Structural Stability and Vibrational Analyses of Haloselenonyl Azides, XSeO2-NNN, where X is F, Cl, Br

Abstract
:Introduction
Ab initio calculations
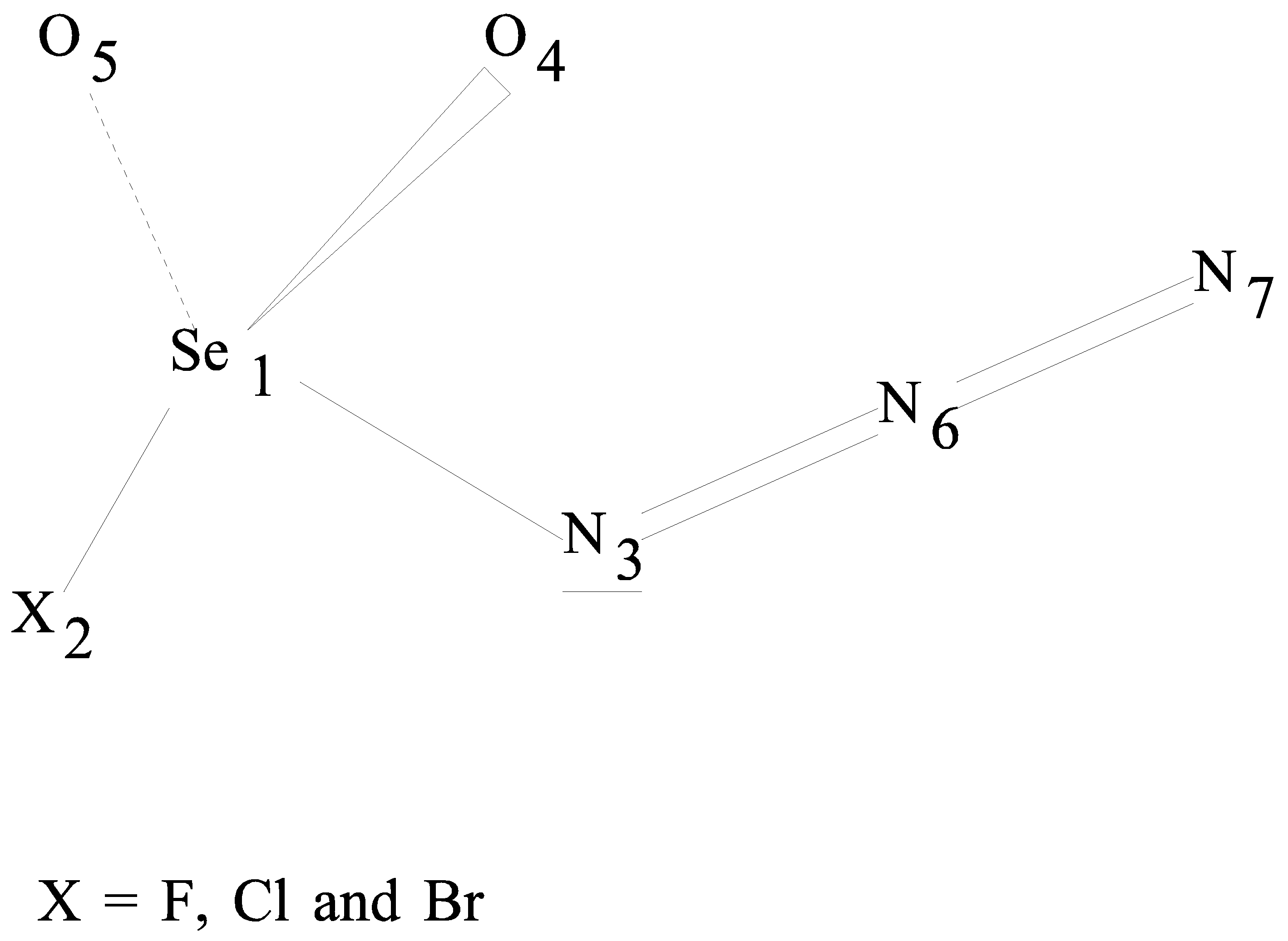
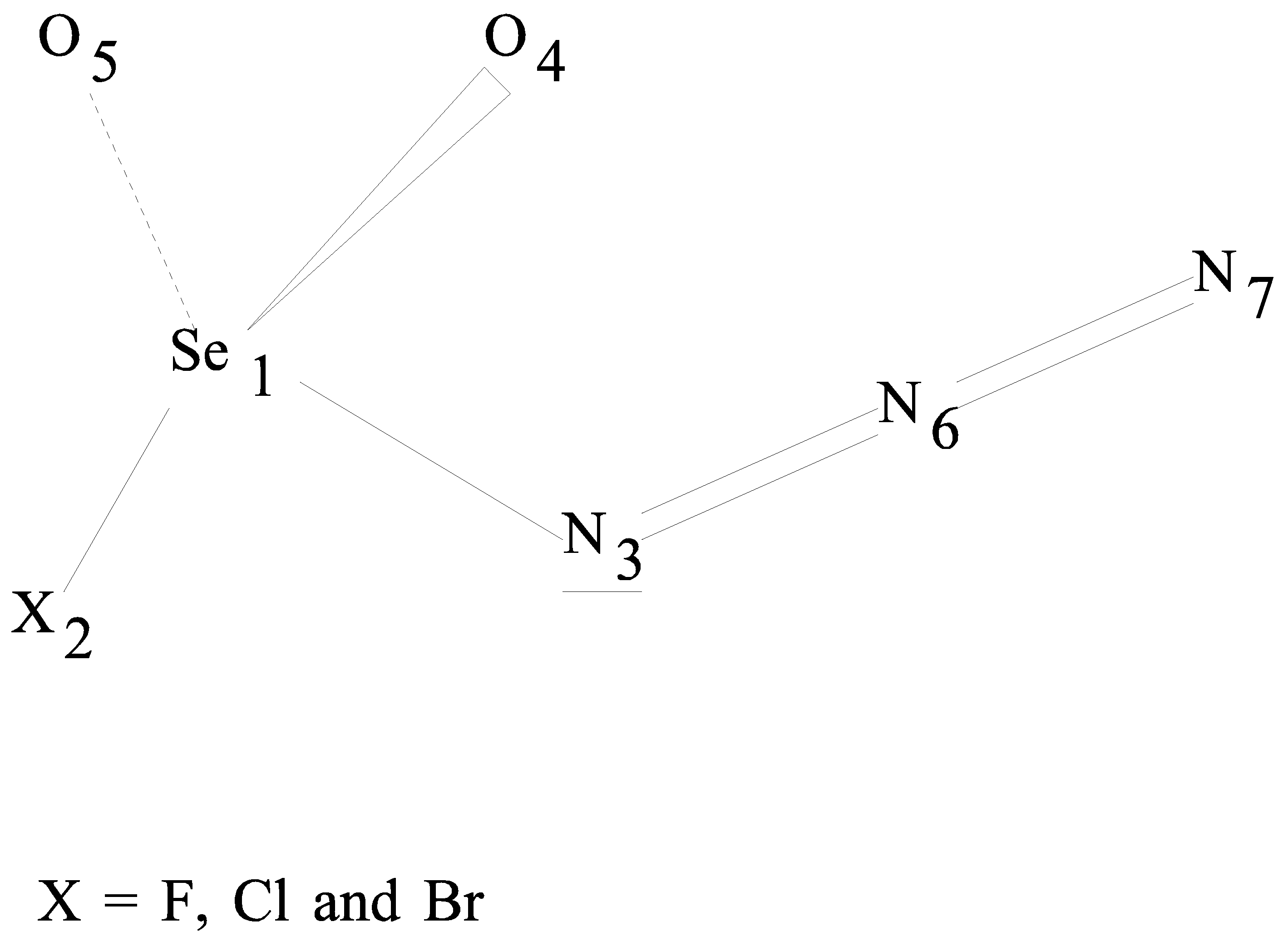
Torsional potential scans


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MP2 | |||
| Parameter | X = F | X = Cl | X = Br |
| bond lengths (Å) | |||
| Se1 - X2 | 1.7747 | 2.1874 | 2.3572 |
| Se1 - N3 | 1.8391 | 1.8599 | 1.8688 |
| Se1 = O4 | 1.5941 | 1.5994 | 1.6018 |
| Se1 = O5 | 1.5847 | 1.5903 | 1.5930 |
| N3 = N6 | 1.2568 | 1.2542 | 1.2532 |
| N6 = N7 | 1.1501 | 1.1518 | 1.1525 |
| bond angles (degree) | |||
| (N3Se1X2) | 96.65 | 99.08 | 99.70 |
| (X2Se1O4) | 104.72 | 106.43 | 107.12 |
| (X2Se1O5) | 106.33 | 107.83 | 108.54 |
| (Se1N3N6) | 111.41 | 110.92 | 110.87 |
| (N3N6N7) | 171.97 | 172.42 | 172.83 |
| torsional angles (degree) | |||
| (N3X2Se1O4) | 113.60 | 113.41 | 113.41 |
| (N3X2Se1O5) | -108.84 | -108.71 | -108.67 |
| (X2Se1N3N6) | 76.18 | 76.18 | 73.87 |
| total energy (H) | |||
| Et | -2813.4541988 | -3173.4367477 | -5286.2910420 |
| total dipole moment (Debye) | |||
| µt | 3.2588 | 3.2584 | 3.4999 |
| rotational constants (GHz) | |||
| A | 3.85980 | 2.30429 | 1.61847 |
| B | 1.47242 | 1.42066 | 1.14392 |
| C | 1.46064 | 1.14140 | 0.81268 |
| DFT/B3LYP | |||
| Parameter | X = F | X = Cl | X = Br |
| bond lengths (Å) | |||
| Se1 - X2 | 1.7869 | 2.2231 | 2.3866 |
| Se1 - N3 | 1.8757 | 1.9001 | 1.9088 |
| Se1 = O4 | 1.6082 | 1.6148 | 1.6176 |
| Se1 = O5 | 1.5990 | 1.6057 | 1.6086 |
| N3 = N6 | 1.2519 | 1.2480 | 1.2466 |
| N6 = N7 | 1.1248 | 1.1267 | 1.1274 |
| bond angles (degrees) | |||
| (N3Se1X2) | 97.13 | 100.30 | 101.22 |
| (X2Se1O4) | 105.03 | 106.97 | 107.70 |
| (X2Se1O5) | 106.44 | 107.72 | 108.25 |
| (Se1N3N6) | 112.34 | 112.15 | 112.16 |
| (N3N6N7) | 172.45 | 173.36 | 173.50 |
| torsional angles (degrees) | |||
| (N3X2Se1O4) | 114.35 | 114.23 | 114.18 |
| (N3X2Se1O5) | -108.91 | -109.12 | -109.18 |
