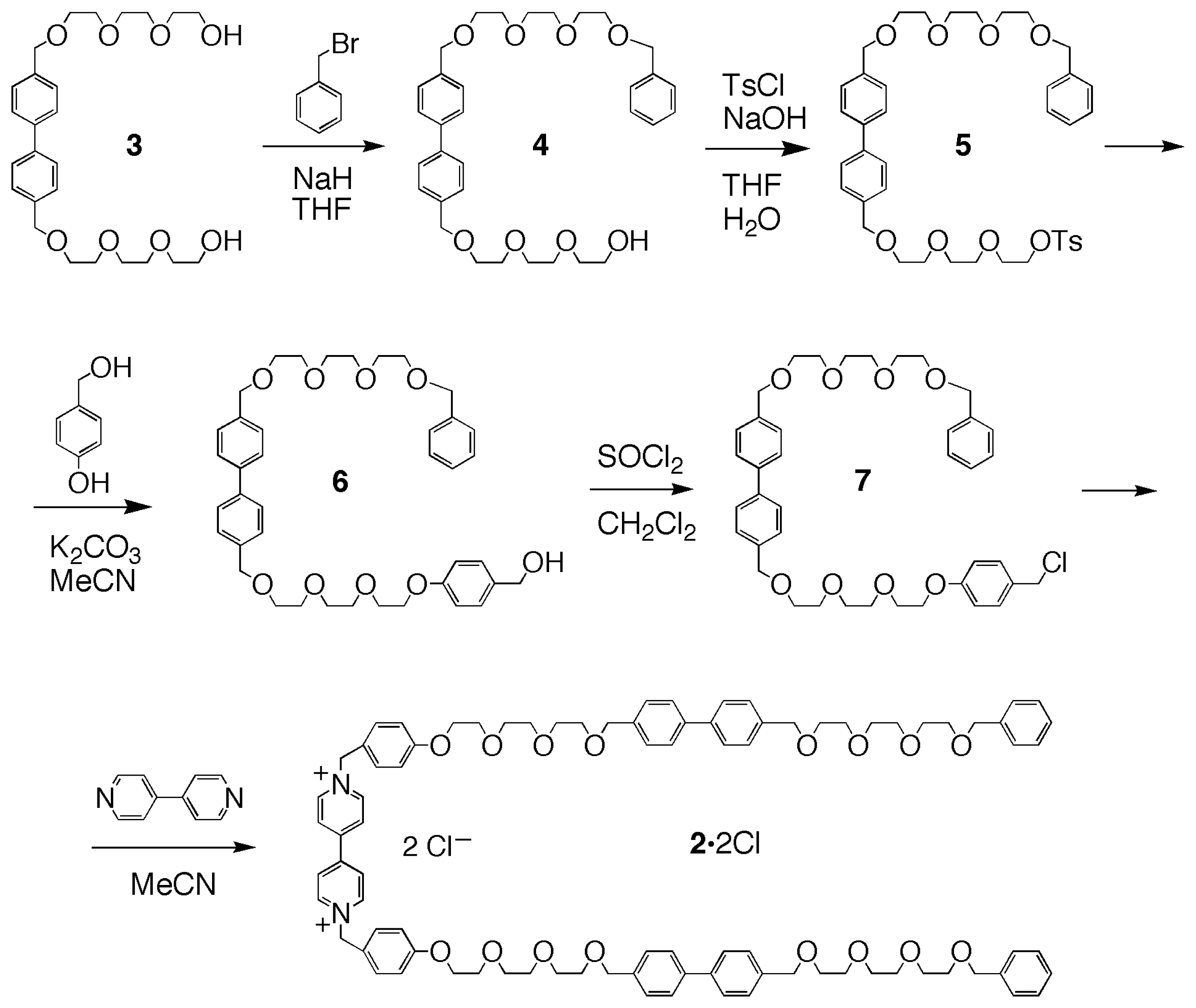
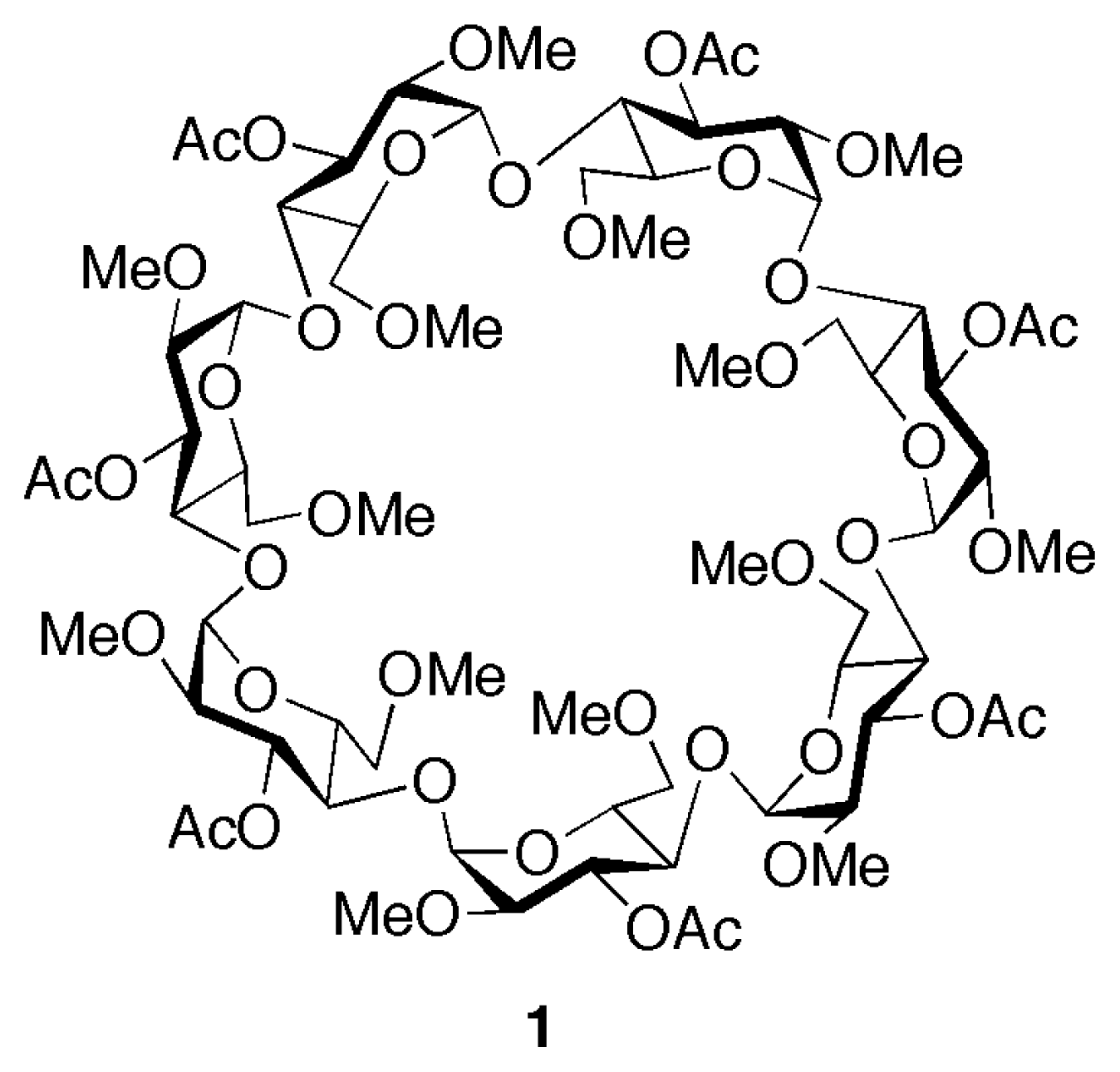
In a previous paper [34], we have demonstrated that by employing a “binary host” approach [35] – i.e., the simultaneous and synergic use of cationic and anionic receptors for the complexation of salt species– it is possible to solubilize paraquat dichloride in chlorinated solvents, with a number of chemically modified CDs, as a result of the breaking up of the chloride-paraquat ion pair. As a follow-up to these findings we have now built a linear component –the “thread”– intended to be recognized and complexed by a suitably substituted cyclodextrin in chloroform. The thread 22+ was designed (Scheme 1) to possess a bipyridinium moiety at its center, along with two additional biphenyl “stations” [36] which would have a lower affinity for the cyclodextrin cavity, with the future aim of developing a controllable molecular machine [19–25]. The choice of building the linear component with two degenerate “stations” flanking the central bipyridinium one was made to avoid the formation of topological isomers deriving from the formation of a pseudorotaxane-like complex between a cyclodextrin and an unsymmetrical guest [14,30,37–41]. The cyclodextrin chosen to probe the threading tendency of 22+ was heptakis-2,6-di-O-methyl-3-O-acetyl-β-cyclodextrin [34,42] (1, see Figure 1), which had been previously investigated and found to form stable complexes with bipyridinium cations in acetonitrile or acetone [32,34].
