Reaction of SO2 with graphite and charcoal in the presence of salts
The reactivity of graphite in the presence of sodium, potassium and calcium salts increased by a factor of 2-5 and the maximum effect was observed for K
2S (
Table 2).
Table 2.
Effect of salts on the reaction of graphite with SO2 at 900 oC a
Table 2.
Effect of salts on the reaction of graphite with SO2 at 900 oC a
| Salt | 109 Ro, mol m-2·s-1·atm-1 | Molar fraction, %b |
|---|
| CO2 | S2 |
|---|
| None | 3.16 | 68.5±1.5 | 31.2±2.1 |
| NaNO3 | 6.79 | 67.2±1.4 | 32.6±1.2 |
| Na2S | 6.38 | 67.9±2.3 | 31.7±2.0 |
| KNO3 | 5.45 | 70.3±3.2 | 29.4±4.5 |
| K2S | 17.73 | 66.5±2.8 | 30.8±3.2 |
| Ca(NO3)2 | 8.46 | 66.5±2.3 | 33.3±2.5 |
| CaS | 11.49 | 66.3±2.0 | 33.6±2.6 |
The average distribution of products, calculated during the steady state period, showed that for the non-catalyzed and catalyzed reaction, the CO
2:S
2 ratio of the products was near two. The molar fraction of the products did not change in the presence of salts. The secondary products CO, COS and CS
2 were ca. 1% or less, suggesting that the catalysis did not involved the secondary reactions that were consecutive to the main reaction [
12,
13].
The effect of the addition of salts on charcoal was lesser than it was for graphite. The rate increased only in the range of 30-60 % (
Table 3). The same reactivity was observed with respect to the carbon conversion for both nitrates and sulfides, increasing slightly in the order Na
+ < K
+ < Ca
2+. This absence of effect of the anion on the catalytic activity was also observed in the reaction of charcoal with sulfur [
4]. Again, the presence of salts did not change the distribution of products. More than 90% of the molar fraction corresponded to the main products CO
2 and S
x.
Table 3.
Effect of salts on the reaction of charcoal with SO2 at 650 oCa
Table 3.
Effect of salts on the reaction of charcoal with SO2 at 650 oCa
| Salt | 108 Ro, mol m-2·s-1·atm-1 | Molar Fraction, %b |
|---|
| CO2 | S2 | COS | CO |
|---|
| None | 4.25 | 62.6±0.5 | 30.5±0.4 | 3.4±0.2 | 2.9±0.5 |
| NaNO3 | 5.39 | 64.2±0.6 | 31.9±0.4 | 2.4±0.3 | 1.6±0.6 |
| Na2S | 5.39 | 65.0±0.2 | 32.6±0.3 | 1.0±0.1 | 1.4±0.2 |
| KNO3 | 6.21 | 64.5±1.3 | 32.1±0.4 | 1.9±1.0 | 1.3±0.5 |
| K2S | 5.88 | 63.3±0.2 | 31.5±0.2 | 2.6±0.2 | 2.6±0.3 |
| Ca(NO3)2 | 6.86 | 62.4±1.0 | 31.7±0.2 | 2.0±0.2 | 4.0±1.2 |
| CaS | 6.53 | 62.8±1.3 | 31.7±0.4 | 2.1±0.5 | 3.5±1.5 |
From thermodynamic analysis [
20] and experimental data [
21], it was concluded that the formation of CaS from CaO and sulfur-containing gases would occur in the presence of the highly reductive carbon. The calcium sulfide formed might produce a polysulfide again, in contact with SO
2 or the intermediates of the reduction. Therefore, the role of the catalyst would be to increase the transport of S
x from the intermediates, similar to other reactions of carbon, where alkaline and alkaline-earth salts are also effective catalysts. This mechanism will be analyzed in detail below.