| (X2Se1N3N6) | 79.03 | 82.75 | 84.06 |
| total energy (H) | |||
| Et | -2816.0122882 | -3176.376035 | -5290.3039151 |
| total dipole moment (Debye) | |||
| µt | 2.2964 | 2.1935 | 2.3910 |
| rotational constants (GHz) | |||
| A | 3.77760 | 2. 28284 | 1.70586 |
| B | 1.44238 | 1.35332 | 1.02641 |
| C | 1.42986 | 1.09894 | 0.77273 |
| Quantity | X = F | X = Cl | X = Br |
| V0 | 2.0910 | 2.2961 | 2.2462 |
| V1 | 0.4423 | 0.7409 x 10-1 | 0.1828 x 10-1 |
| V2 | -1.9892 | -2.0817 | -2.0485 |
| V3 | -0.4604 | -0.4314 | -0.3915 |
| V4 | 0.2529 x 10-1 | -0.3348 x 10-1 | -0.4481 x 10-1 |
| V5 | 0.7158 x 10-3 | -0.3756 x 10-1 | -0.3175 x 10-1 |
| V6 | -0.5353 x 10-2 | -0.1161 x 10-2 | 0.5389 x 10-2 |
| rms | 0.1368 x 10-1 | 0.3625 x 10-2 | 0.4879 x 10-2 |
| E0 | -2816.012294 | -3176.376037 | -5290.303921 |
Vibrational modes and normal coordinate analyses
| No. | Coordinate | Definition | |
| 1 | Se1-X2 | stretch | Q |
| 2 | Se1-N3 | stretch | T |
| 3 | Se1-O4 | stretch | X1 |
| 4 | Se1-O5 | stretch | X2 |
| 5 | N3-N6 | stretch | R1 |
| 6 | N6-N7 | stretch | R2 |
| 7 | N3Se1X2 | bend | θ |
| 8 | X2Se1O4 | bend | ε1 |
| 9 | X2Se1O5 | bend | ε2 |
| 10 | O4Se1O3 | bend | π1 |
| 11 | O5Se1O3 | bend | π2 |
| 12 | O4Se1O5 | bend | δ |
| 13 | Se1N3N6 | bend | γ |
| 14 | N3N6N7 | bend | σ1 |
| 15 | N7N6N3Se1 | wag | σ2 |
| 16 | N6N3Se1O4 + N6N3Se1O5 | torsion | τ |
| Description | Symmetry coordinate |
| X - Se stretch | S1 = Q |
| Se - N stretch | S2 = T |
| NNN symmetric stretch | S3 = R1 + R2 |
| SeO2 symmetric stretch | S4 = X1 + X2 |
| NNN antisymmetric stretch | S5 = R1 - R2 |
| SeO2 antisymmetric stretch | S6 = X1 - X2 |
| XSeN bend | S7 = 5θ-ε1-ε2-π1-π2-δ |
| SeNN bend | S8 = γ |
| NNN in-plane bend | S9 = σ1 |
| NNN out-of-plane bend | S10 = σ2 |
| SeO2 deformation | S11 = 4δ- ε1-ε2-π1-π2 |
| SeO2 rock | S12 = ε1-ε2+π1-π2 |
| SeO2 twist | S13 = ε1-ε2-π1+π2 |
| SeO2 wag | S14 = ε1+ε2-π1-π2 |
| NNN torsion | S15 = τ |
| ki | Ii | Si | ρi | PED |
| Fluoroselenonyl azide (X = F) | ||||