In order to gather preliminary evidence on the possibility of building a supramolecular switching system, the binding constants for the complexes of cyclodextrin 1 and two different guest molecules – chosen as prototype stations– were determined by 1H NMR dilution experiments [15,43], both in aqueous and in organic media. The binding experiments were carried out by progressively diluting equimolar solutions of 1 with each of the two guests, namely the biphenyl derivative 3[15] (vide infra, Scheme 1) and N,N′-dibenzyl-4,4′-bipyridinium (82+), gave the expected results (Table 1), proving that CD 1 displays a good affinity for both the dicationic guest 82+ and the neutral aromatic guest 3 in acetone and in water-rich media, respectively.
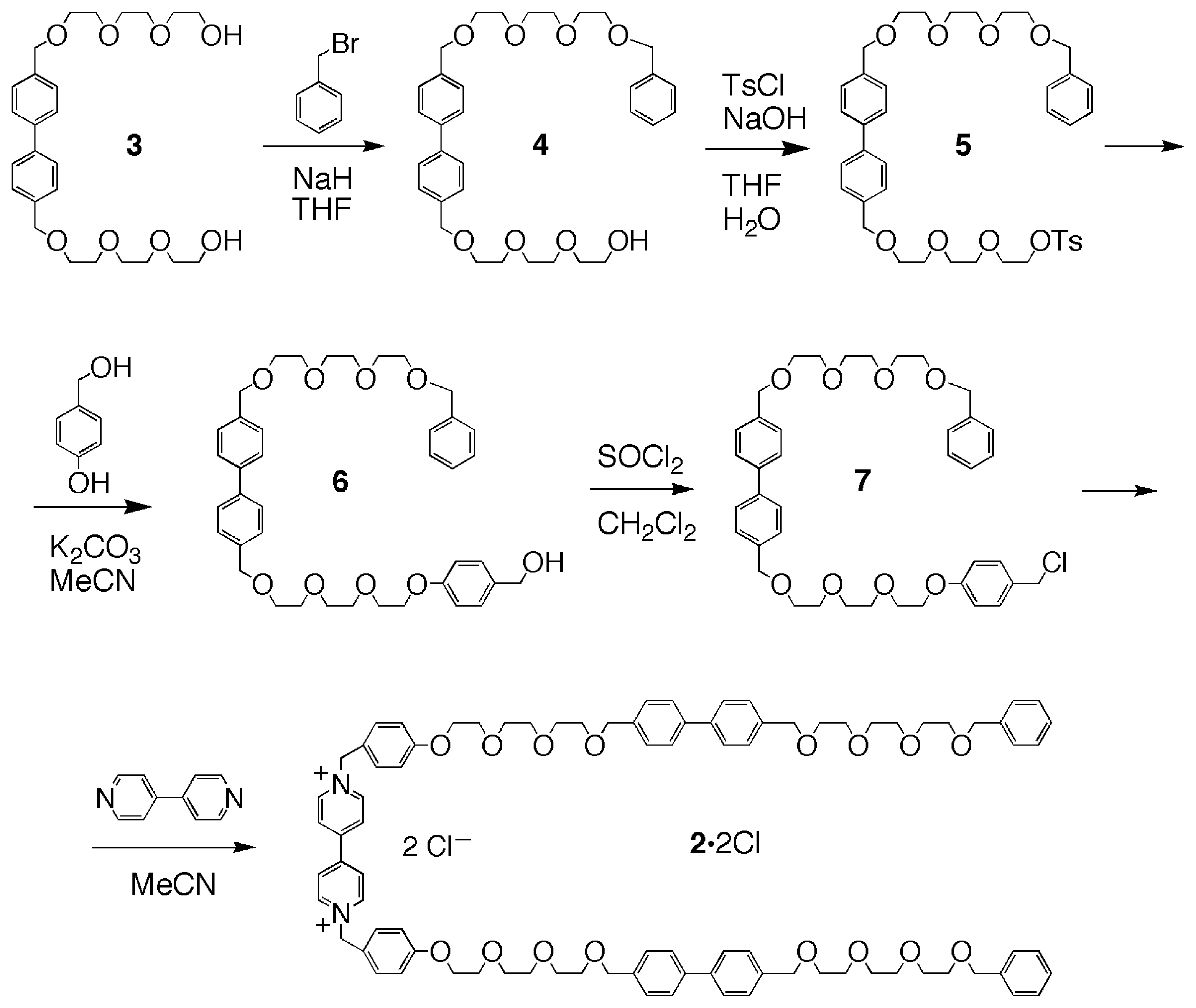
On the basis of these preliminary binding experiments, the synthesis of the linear component 22+ – containing a dibenzyl-bipyridinium moiety flanked by two biphenyl ones, as a potential three-station thread– was accomplished in five steps, starting from the known diol 3[15] (Scheme 1). An excess of 3 was treated with benzyl bromide, in THF with sodium hydride as the base, to produce, after chromatographic purification, the benzyl-monoprotected derivative 4 in 18% yield. The alcohol 4 was treated with tosyl chloride under standard conditions (NaOH, H2O/THF), to give the tosylate 5 (55%), which was then treated with p-hydroxybenzyl alcohol and potassium carbonate in refluxing acetonitrile, to provide alcohol 6 (59%). Compound 6 was reacted with thionyl chloride in dichloromethane, affording chloro-derivative 7 (80%). The final product 2·2Cl was prepared by reacting chloro-derivative 7 with 4,4′-bipyridine, and obtained, after chromatography, in 30% yield as the dichloride salt.