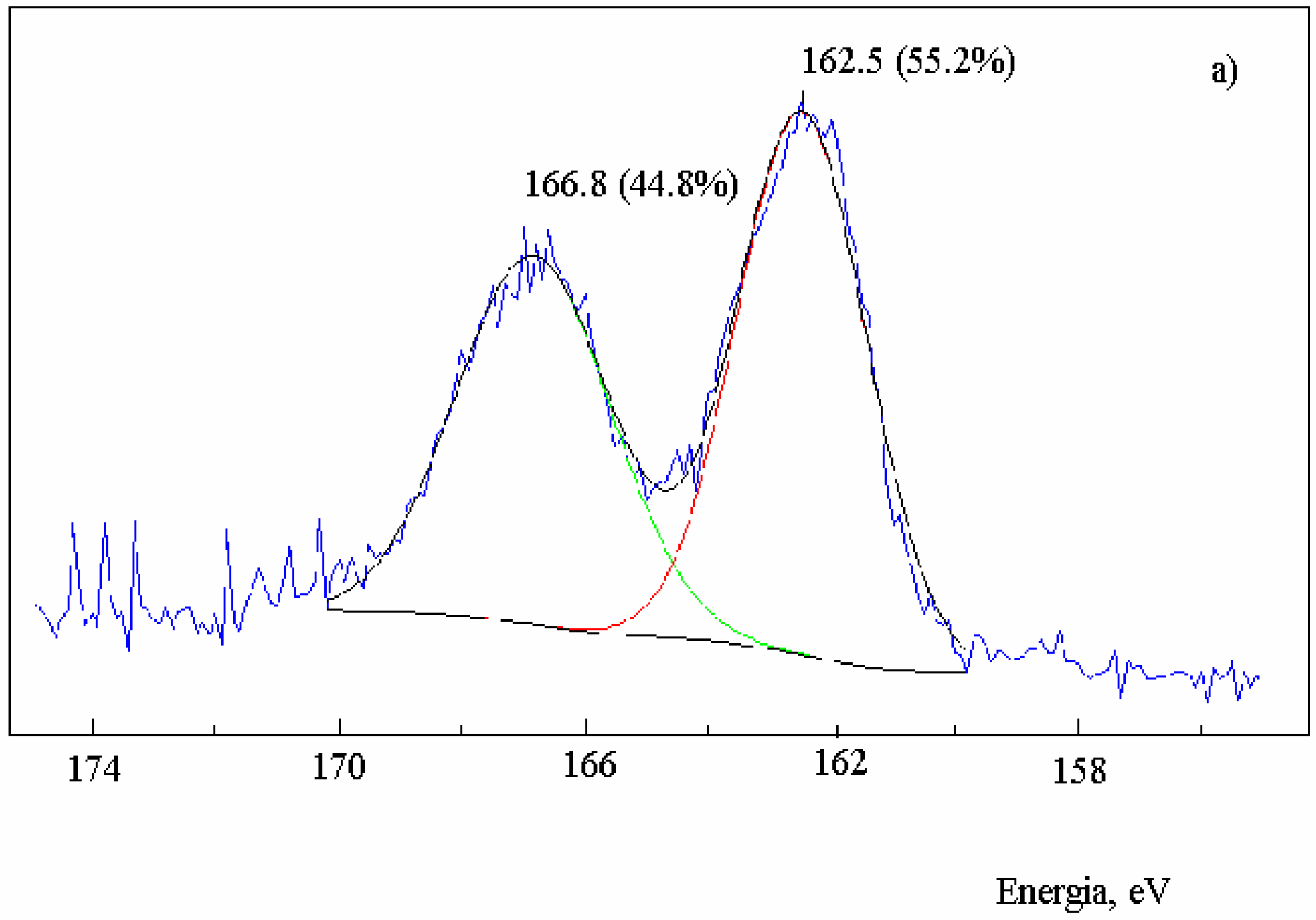
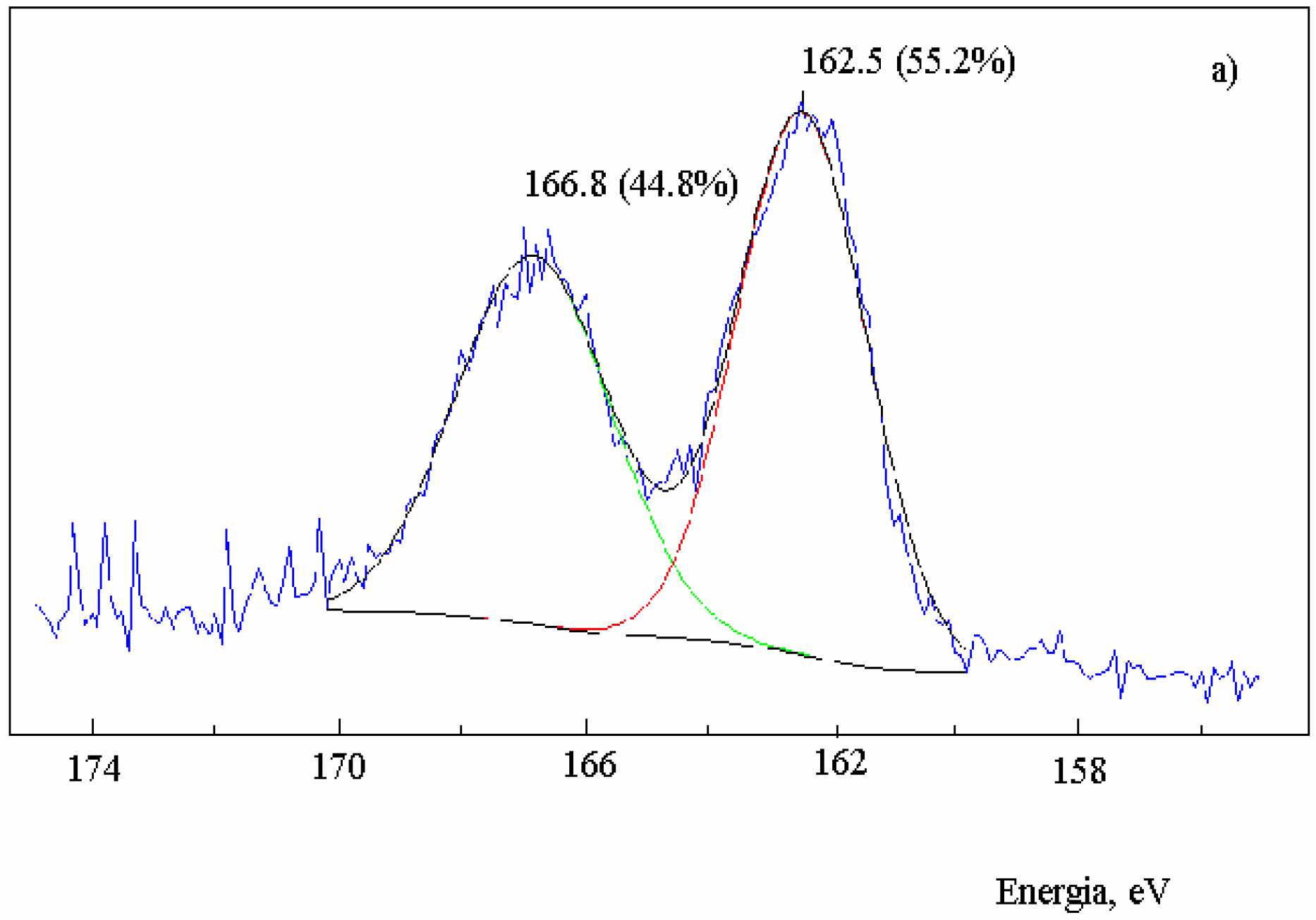
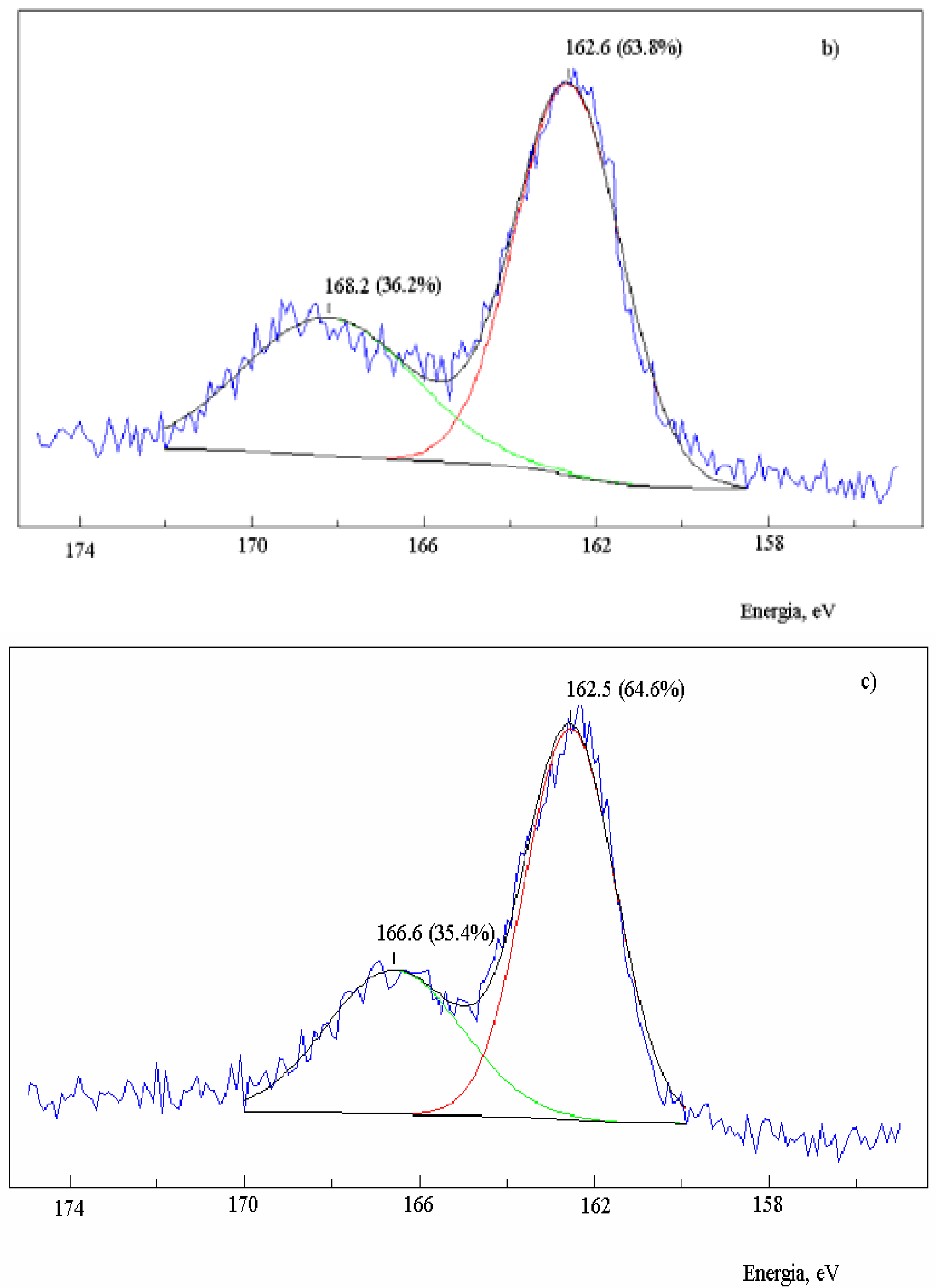
Figure 2.
XPS spectra in the S2p region of activated carbon after the reaction with SO2 at 630 oC and extraction with CS2. Sample: 2.0 g; salt 1.2x10-3 mol of metal; total flow 95 mL·min-1; PSO2 0.2 atm; a) no salt added; b) in the presence of NaNO3; c) in the presence of Na2S.
Figure 2.
XPS spectra in the S2p region of activated carbon after the reaction with SO2 at 630 oC and extraction with CS2. Sample: 2.0 g; salt 1.2x10-3 mol of metal; total flow 95 mL·min-1; PSO2 0.2 atm; a) no salt added; b) in the presence of NaNO3; c) in the presence of Na2S.
In the absence of salts.
The reduction of SO
2 on activated carbon in the absence of salt showed that the sulfur content of the carbon increased until a constant value when the reaction reached the steady-state condition. The XPS spectra in the S
2p region of the residual carbon after the reaction with SO
2 (Fig. 2a) showed that the band at 162.5 eV assigned to a non-oxidized sulfur C(S) had 55.2% weight of the total sulfur and the band at 166.8 eV related to an oxidized sulfur bond C(SO) had 44.8% weight [
13]. Therefore, the ratio C(S): C(SO) was 1.2.