| 2237 | 411.8 | 133.6 | 0.36 | 88% S5, 12% S3 |
| 1214 | 206.6 | 3.4 | 0.72 | 85% S3, 13% S5 |
| 996 | 99.7 | 12.7 | 0.64 | 98% S6 |
| 912 | 75.2 | 28.7 | 0.07 | 97% S4 |
| 675 | 7.1 | 9.1 | 0.20 | 49% S9, 31% S8, 17% S2 |
| 591 | 120.4 | 15.1 | 0.09 | 97% S1 |
| 554 | 3.1 | 0.9 | 0.67 | 96% S10 |
| 441 | 90.1 | 25.8 | 0.12 | 68% S2, 17% S9 |
| 329 | 40.2 | 5.0 | 0.44 | 30% S12, 22% S11, 18% S14, 13% S9 |
| 311 | 22.2 | 6.0 | 0.62 | 70% S14, 15% S12 |
| 308 | 21.7 | 6.7 | 0.53 | 47% S11, 35% S12 |
| 251 | 0.8 | 6.4 | 0.62 | 78% S13 |
| 231 | 0.7 | 1.6 | 0.74 | 78% S7, 17% S11 |
| 130 | 1.4 | 4.0 | 0.52 | 54% S8, 15% S12, 15% S13, 14% S9 |
| 52 | 0.0 | 3.3 | 0.75 | 95% S15 |
| Chloroselenonyl azide (X = Cl) | ||||
| 2221 | 444.1 | 154.9 | 0.37 | 88% S5, 12% S3 |
| 1220 | 201.1 | 4.5 | 0.75 | 86% S3, 13% S5 |
| 977 | 92.6 | 15.9 | 0.64 | 97% S6 |
| 899 | 77.2 | 30.2 | 0.05 | 97% S4 |
| 669 | 4.8 | 9.5 | 0.19 | 51% S9, 32% S8, 14% S2 |
| 550 | 4.8 | 0.9 | 0.63 | 97% S10 |
| 420 | 86.6 | 29.7 | 0.15 | 72% S2, 16% S9 |
| 378 | 123.1 | 15.6 | 0.11 | 67% S1, 17% S14, 12% S11 |
| 311 | 29.6 | 6.1 | 0.36 | 42% S11, 24% S14, 11% S12 |
| 289 | 5.3 | 18.6 | 0.36 | 43% S14, 32% S1, 22% S11 |
| 281 | 4.8 | 10.6 | 0.53 | 47% S12, 11% S11 |
| 211 | 1.2 | 4.8 | 0.70 | 76% S13, 18% S12 |
| 188 | 1.4 | 2.8 | 0.75 | 83% S7 |
| 126 | 1.2 | 5.2 | 0.47 | 53% S8, 17% S12, 14% S9, 11% S13 |
| 38 | 0.1 | 6.6 | 0.74 | 95% S15 |
| Bromoselenonyl azide (X = Br) | ||||
| 2215 | 458.4 | 167.5 | 0.37 | 88% S5, 12% S3 |
| 1223 | 199.7 | 5.3 | 0.75 | 86% S3, 13% S5 |
| 970 | 90.6 | 18.5 | 0.65 | 97% S6 |
| 893 | 78.5 | 31.9 | 0.05 | 97% S4 |
| 668 | 4.6 | 9.4 | 0.20 | 52% S9, 32% S8, 13% S2 |
| 550 | 4.4 | 1.0 | 0.56 | 97% S10 |
| 412 | 85.2 | 34.0 | 0.16 | 74% S2, 15% S9 |
| 341 | 109.8 | 2.5 | 0.21 | 40% S14, 32% S11, 20% S1 |
| 306 | 27.1 | 5.5 | 0.36 | 39% S11, 30 S14 |
| 275 | 3.3 | 1.7 | 0.49 | 42% S12, 13% S13, 11% S11 |
| 222 | 1.1 | 23.1 | 0.25 | 78% S1, 15% S14 |