2.2. Self-assembly of the [2]Pseudorotaxane 1⊃2·2Cl
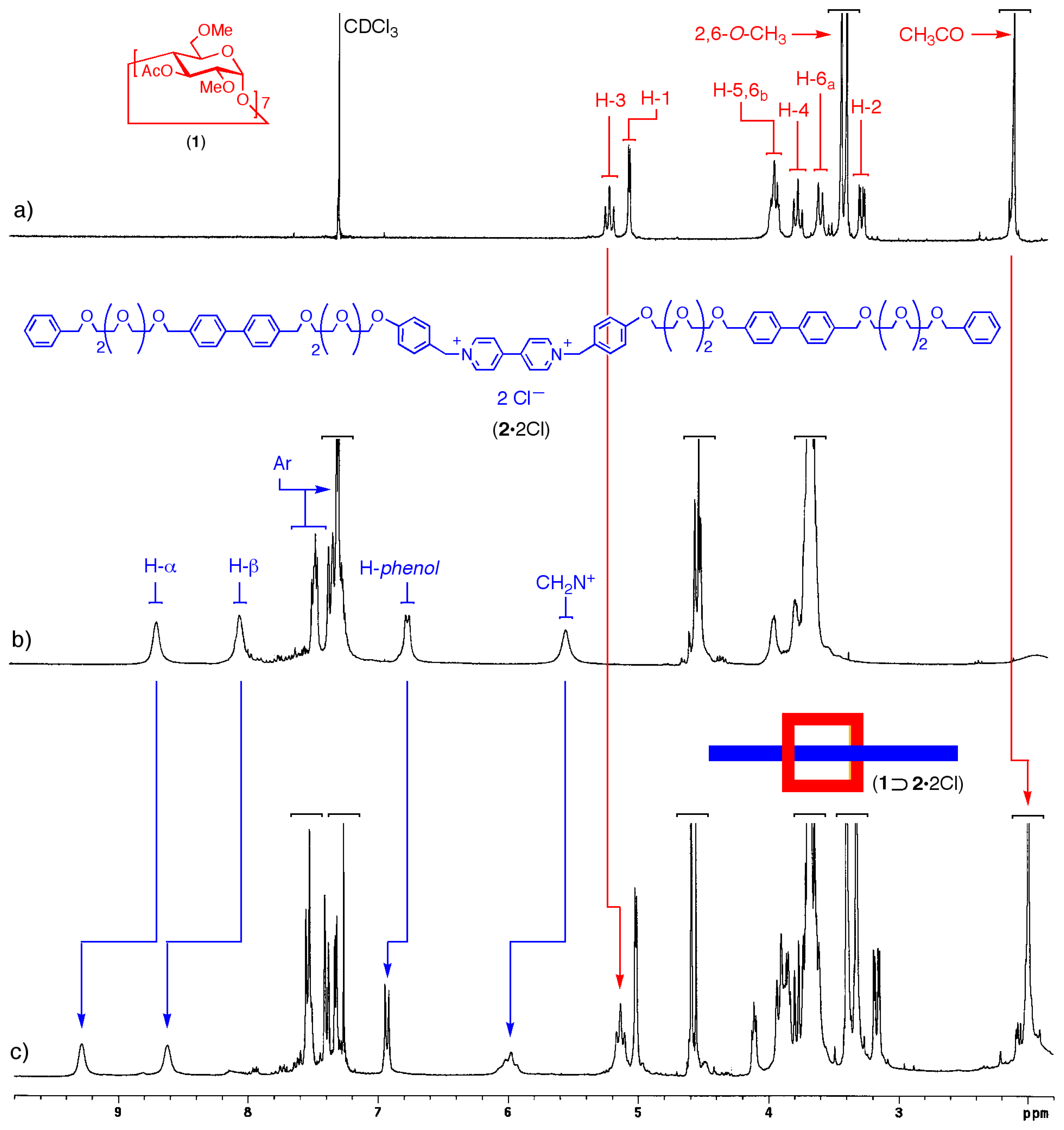
The original design of the linear component 22+ was based upon the assumption that a final anionexchange step (e.g. Cl− to PF6−) would have to be carried out in order to provide a saline species that could more easily dissolve in organic solvents. However, the dichloride salt 2·2Cl was found to be soluble in chloroform as such and it was therefore possible to proceed with the threading of the pseudorotaxane without any further transformation. Addition of a stoichiometric amount of cyclodextrin 1 resulted in the immediate formation of the expected supermolecule, which was then conveniently monitored by 1H NMR spectroscopy (Figure 2).
A comparison of the 1H NMR spectra of the two components 1 and 2·2Cl separately and after mixing provides a useful insight into the assembly process. Upon threading (Figure 2, trace c) the CD component of the pseudorotaxane lies on the central bipyridinium station of the thread component and, as a result, the resonances of this moiety as well as those of the neighboring groups undergo distinctive complexation-induced shifts (CISs).
The α- and β-bipyridinium proton signals experience a strong downfield shift, from 8.66 to 9.28 and 8.03 to 8.62 ppm, respectively, whereas the doublet belonging to a pair of hydrogen atoms of the adjacent phenolic rings moves from 6.75 to 6.94 ppm. The broad singlet assigned to the methylene groups bound to the positively charged nitrogens, shows a CIS of 0.49 ppm (from 5.51 ppm in the free species to 6.00 ppm in the pseudorotaxane), and in addition, it becomes a broad AB system. This behavior, typical for enantiotopic hydrogens that become diastereotopic upon inclusion in the chiral cavity of the cyclodextrin [15], provides further evidence that complexation is taking place in the expected manner. Small but significant CIS are also observed for some of the peaks belonging to the receptor. In particular, both the H-3 of the CD ring and the methyl group of the 3-O-acetyl substituents show upfield shifts of ca. 0.1 ppm, suggesting that the carboxyl oxygens are responsible for the complexation of the bipyridinium cation.
The association constant for 1⊃2·2Cl was determined by 1H NMR by means of a dilution experiment, and was found to be 410 ± 30 M−1 in chloroform. Although this value is lower than the one measured for the complexation of the test cationic guest 82+ in CD3COCD3, it is quite promising in view of a future rotaxane preparation. In the case of 2·2Cl, in addition to an increased molecular complexity, ion-pairing effects (between the cationic bipyridinium moiety and the chloride counterions) play, in a low-polarity solvent such as CDCl3, an unfavorable role in the complexation process and cause the thread component to be less available for inclusion into the cavity [34,35].
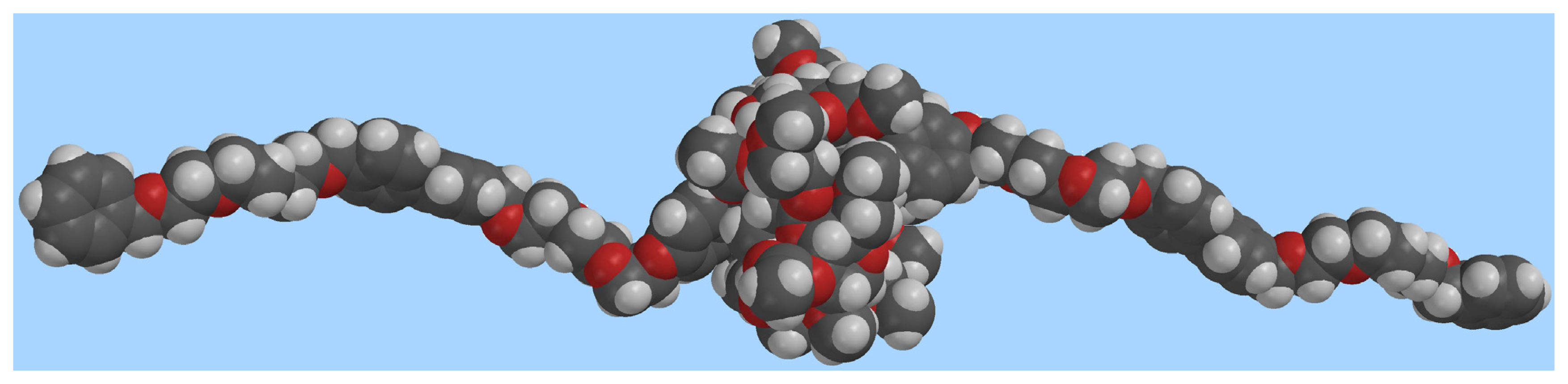
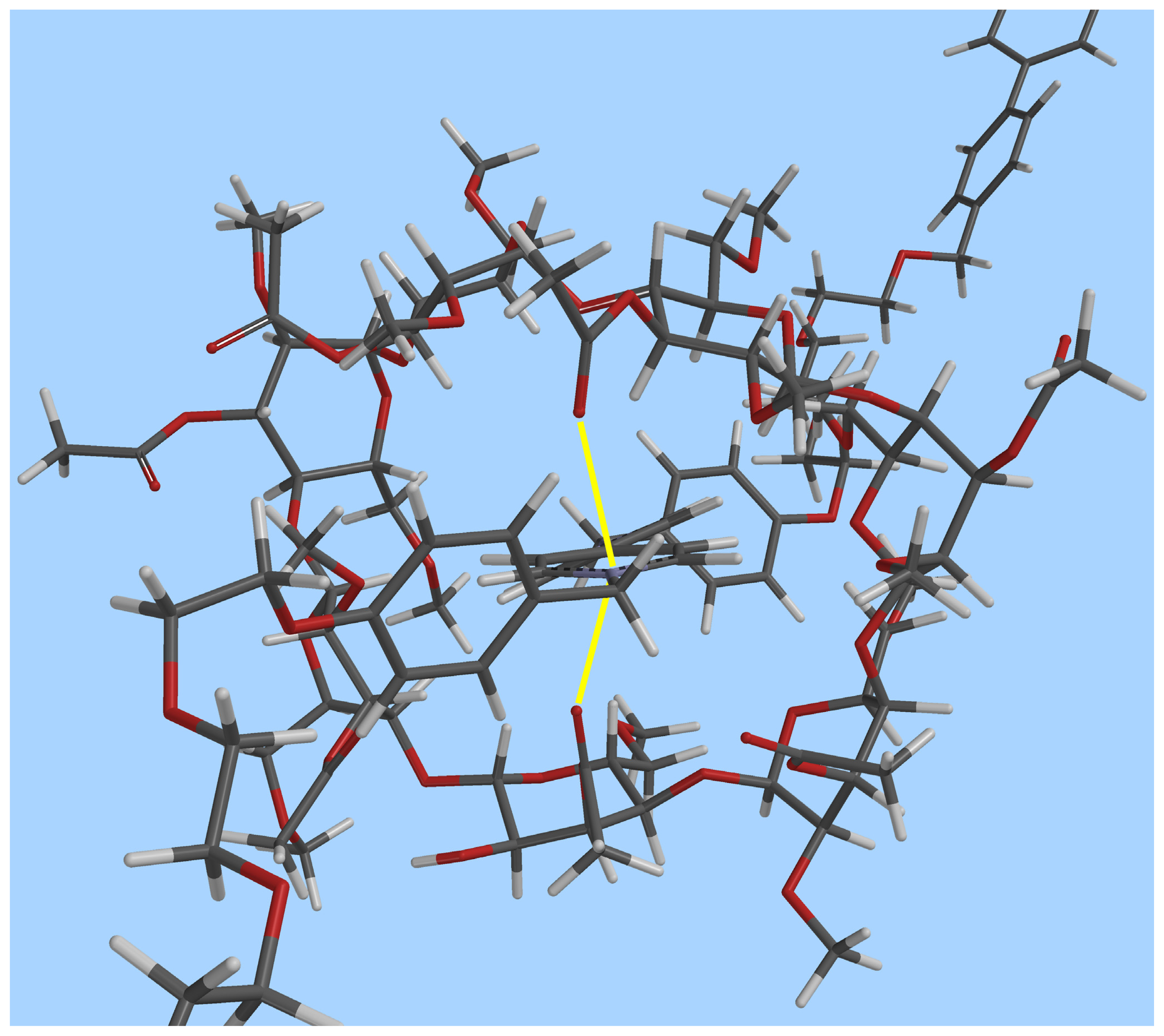
Molecular modeling provides additional insight into the geometry of the complex (Figure 3) and on the mode of binding of the dication inside the CD cavity (Figure 4). Figure 3 shows the overall geometry of the complex, obtained by MMFF geometry optimization. The linear component 22+, as expected is threaded through the cyclodextrin cavity, which, in turn rests on the dicationic moiety.
Figure 4 features a top-down view of the CD cavity. It is possible to observe that at least two of the carboxyl groups (perpendicular to the bipyridinium plane) are pointing towards the positively charged nitrogen atom, their O···N+ distances being 2.74 and 3.02 Å, respectively.
Conclusions
A novel [2]pseudorotaxane composed of a chemically modified acyl-bearing cyclodextrin and a three station thread has been reported. These two components surprisingly self-assemble in a lowpolarity organic solvent such as chloroform to give rise to a supermolecule in which the doubly charged station is encircled by the macrocyclic component. The pseudorotaxane possesses two additional degenerate stations. Future work will involve the preparation of derivatives that can be subjected to stoppering reactions. In the resulting rotaxanes, the two additional stations will hopefully provide efficient alternative binding sites to which the cyclodextrin macrocycle will migrate upon a change in the polarity of the solvent or as a result of an electrochemical reduction of the bipyridinium dication.
3. Experimental Section
3.1. General Methods
All reagents employed were commercially available. Solvents were dried according to literature procedures [44]. Glassware was dried prior to use (T>140 °C, 12–14 h). Thin layer chromatography was performed on silica gel plates with fluorescence indicator (Polygram SIL G/UV254, Macherey- Negel) and compounds were detected either under UV light (λ=254 nm) or by heating with sulphuric acid (5% v/v in EtOH). Column chromatography was performed on silica gel (230–400 mesh, 60 Å, Merck). 1H and 13C NMR spectra were recorded on a Varian Mercury 300 spectrometer at 300 and 75 MHz, respectively. Unless otherwise stated, spectra were recorded in CDCl3. Chemical shifts (δH) are reported in ppm from TMS as an internal standard. Coupling constants (J), where not specified, are 3J. Mass spectra (FAB-MS) were recorded on a Finnigan Mat 90 spectrometer using 3-nitrobenzyl alcohol as a matrix. Molecular modeling was performed by running the MMFF94 force field included in the Spartan-04 package (Wavefunction, Inc.) on a SUN workstation.
3.2. 1H NMR Binding experiments
Binding constants (Ka) were obtained from 1H NMR dilution experiments [43]. The dilution procedure is based on the change in chemical shift for a probe proton of either host or guest upon varying the concentration of an equimolar solution of the two. In a typical experiment, a stock solution of host and guest (10 mM) was prepared by dissolving a weighed equimolar amount of guest (either 2·2Cl, 3, 8·2Cl or 8·2PF6) and host (CD 1) in the appropriate volume of deuterated solvent(s) (CDCl3, CD3COCD3 or D2O/CD3COCD3 4:1 v/v). Five different solutions (5.0, 3.0, 1.5, 0.5, 0.3 mM) were first prepared by diluting the original stock solution and then examined by 1H NMR (300 MHz at 22±1 °C ). Ka values (M−1) were calculated from equation (1)[15] by following the resonances of either the α-bipyridyl protons (for 22+ and 82+) or the aromatic ring protons (for 3):
where Δδobs is the observed difference in chemical shift between the complex and the free species for a probe proton of either host or guest at a given concentration, Δδmax is the extrapolated value corresponding to the maximum CIS, and c is the concentration of the equimolar solution of host and guest.