| 188 | 1.0 | 2.9 | 0.71 | 70% S13, 21% S12 |
| 165 | 1.6 | 2.6 | 0.74 | 75% S7 |
| 123 | 1.2 | 5.5 | 0.43 | 50% S8, 16% S12, 14% S7, 13% S9 |
| 33 | 0.1 | 7.2 | 0.73 | 94% S15 |
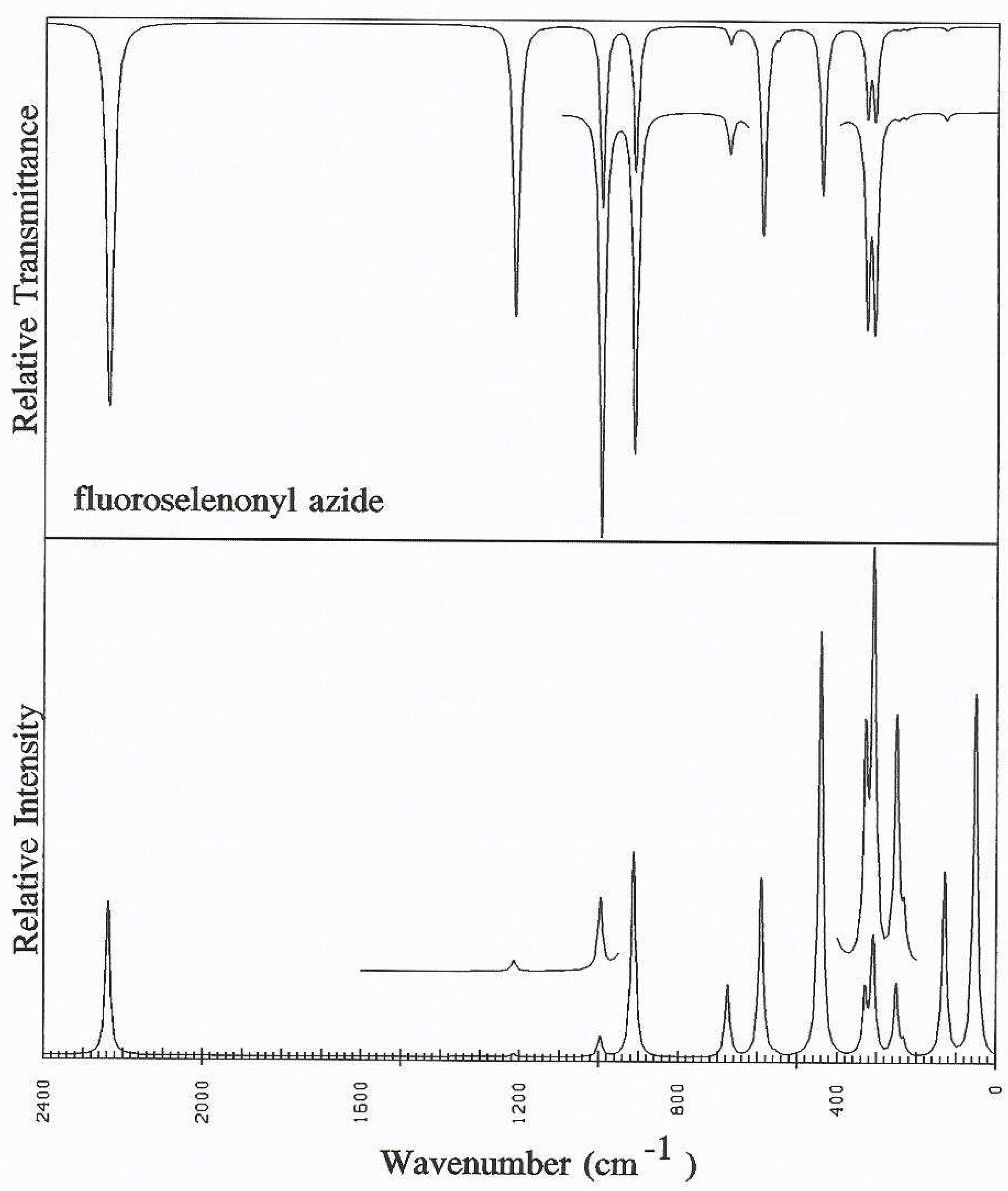
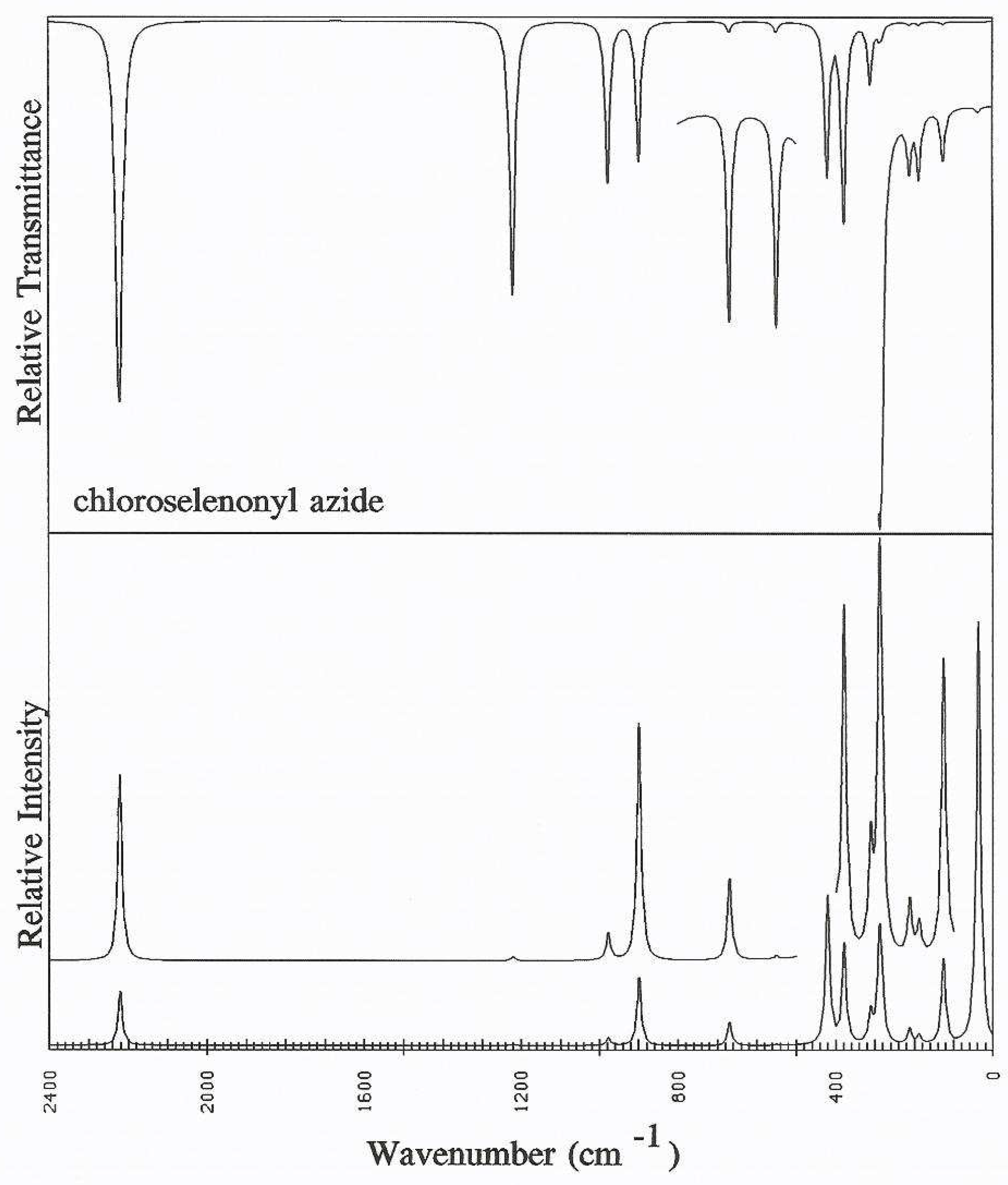
Discussion
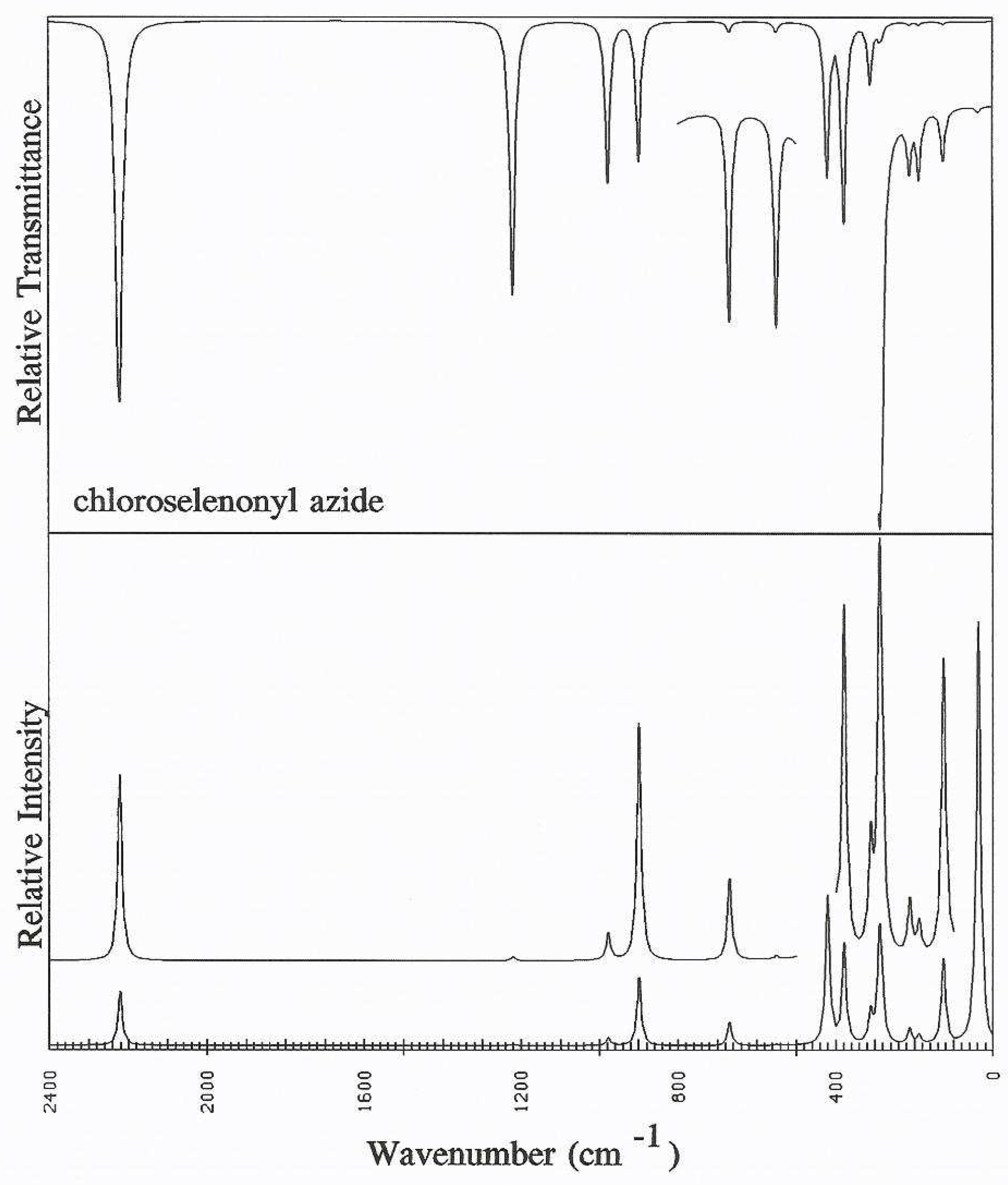
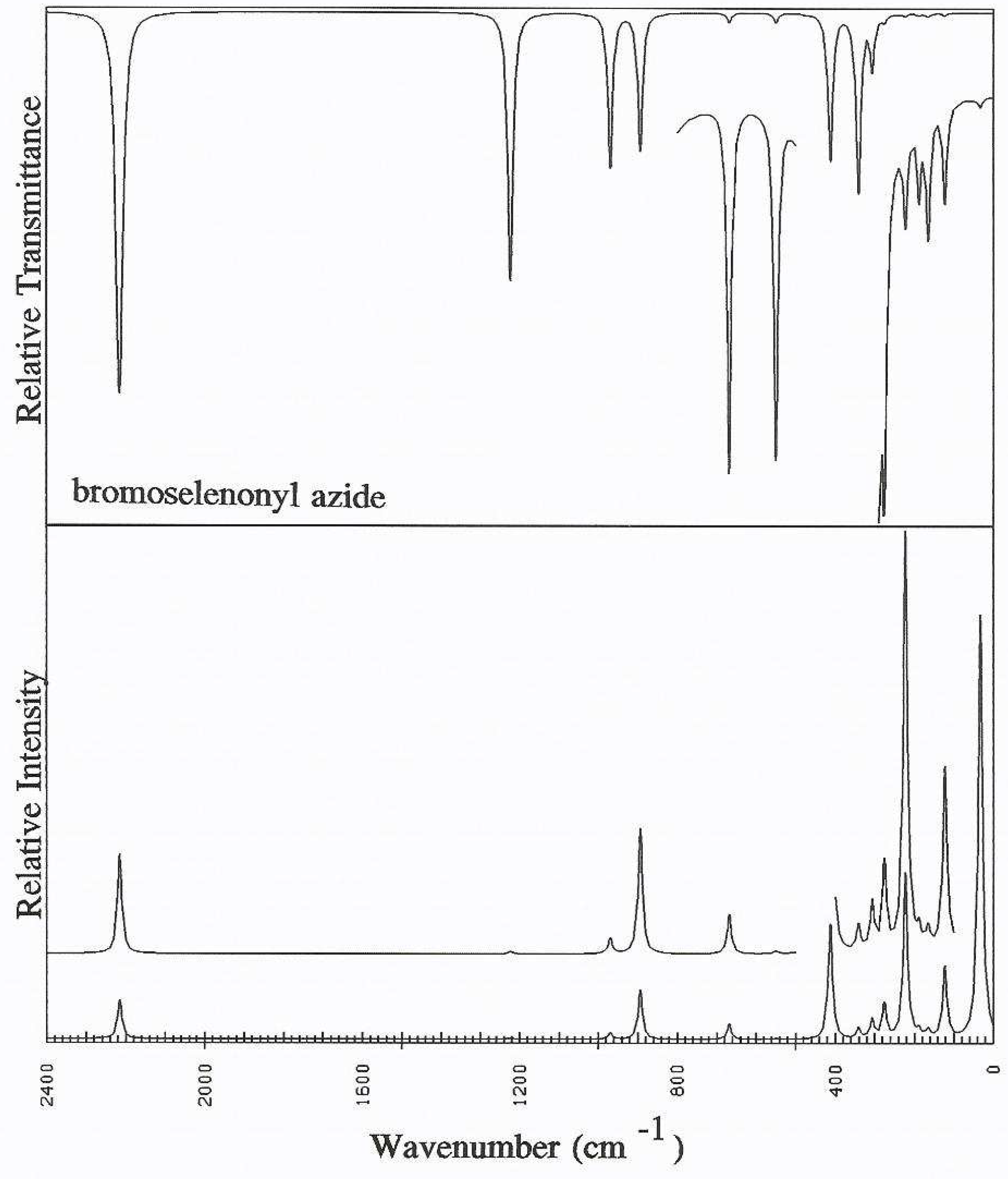
Acknowledgements
References
- Brunvoll, J.; Hargittai, I.; Ragnhild, S. Electron-diffraction Investigation of the Molecular Structure of Sulfonyl Chloride Isocyanate. J. Chem. Soc. Dalton Trans. 1978, 299–302. [Google Scholar] [CrossRef]
- Jo, O. L.; Graybeal, J. D.; Lovas, F. J.; Suenram, R. D. Determination of the Molecular Structure of Sulfonyl Chloride Isocyanate Using Microwave Spectroscopy. J. Mol. Spectrosc. 1992, 152, 261–273. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Hajnal, M.R.; Vanquickenborne, L.G.; Ha, T.K.; Stohner, J. Structure and conformation of chlorosulfonyl isocyanate and cyclopropyl isocyanate. J. Chem. Soc. Faraday Trans. 1993, 2381–2384. [Google Scholar] [CrossRef]
- Alvarez, R. M. S.; Cutin, E. H.; Della Vedova, C. O. Conformational and bond properties of chlorosulfonyl isocyanate, ClSO2NCO. Spectrochim. Acta 1995, A 51, 555–561. [Google Scholar] [CrossRef]
- Guirgis, G. A.; Zhou, L.; Gounev, T. K.; Durig, J. R. Vibrational spectra, conformational stability and ab initio calculations of fluorosulfonyl isocyanate. Vib. Spectrosc. 1996, 12, 177–188. [Google Scholar] [CrossRef]
- Durig, J. R.; Zhou, L.; Gounev, T. K.; Guirgis, G. A. Vibrational spectra, conformational stability and ab initio calculations of chlorosulfonyl isocyanate. Spectrochim. Acta 1997, A 53, 1581–1593. [Google Scholar] [CrossRef]
- Lysek, R.; Kaluza, Z.; Furman, B.; Chmielewski, M. [2+2] Cycloaddition of chlorosulfonyl isocyanate to (Z)-3-O-(2’-silvylvinyl) ethers of 1,2-O-isopropylidene-5-O-trityl-α-D-xylo-furanose. Tetrahedron 1998, 54, 14065–14080. [Google Scholar] [CrossRef]