3.3. Synthetic Procedures
4-{2-[2-(2-benzyloxyethoxy)ethoxy]ethoxymethyl}-4′-{2-[2-(2-hydroxyethoxy)ethoxy]ethoxymethyl} biphenyl (4). A solution of benzyl bromide (1.92 g, 10.27 mmol) in anhydrous THF (5 mL) was added, under nitrogen, to a stirred mixture of 3 (7.72 g, 16.16 mmol) and NaH (0.377 g, 15.7 mmol) in anhydrous THF (200 mL). The mixture was refluxed for 48 h, the solvent was evaporated under reduced pressure, and the residue was then partitioned between CH2Cl2 (50 mL) and water (100 mL). The aqueous layer was washed with CH2Cl2 (2 × 50 mL). The combined organic layers were dried (MgSO4) and concentrated. The crude product was purified by column chromatography (SiO2, AcOEt/n-hexane 95:5, then AcOEt) to give 4 (1.69 g, 18%). 1H NMR: δ = 3.59–3.75 (m, 24 H, OCH2), 4.57 (s, 2 H, CH2-Ph), 4.61 (s, 4 H, CH2-C6H4-), 7.28–7.59 (m, 13 H, Ar) ppm. 13C NMR: δC = 61.3, 70.0, 70.3, 70.3, 70.4, 70.5, 73.0, 127.4, 127.8, 127.9, 136.4, 137.5, 139.9 ppm. FAB-MS: m/z 591 [M+Na]+. Elemental analysis calcd (%) for C33H44O8: C, 69.69; H, 7.80; found: C, 69.17; H, 7.65.
4-{2-[2-(2-benzyloxyethoxy)ethoxy]ethoxymethyl}-4′-{2-[2-(2-p-toluensulfonyloxyethoxy) ethoxy]ethoxymethyl}biphenyl (5). A solution of NaOH (5 M, 2.5 mL) was added dropwise to a stirred mixture of 4 (1.65 g, 2.90 mmol) and tosyl chloride (1.71 g, 8.99 mmol) in THF (100 mL) at 0 °C. The reaction was allowed to reach room temperature and then stirred for 24 h. The solvent was evaporated under reduced pressure. The residue was partitioned between CH2Cl2 (50 mL) and water (100 mL), and the organic layer washed with water (2 × 100 mL). The organic layer was then dried (MgSO4) and concentrated. The crude product was purified by column chromatography (SiO2, AcOEt/n-hexane 60:40) to yield 5 (1.29 g, 55%). 1H NMR: δH = 2.42 (s, 3 H, CH3), 3.60–3.71 (m, 22 H, O-CH2), 4.14–4.17 (m, 2 H, CH2OTs) 4.56 (s, 2H, CH2-Ph), 4.60 (s, 4 H, CH2-C6H4-), 7.30–7.80 (m, 17 H, Ar) ppm. 13C NMR: δC = 21.6, 68.6, 69.2, 69.4, 70.5, 70.6, 70.7, 72.9, 73.2, 127.0, 127.5, 127.7, 127.9, 128.2, 128.3, 129.8, 132.9, 137.2, 137.3, 138.2, 140.2, 144.7 ppm. FAB-MS: m/z 745 [M+Na]+. Elemental analysis calcd (%) for C40H50O10S: C, 66.46; H, 6.97; found: C, 66.01; H, 6.73.
4-{2-[2-(2-benzyloxyethoxy)ethoxy]ethoxymethyl}-4′-{2-[2-(2-(4-hydroxymethylphenyloxy) ethoxy)ethoxy]ethoxymethyl}biphenyl (6). A stirred mixture of 5 (1.22 g, 1.70 mmol) p-hydroxybenzyl alcohol (0.412 g, 3.33 mmol) and K2CO3 (0.459 g, 3.33 mmol) in anhydrous CH3CN (150 mL) was refluxed under nitrogen for 48 h. The solvent was evaporated under reduced pressure. The residue was partitioned between CH2Cl2 (50 mL) and water (100 mL), and the organic layer washed with water (2 × 100 mL). The organic layer was then dried (MgSO4) and concentrated. The crude product was purified by column chromatography (SiO2, AcOEt/n-hexane 60:40) to give 6 (0.678 g, 59%). 1H NMR: δH = 3.61–3.76 (m, 20 H, O-CH2), 3.85–4.12 (m, 4 H, CH2-O-Ar), 4.56, 4.59, 4.61 (s, 8 H, 4 × CH2-Ar), 6.87–7.56 (m, 17 H, Ar) ppm. 13C NMR: δC = 64.7, 69.3, 69.9, 70.0, 70.3, 70.4, 70.5, 73.0, 114.6, 127.4, 127.8, 127.9, 128.0, 128.6, 128.9, 132.8, 136.4, 137.5, 139.9, 158.3 ppm. FAB-MS: m/z 697 [M+Na]+. Elemental analysis calcd (%) for C40H50O9: C, 71.19; H, 7.47; found: C, 69.85; H, 7.29.
4-{2-[2-(2-benzyloxyethoxy)ethoxy]ethoxymethyl}-4′-{2-[2-(2-(4-cholormethylphenyloxy) ethoxy)ethoxy]ethoxymethyl}biphenyl (7). A stirred mixture of 6 (0.638 g, 0.95 mmol) and SOCl2 (83 μL, 1.14 mmol) in anhydrous CH2Cl2 (150 mL) was stirred under nitrogen for 16 h. The solvent was evaporated under reduced pressure and the remaining residue was purified by column chromatography (SiO2, AcOEt/n-hexane 50:50) to yield 7 (0.522 g, 0.76 mmol, 80%). 1H NMR: δH = 3.63–3.74 (m, 20 H, O-CH2), 3.84–4.14 (m, 4 H, CH2-O-Ar), 4.54, 4.56, 4.60 (s, 8 H, 4 × CH2-Ar), 6.86–7.56 (m, 17 H, Ar). 13C NMR: δC = 67.5, 69.4, 69.7, 70.6, 70.8, 72.9, 73.2, 114.8, 127.0, 127.5, 127.7, 128.2, 128.3, 129.8, 130.0, 137.3, 138.1, 140.2, 158.9. FAB-MS: m/z 715 [M+Na]+. Elemental analysis calcd (%) for C40H49ClO8: C, 69.30; H, 7.12; found: C, 69.05; H, 6.99.
N,N′-Bis{4-[2-(2-(2-(4-(4-(2-(2-(2-benzyloxyethoxy)ethoxy]ethoxymethylphenyl)benzyloxy) ethoxy)ethoxy)ethoxy]benzyl}-4,4′-bipyridinium dichloride (2·2Cl). A stirred mixture of 7 (0.224 g, 0.32 mmol) and 4,4′-bipyridine (0.024 g, 0.154 mmol) in anhydrous CH3CN (15 mL) was refluxed under nitrogen for 48 h. The solvent was evaporated under reduced pressure, the residue partitioned between CH2Cl2 (20 mL) and water (20 mL). The organic layer was washed with water (2 × 20 mL), dried (MgSO4) and concentrated. The crude product was purified by column chromatography (SiO2, MeOH/NH4Cl(aq.) (2 M)/MeNO2 10:0.2:1), to give 2·2Cl (0.071 g, 30%). 1H NMR: δH = 3.59.378 (m, 44 H, O-CH2), 3.93 (s broad, 4 H, CH2-CH2-O-Ar), 4.49, 4.50, 4.52 (3s, 12 H, CH2-Ar), 5.51 (s broad, 4 H, CH2-N+), 6.75 (part of an AB system, JAB = 8.0 Hz, 4 H, Ar), 7.19–7.42 (m, 30 H, Ar), 8.03 (s broad, 4 H, H-β bipyridinium), 8.66 (s broad, 4 H, H-α bipyridinium). 13C NMR: δC = 64.1, 64.3, 67.5, 69.2, 69.4, 69.5, 69.6, 70.6, 72.8, 72.9, 73.2 (CH2), 115.7, 123.7, 125.8, 126.9, 127.2, 127.6, 127.7, 128.3, 128.4, 129.6, 130.2, 130.3, 131.2, 133.0, 137.3, 137.4, 138.1, 139.9, 141.6, 144.5, 153.7, 160.1 ppm. FAB-MS: m/z 1506 [M–Cl]+. Elemental analysis calcd (%) for C90H106Cl2N2O16: C, 70.07; H, 6.93; N, 1.82; found: C, 69.84; H, 7.12; N, 1.86.
Figure 1.
Structure of heptakis-2,6-di-O-methyl-3-O-acetyl-β-cyclodextrin (1).
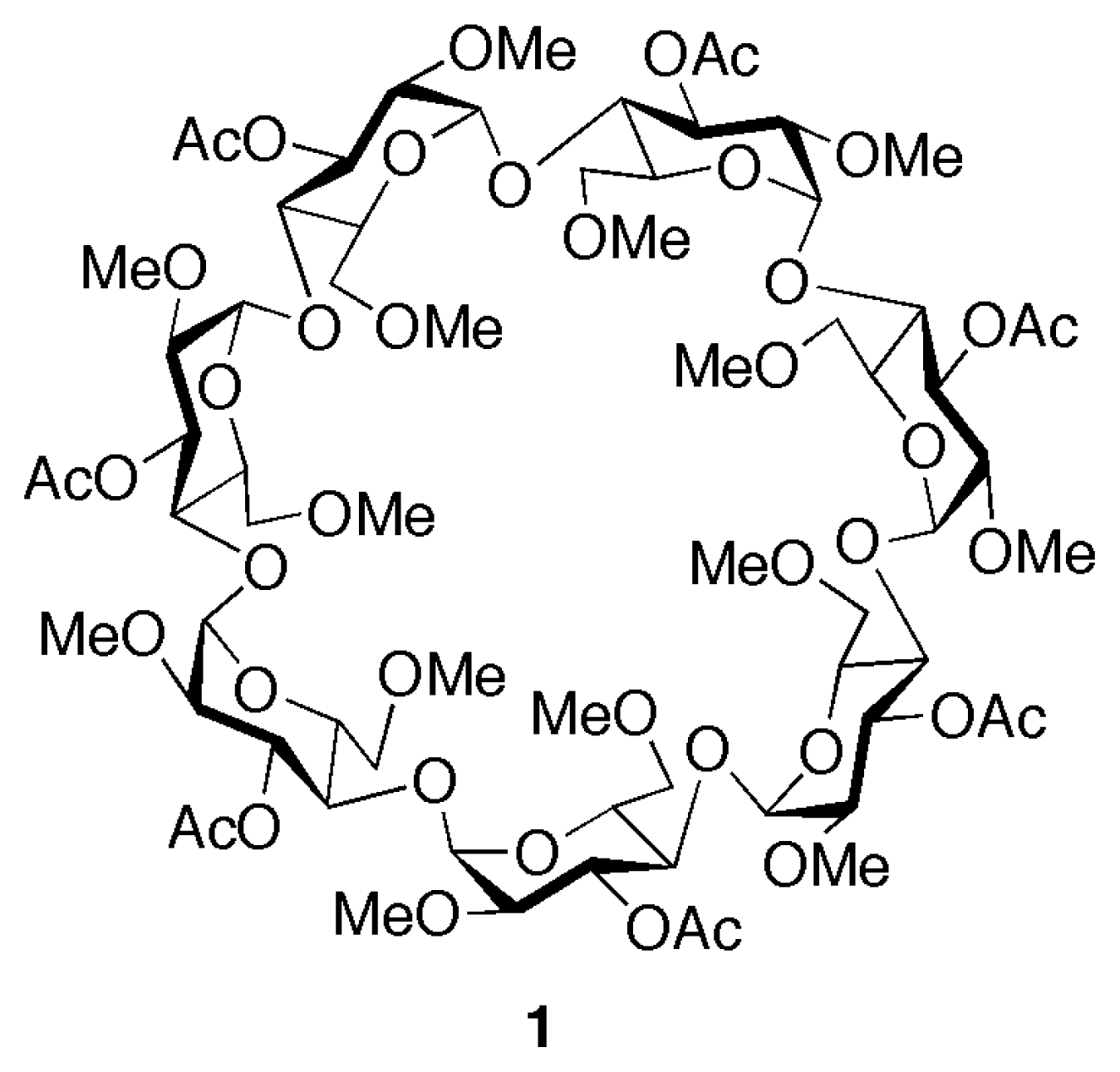
Figure 2.
1H NMR spectra (300 MHz, CDCl3, 22 °C) of 5 mM samples of: a) cyclodextrin 1; b) linear component 2·2Cl; c) [2]pseudorotaxane 1⊃2·2Cl.
Figure 2.
1H NMR spectra (300 MHz, CDCl3, 22 °C) of 5 mM samples of: a) cyclodextrin 1; b) linear component 2·2Cl; c) [2]pseudorotaxane 1⊃2·2Cl.
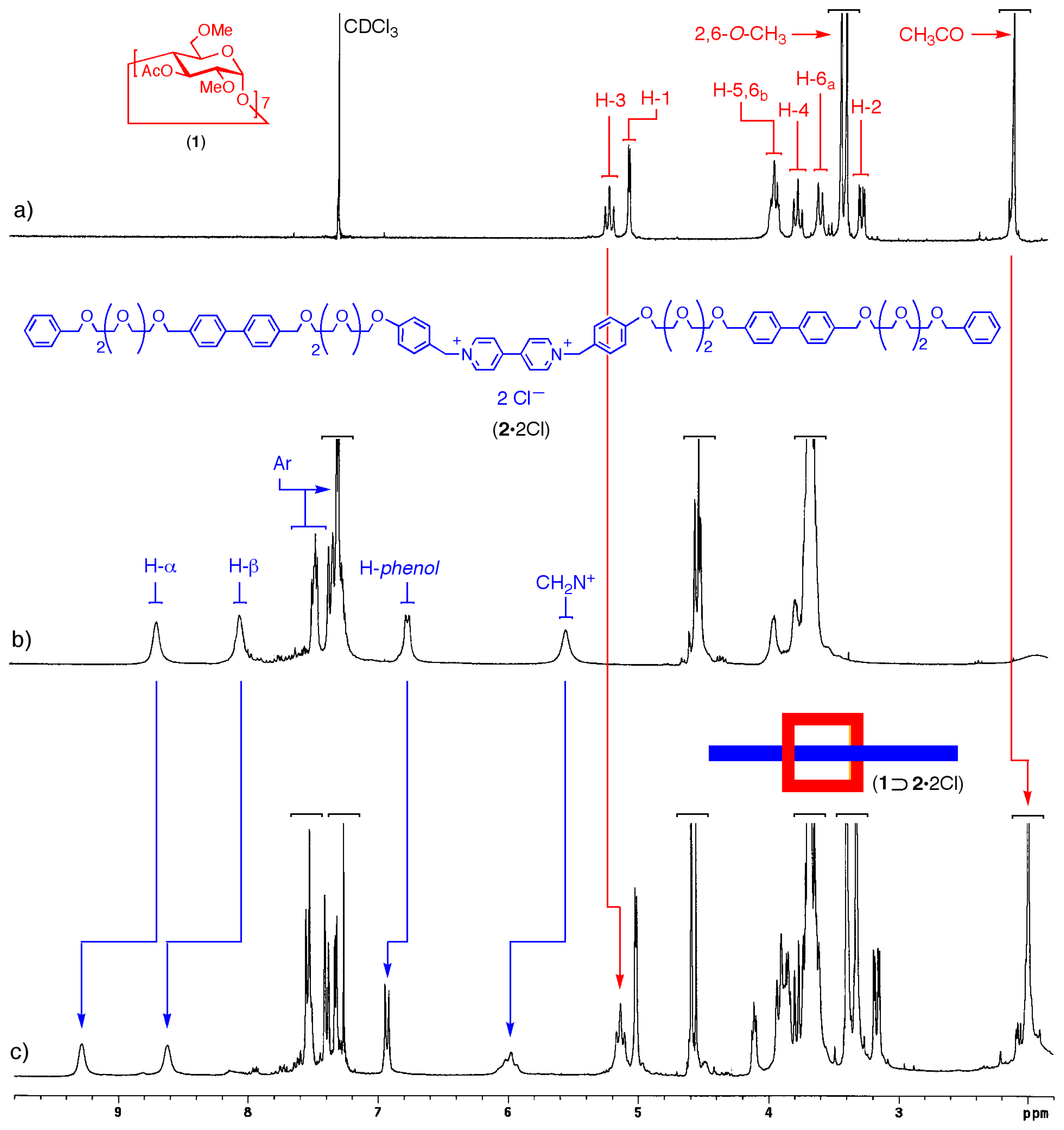
Figure 3.