- Furman, B.; Kaluza, Z.; Lysek, R.; Chmielewski, M. [2+2] cycloaddition of chlorosulfonyl isocyanate to chiral vinyl ethers. Polish J. Chem. 1999, 73, 43–54. [Google Scholar]
- Isaka, M.; Williard, P. G.; Nakamura, E. [3+2] Cycloaddition of Allylic Silane with N-Chloro-sulfonyl Isocyanate. Bull. Chem. Soc. Jpn. 1999, 72, 2115–2116. [Google Scholar] [CrossRef]
- Furman, B.; Kaluza, Z.; Chmielewski, M. Stereochemical course of [2+2] cycloaddition of chlorosulfonyl isocyanate to cis and trans 3-O-but-1’-enyl-1,2-O-isopropylidene-α-D-xylo-furanose. Tetrahedron 1996, 52, 6019–6024. [Google Scholar] [CrossRef]
- Turnbull, K.; Gross, K. C.; Hall, T. A. Direct carboxamidation of sydnones with chlorosulfonyl isocyanate. Synth. Commun. 1998, 28, 931–937. [Google Scholar] [CrossRef]
- Kumar, E. S.; Dhar, D. N. Reaction of styryl cyclopropyl ketones with chlorosulfonyl isocyanate: formation of iminosulfonyl chloride. Ind. J. Chem. 1995, 34B, 63–64. [Google Scholar]
- Badawi, H. M.; Föِrner, W.; Al-Ghamdi, K. S. Structural Stability and Vibrational Assignments of Halosulfonylazides. J. Mol. Struct. (Theochem) 2003, 624, 225–232. [Google Scholar] [CrossRef]
- Skopenko, V. V.; Tsintsadze, G. V.; Ivanova, E. I. Metal selenocyanates. Russ. Chem. Rev. 1982, 51, 40–56. [Google Scholar] [CrossRef]
- Mark, S. D.; Qiao, Y. L.; Dawsey, S. M.; Wu, Y. P.; Katki, H.; Gunter, E. W.; Fraumeni, J. F., Jr; Blot, W. J.; Dong, Z. W.; Taylor, P. R. R. Prospective Study of Serum Selenium Levels and Incident Esophageal and Gastric Cancers. J. Natl. Cancer Inst. 2000, 92, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T. Organoselenchemie in der stereoselektiven Synthese. Angew. Chem. 2000, 112, 3890–3900. [Google Scholar] [CrossRef]
- Arner, E. S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Jiang, W.; Ganther, H. E.; Ip, C.; Thompson, H. J. In vitro effects of Se-allyl-selenocysteine and Se-propylselenocysteine on cell growth, DNA integrity, and apoptosis. Pharmacology 2000, 60, 1467–1473. [Google Scholar]