Space-filling molecular model of the [2]pseudorotaxane 1⊃2·2Cl (grey: carbon, white: hydrogen, red: oxygen; chloride ions omitted for clarity).
Figure 3.
Space-filling molecular model of the [2]pseudorotaxane 1⊃2·2Cl (grey: carbon, white: hydrogen, red: oxygen; chloride ions omitted for clarity).
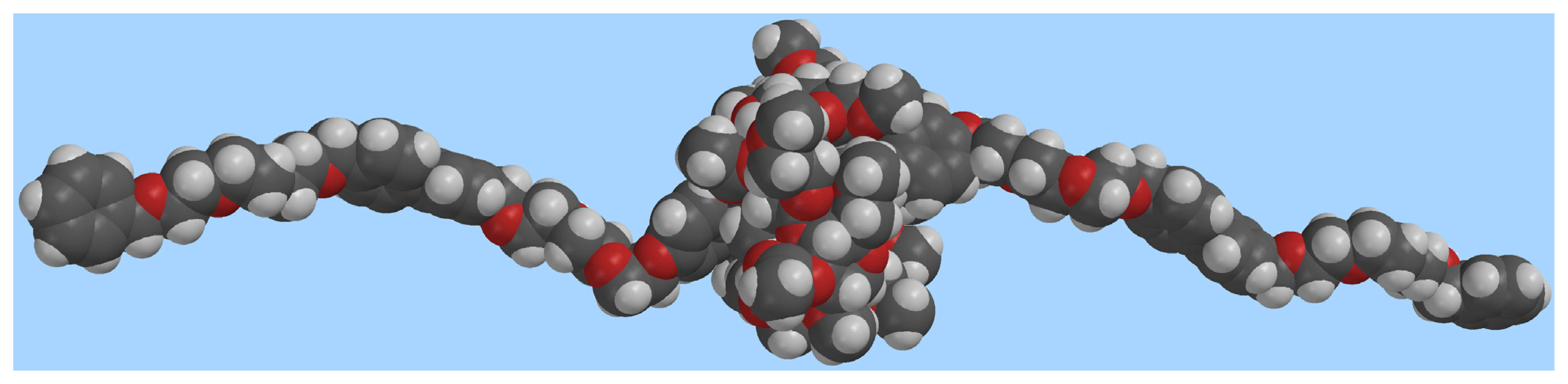
Figure 4.
Top-down view of the CD cavity in the [2]pseudorotaxane 1⊃2·2Cl. O···N+ interactions are highlighted in yellow (grey: carbon, white: hydrogen, red: oxygen; chloride ions omitted for clarity).
Figure 4.
Top-down view of the CD cavity in the [2]pseudorotaxane 1⊃2·2Cl. O···N+ interactions are highlighted in yellow (grey: carbon, white: hydrogen, red: oxygen; chloride ions omitted for clarity).
Scheme 1.
Synthesis of the linear component 2·2Cl.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Binding constants for the receptor 1 and the test guest species simulating the two stations of the pseudorotaxane. Ka values (M−1) were determined by 1H NMR (300 MHz, 22±1 °C).
| Ka in D2O/CD3COCD3 (4:1 v/v) | Ka in CD3COCD3 | |
|---|---|---|
| 1⊃3 | 2900 ± 250 | no binding |
| 1⊃82+ | no bindingA | 13200 ± 1100 B |
A8·2Cl was used as the guest.
B8·2PF6 was used as the guest.
Acknowledgements
This work was supported by MiUR.
References and Notes
- Armspach, D.; Gattuso, G.; Koeniger, R.; Stoddart, J. F. Bioorganic Chemistry: Carbohydrates; Hecht, S.M., Ed.; Oxford University Press: New York, 1999; pp. 458–488. [Google Scholar]
- Szejtli, J. Introduction and general overview of cyclodextrin chemistry. Chem. Rev 1998, 98, 1743–1754. [Google Scholar]
- Comprehensive Supramolecular Chemistry: Cyclodextrins; Szejtli, J.; Osa, T. (Eds.) Elsevier: Oxford, 1996; Volume 3.
- Hedges, A.R. Industrial applications of cyclodextrins. Chem. Rev 1998, 98, 2035–2044. [Google Scholar]
- Del Valle, E.M.M. Cyclodextrins and their uses: a review. Process Biochem 2004, 39, 1033– 1046. [Google Scholar]
- Uekama, K.; Hirayama, F.; Irie, T. Cyclodextrin drug carrier systems. Chem. Rev 1998, 98, 2045–2076. [Google Scholar]
- Szente, L.; Szejtli, J. Highly soluble cyclodextrin derivatives: chemistry, properties, and trends in development. Adv. Drug Deliver. Rev 1999, 36, 17–28. [Google Scholar]
- Stoddart, J.F. Cyclodextrins, off-the-shelf components for the construction of mechanically interlocked molecular-systems. Angew. Chem., Int. Ed. Engl 1992, 31, 846–848. [Google Scholar]
- Wenz, G. Cyclodextrins as building-blocks for supramolecular structures and functional units. Angew. Chem., Int. Ed. Engl 1994, 33, 803–822. [Google Scholar]
- Wenz, G.; Han, B.H.; Muller, A. Cyclodextrin rotaxanes and polyrotaxanes. Chem. Rev 2006, 106, 782–817. [Google Scholar]
- Harada, A. Cyclodextrin-based molecular machines. Acc. Chem. Res 2001, 34, 456–464. [Google Scholar]
- Nepogodiev, S.A.; Stoddart, J.F. Cyclodextrin-based catenanes and rotaxanes. Chem. Rev 1998, 98, 1959–1976. [Google Scholar]
- Onagi, H.; Blake, C.J.; Easton, C.J.; Lincoln, S.F. Installation of a ratchet tooth and pawl to restrict rotation in a cyclodextrin rotaxane. Chem. Eur. J 2003, 9, 5978–5988. [Google Scholar]
- Stanier, C.A.; Alderman, S.J.; Claridge, T.D.W.; Anderson, H.L. Unidirectional photoinduced shuttling in a rotaxane with a symmetric stilbene dumbbell. Angew. Chem. Int. Ed 2002, 41, 1769–1772. [Google Scholar]
- Armspach, D.; Ashton, P.R.; Ballardini, R.; Balzani, V.; Godi, A.; Moore, C.P.; Prodi, L.; Spencer, N.; Stoddart, J.F.; Tolley, M.S.; Wear, T.J.; Williams, D.J. Catenated cyclodextrins. Chem. Eur. J 1995, 1, 33–55. [Google Scholar]
- Harada, A.; Hashidzume, A.; Takashima, Y. Cyclodextrin-based supramolecular polymers. Polym. Sci 2006, 201, 1–43. [Google Scholar]
- Suh, J. Synthetic artificial peptidases and nucleases using macromolecular catalytic systems. Acc. Chem. Res 2003, 36, 562–570. [Google Scholar]
- Breslow, R.; Dong, S.D. Biomimetic reactions catalyzed by cyclodextrins and their derivatives. Chem. Rev 1998, 98, 1997–2012. [Google Scholar]
- Saha, S.; Stoddart, J.F. Photo-driven molecular devices. Chem. Soc. Rev 2007, 36, 77–92. [Google Scholar]
- Tian, H.; Wang, Q.C. Recent progress on switchable rotaxanes. Chem. Soc. Rev 2006, 35, 361– 374. [Google Scholar]
- Mirzoian, A.; Kaifer, A.E. Reactive pseudorotaxanes: Inclusion complexation of reduced viologens by the hosts β-cyclodextrin and heptakis(2,6-di-O-methyl)-β-cyclodextrin. Chem. Eur. J 1997, 3, 1052–1058. [Google Scholar]
- Murakami, M.; Kawabuchi, A.; Matsumoto, R.; Ido, T.; Nakashima, N. A multi-mode-driven molecular shuttle: photochemically and thermally reactive azobenzene rotaxanes. J. Am. Chem. Soc 2005, 127, 15891–15899. [Google Scholar]
- Murakami, M.; Kawabuchi, A.; Kotoo, K.; Kunitake, M.; Nakashima, N. A light-driven molecular shuttle based on a rotaxane. J. Am. Chem. Soc 1997, 119, 7605–7606. [Google Scholar]