- Finley, J. W.; Davis, C. D.; Feng, Y. Selenium from High Selenium Broccoli Protects Rats from Colon Cancer. J. Nutrit. 2000, 130, 2384–2389. [Google Scholar]
- Rayman, M. P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Hassner, A.; Stern, M. Synthesis of Alkylazides with a Polymeric Reagent. Angew. Chem. Int. Ed. 1986, 25, 478–479. [Google Scholar] [CrossRef]
- Yu, K. L.; Johnson, R. L. Synthesis and Chemical Properties of Tetrazole Peptide Analogues. J. Org. Chem 1987, 52, 2051–2059. [Google Scholar] [CrossRef]
- Scriven, E. F. V.; Turnbull, K. Azides: Their Preparation and Synthetic Uses. Chem. Rev. 1988, 88, 297–362. [Google Scholar] [CrossRef]
- Lowe-Ma, C. K.; Nissan, R. A.; Wilson, W. S. Tetrazolo[1,5-a]pyridines and Furazano[4,5-b]pyridine 1-Oxides . J. Org. Chem. 1990, 55, 3755–3761. [Google Scholar] [CrossRef]
- Kamiya, N.; Shiro, Y.; Iwata, T.; Iizulka, T.; Iwasaki, H. Heme Environmental Structure of a Novel Artificial Myoglobin with Closed Heme Pocket: Site-Specific Chemical Modi-fication Producing Distal N-Tetrazolylhistidine E7 by Cyanogen Bromide and Azide Ion. J. Amer. Chem. Soc. 1991, 113, 1826–1829. [Google Scholar] [CrossRef]
- Aube, J.; Milligan, G. L. Intramolecular Schmidt Reaction of Alkyl Azides. J. Amer. Chem. Soc. 1991, 113, 8965–8966. [Google Scholar] [CrossRef]
- Norris, P.; Horton, D.; Levine, B. R. Synthesis of 1,5-Dideoxy-1,5-Imino-D-Xylonolactam via Acid-Catalyzed Intramolecular Schmidt Rearrangement. Tetrahedron Lett 1995, 36, 7811–7841. [Google Scholar] [CrossRef]
- Milligan, G. L.; Mossman, C. J.; Aube, J. Intramolecular Schmidt Reactions of Alkyl Azides with Ketones: Scope and Stereochemical Studies. J. Amer. Chem. Soc. 1995, 117, 10449–10459. [Google Scholar] [CrossRef]
- Badawi, H. M.; Fِörner, W.; Al-Saadi, A. An Investigation of Structural Stability and Analysis of Vibrational Spectra of Formyl Ketene Based on Ab Initio Calculations. J. Mol. Struct. (Theochem) 2000, 505, 19–30. [Google Scholar] [CrossRef]