- Ma, X.; Wang, Q.C.; Qu, D.H.; Xu, Y.; Ji, F.Y.; Tian, H. A light-driven pseudo[4]rotaxane encoded by induced circular dichroism in a hydrogel. Adv. Funct. Mater 2007, 17, 829–837. [Google Scholar]
- Liu, Y.; Li, X.-Y.; Zhang, H.-Y.; Li, C.-J.; Ding, F. Cyclodextrin-driven movement of cucurbit[7]uril. J. Org. Chem 2007, 72, 3640–3645. [Google Scholar]
- Saenger, W. Inclusion Compounds; Atwood, J.L., Davies, J.E.D., MacNicol, D.D., Eds.; Academic Press: London, 1984; Volume 2, pp. 231–259. [Google Scholar]
- Engeldinger, E.; Armspach, D.; Matt, D. Capped cyclodextrins. Chem. Rev 2003, 103, 4147– 4173. [Google Scholar]
- Rauf Khan, A.; Forgo, P.; Stine, K.J.; D’Souza, V.T. Methods for selective modifications of cyclodextrins. Chem. Rev 1998, 98, 1977–1996. [Google Scholar]
- Ashton, P.R.; Boyd, S. E.; Gattuso, G.; Hartwell, E. Y.; Koeniger, R.; Spencer, N.; Stoddart, J. F. A novel approach to the synthesis of some chemically-modified cyclodextrins. J. Org. Chem 1995, 60, 3898–3903. [Google Scholar]
- Morozumi, T.; Sato, N.; Nakamura, H. Selective pseudo-rotaxane type complex formation of zinc(II) (t-butylatedtetraphenyl) porphyrin-viologen linked compounds with tri-O-methyl-β-cyclodextrin. J. Incl. Phenom. Macro 2006, 56, 141–148. [Google Scholar]
- Wenz, G.; Wolf, F.; Wagner, M.; Kubik, S. Topology and molecular chemistry – synthesis, topography and stability of a [2]-rotaxane derived from a lipophilic cyclodextrin derivative. New J. Chem 1993, 17, 729–738. [Google Scholar]
- Wenz, G.; Vonderbey, E.; Schmidt, L. Synthesis of a lipophilic cyclodextrin-[2]-rotaxane. Angew. Chem., Int. Ed. Engl 1992, 31, 783–785. [Google Scholar]
- Sliwa, W.; Bachowska, B. Supramolecular species bearing quaternary azaaromatic moieties. Heterocycles 2006, 68, 1467–1500. [Google Scholar]
- Cafeo, G.; Gargiulli, C.; Gattuso, G.; Kohnke, F.H.; Notti, A.; Occhipinti, S.; Pappalardo, S.; Parisi, M.F. Recognition and binding of paraquat dichloride by cyclodextrin/calix[6]pyrrole binary host systems. Tetrahedron Lett 2002, 43, 8103–8106. [Google Scholar]
- Cafeo, G.; Gattuso, G.; Kohnke, F.H.; Notti, A.; Occhipinti, S.; Pappalardo, S.; Parisi, M.F. Remarkable boosting of the binding of ion-paired organic salts by binary host systems. Angew. Chem. Int. Ed 2002, 41, 2122–2126. [Google Scholar]
- Giastas, P.; Mourtzis, N.; Yannakopoulou, K.; Mavridis, I.M. Pseudorotaxanes of β-cyclodextrin with diamino end-functionalized oligo-phenyl and -benzyl compounds in solution and in the solid state. J. Incl. Phenom. Macro 2002, 44, 247–250. [Google Scholar]
- Park, J.W.; Song, H.J.; Chang, H.J. Unidirectional α-cyclodextrin-based [2]rotaxanes bearing viologen unit on axle. Tetrahedron Lett 2006, 47, 3831–3834. [Google Scholar]
- Oshikiri, T.; Takashima, Y.; Yamaguchi, H.; Harada, A. Kinetic control of threading of cyclodextrins onto axle molecules. J. Am. Chem. Soc 2005, 127, 12186–12187. [Google Scholar]
- Park, J.W.; Song, H.J. Isomeric [2]rotaxanes and unidirectional [2]pseudorotaxane composed of α-cyclodextrin and aliphatic chain-linked carbazole-viologen compounds. Org. Lett 2004, 6, 4869– 4872. [Google Scholar]
- Baer, A.J.; Macartney, D.H. Orientational isomers of α-cyclodextrin [2]semi-rotaxanes with asymmetric dicationic threads. Org. Biomol. Chem 2005, 3, 1448–1452. [Google Scholar]
- Park, J.W.; Song, H.J.; Lee, S.Y. Face selectivity of inclusion complexation of viologens with β-cyclodextrin and 6-O-(2-sulfonato-6-naphthyl)-β-cyclodextrin. J. Phys. Chem. B 2002, 106, 7186–7192. [Google Scholar]
- Cyclodextrin 1 was prepared according to a literature procedure and its spectroscopic data were in full agreement with those previously published. See: Hirayama, F.; Mieda, S.; Miyamoto, Y.; Arima, H.; Uekama, K. Heptakis(2,6-di-O-methyl-3-O-acetyl)-β-cyclodextrin: a water-soluble cyclodextrin derivative with low hemolytic activity. J. Pharm. Sci 1999, 88, 970–975. [Google Scholar]
- Alston, D.R.; Lilley, T.H.; Stoddart, J.F. The binding of cyclobutane-1,1- dicarboxylatodiamineplatinum(II) by α-cyclodextrin in aqueous solution. J. Chem. Soc. Chem. Commun 1985, 1600–1602. [Google Scholar]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals; Pergamon Press: Oxford, 1989. [Google Scholar]
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Gattuso, G.; Gargiulli, C.; Parisi, M.F. Threading Cyclodextrins in Chloroform: A [2]Pseudorotaxane. Int. J. Mol. Sci. 2007, 8, 1052-1063. https://doi.org/10.3390/i8101052
AMA Style
Gattuso G, Gargiulli C, Parisi MF. Threading Cyclodextrins in Chloroform: A [2]Pseudorotaxane. International Journal of Molecular Sciences. 2007; 8(10):1052-1063. https://doi.org/10.3390/i8101052
Chicago/Turabian StyleGattuso, Giuseppe, Claudia Gargiulli, and Melchiorre F. Parisi. 2007. "Threading Cyclodextrins in Chloroform: A [2]Pseudorotaxane" International Journal of Molecular Sciences 8, no. 10: 1052-1063. https://doi.org/10.3390/i8101052