- Haist, R.; Mack, H. G.; Vedova, C. O.; Cutin, E. H.; Oberhammer, H. Structures and conformational properties of trifluoromethylsulfonyl isocyanate, CF3SO2NCO, and trifluoromethyl-sulfonyl azide, CF3SO2N3. J. Mol. Struct. (Theochem) 1998, 445, 197–205. [Google Scholar] [CrossRef]
- Dyke, J. M.; Groves, A. P.; Morris, A.; Ogden, J. S.; Catarino, M. I.; Dias, A. A.; Oliveira, A. M. S.; Costa, M. L.; Barros, M. T.; Cabral, M. H.; Moutinho, A. M. C. A Study of the Thermal Decomposition of Azidoacetone by Photoelectron and Matrix Isolation Spectroscopy. J. Phys. Chem. 1999, A 103, 8239–8245. [Google Scholar] [CrossRef]
- Rauhut, G.; Eckert, F. A Computational Study on the Mechanism and Kinetics of the Pyrolysis of 2-Nitrophenyl Azide. J. Phys. Chem. 1999, A 103, 9086–9092. [Google Scholar] [CrossRef]
- McIntosch, M. B.; Hartle, T. J.; Allcock, H. R. Synthesis and Reactivity of Alkoxy, Aryloxy, and Dialkylamino Phosphazene Azides. J. Amer. Chem. Soc. 1999, 121, 884–885. [Google Scholar] [CrossRef]
- McClelland, R. A.; Ahmed, A.; Dicks, A. P.; Licence, V. E. Spectroscopic Characterization of the Initial C8 Intermediate in the Reaction of the 2-Fluoronitrenium Ion with 2’-Deoxygunaosine. J. Amer. Chem. Soc. 1999, 121, 3303–3310. [Google Scholar] [CrossRef]
- Chahonua, L.; Cai, H.; Fishbein, J. C. Cyclic α-Acetoxynitrosamines: Mechanisms of Decomposition and Stability of α-Hydroxynitrosamine and Nitrosiminium Ion Reactive Intermediates. J. Amer. Chem. Soc. 1999, 121, 5161–5169. [Google Scholar] [CrossRef]
- Varotsis, C.; Vamvouka, M. Resonance Raman and Fourier Transform Infrared Detection of Azide Binding to the Binuclear Center of Cytochrome bo3 Oxidase from Escherichia coli. J. Phys. Chem. 1999, B 103, 3942–3946. [Google Scholar] [CrossRef]
- Badawi, H. M. Structural stability, vibrational assignments and C-N rotational barrier in vinyl azide. J. Mol. Struct. (Theochem) 2002, 579, 11–19. [Google Scholar] [CrossRef]
- Badawi, H. M. Theoretical study of the structure and vibrational spectra of formyl and methyl azides. J. Mol. Struct. (Theochem) 2002, 583, 89–97. [Google Scholar] [CrossRef]
- Frisch, J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. R.; Cheeseman, J. R.; Za- krzewski, V. G.; Montgomery, J. A., Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Frakas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. T.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.; Chen, W.; Wong, W.; Andres, J. L.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98; Gaussian Inc.: Pitts- burgh PA, 1998. [Google Scholar]
- Wilson, E. B.; Decius, J. C.; Cross, P. C. Molecular vibrations; McGraw-Hill: New York, 1995. [Google Scholar]
- Förner, W.; Badawi, H. M. Theoretical Vibrational Spectra of Cyclohexanecarboxaldehyde. J. Mol. Model. 2001, 7, 288–305. [Google Scholar]
- Chantry, G. W. Polarizability Theory of the Raman Effect. In The Raman Effect; Anderson, A., Ed.; Marcel Dekker: New York, 1997; Vol. 1, Chapter 2; pp. 49–94. [Google Scholar]
© 2005 by MDPI (http://www.mdpi.org).
Share and Cite
Förner, W.; Badawi, H.M.; Seddigi, Z.S. Structural Stability and Vibrational Analyses of Haloselenonyl Azides, XSeO2-NNN, where X is F, Cl, Br. Int. J. Mol. Sci. 2005, 6, 230-244. https://doi.org/10.3390/i6090230
Förner W, Badawi HM, Seddigi ZS. Structural Stability and Vibrational Analyses of Haloselenonyl Azides, XSeO2-NNN, where X is F, Cl, Br. International Journal of Molecular Sciences. 2005; 6(9):230-244. https://doi.org/10.3390/i6090230
Chicago/Turabian StyleFörner, Wolfgang, Hassan M. Badawi, and Zaki S. Seddigi. 2005. "Structural Stability and Vibrational Analyses of Haloselenonyl Azides, XSeO2-NNN, where X is F, Cl, Br" International Journal of Molecular Sciences 6, no. 9: 230-244. https://doi.org/10.3390/i6090230
APA StyleFörner, W., Badawi, H. M., & Seddigi, Z. S. (2005). Structural Stability and Vibrational Analyses of Haloselenonyl Azides, XSeO2-NNN, where X is F, Cl, Br. International Journal of Molecular Sciences, 6(9), 230-244. https://doi.org/10.3390/i6090230