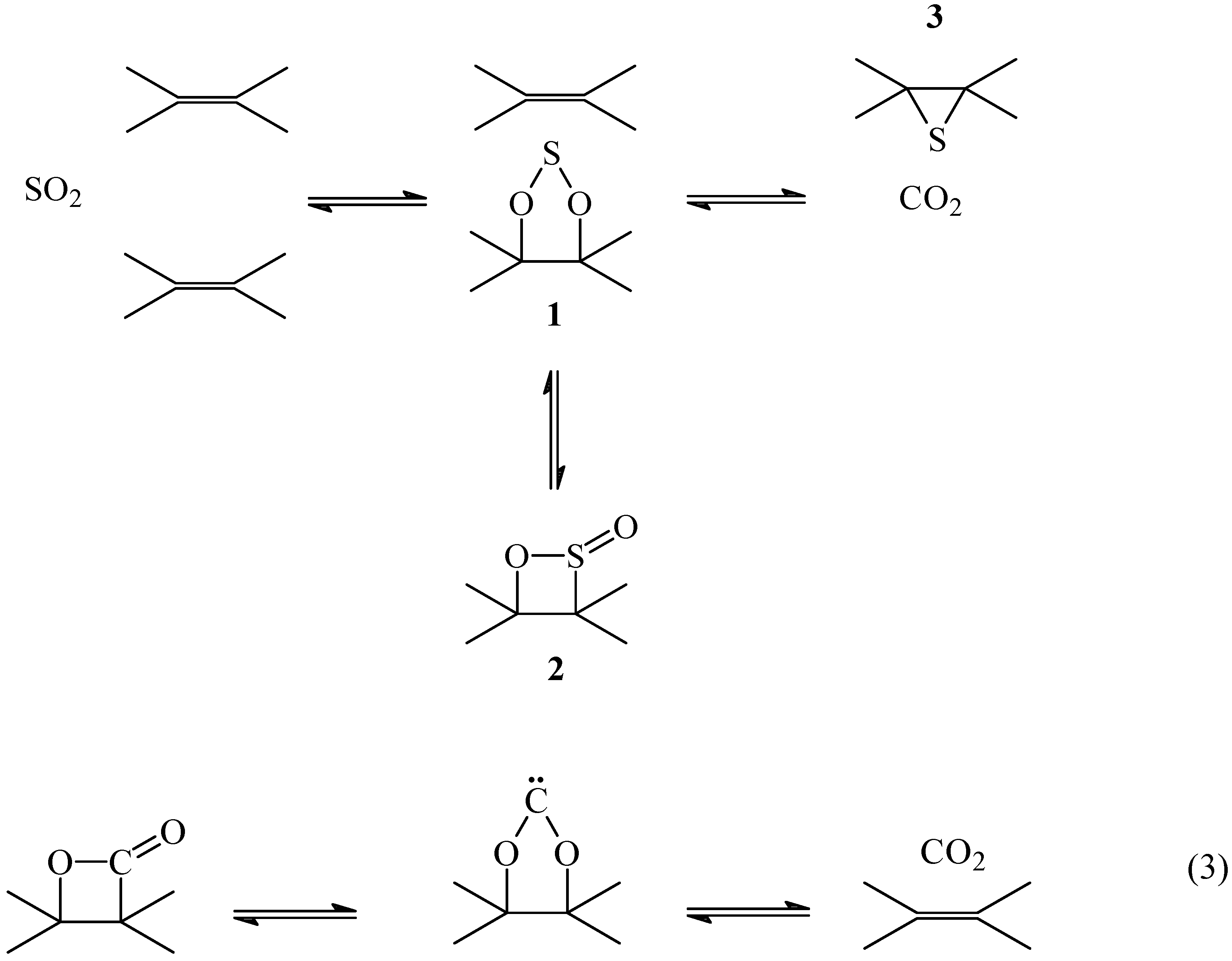
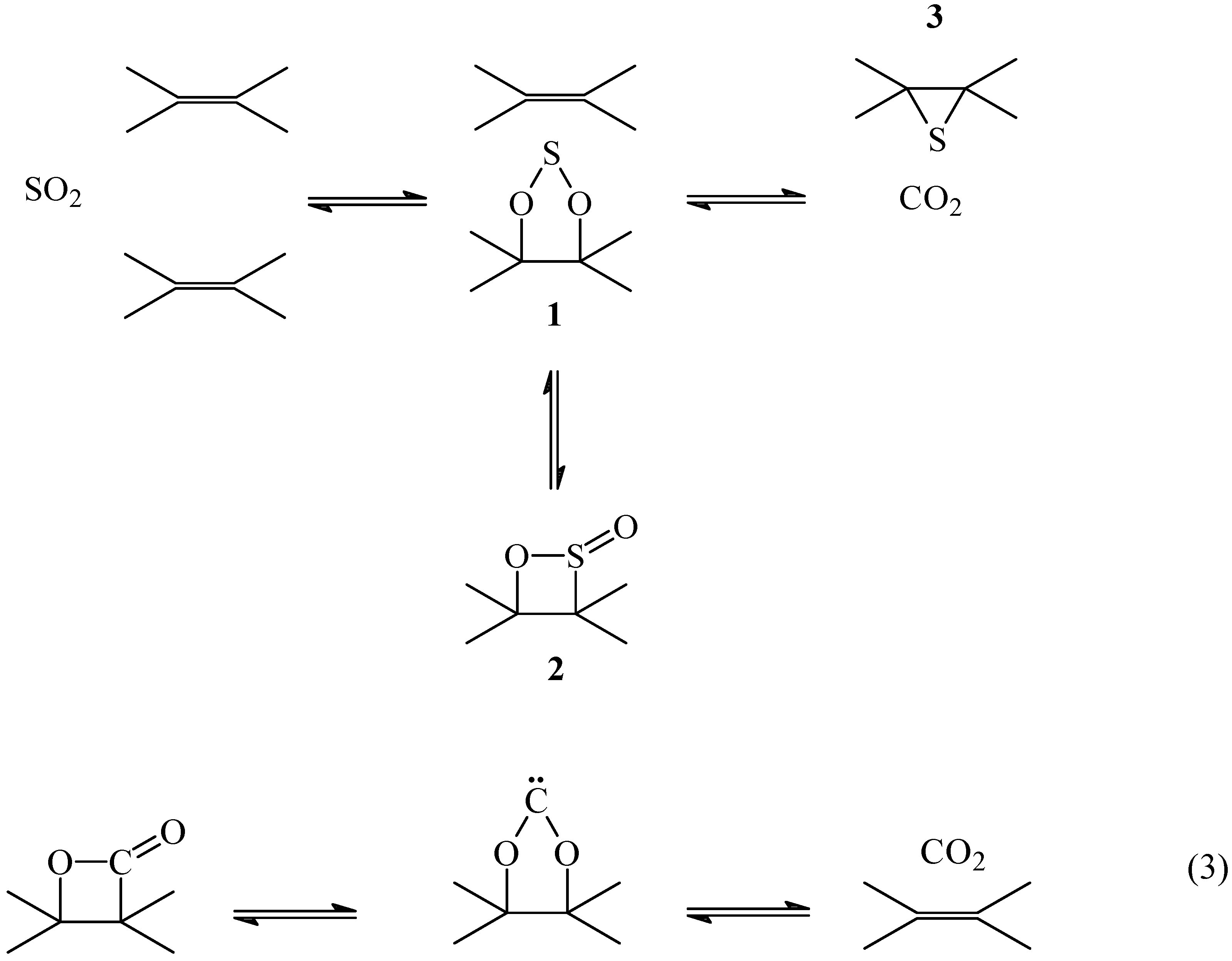
A detailed interpretation of the mechanism of the primary reaction C + SO
2 is shown in
Scheme 1. The reaction of SO
2 with a double bond would form a 1, 3, 2-dioxathiolane
1 and/or a 1,2-oxathietene 2-oxide,
2. This is the reverse reaction of the thermal extrusion of SO
2 from
1 [
22] or
2 [
23] that has been proposed to occur by a concerted path to form a double bond. The reaction is similar to the thermal decomposition of β-lactones to produce CO
2 and an olefin through a carbene (eq 3) [
24].
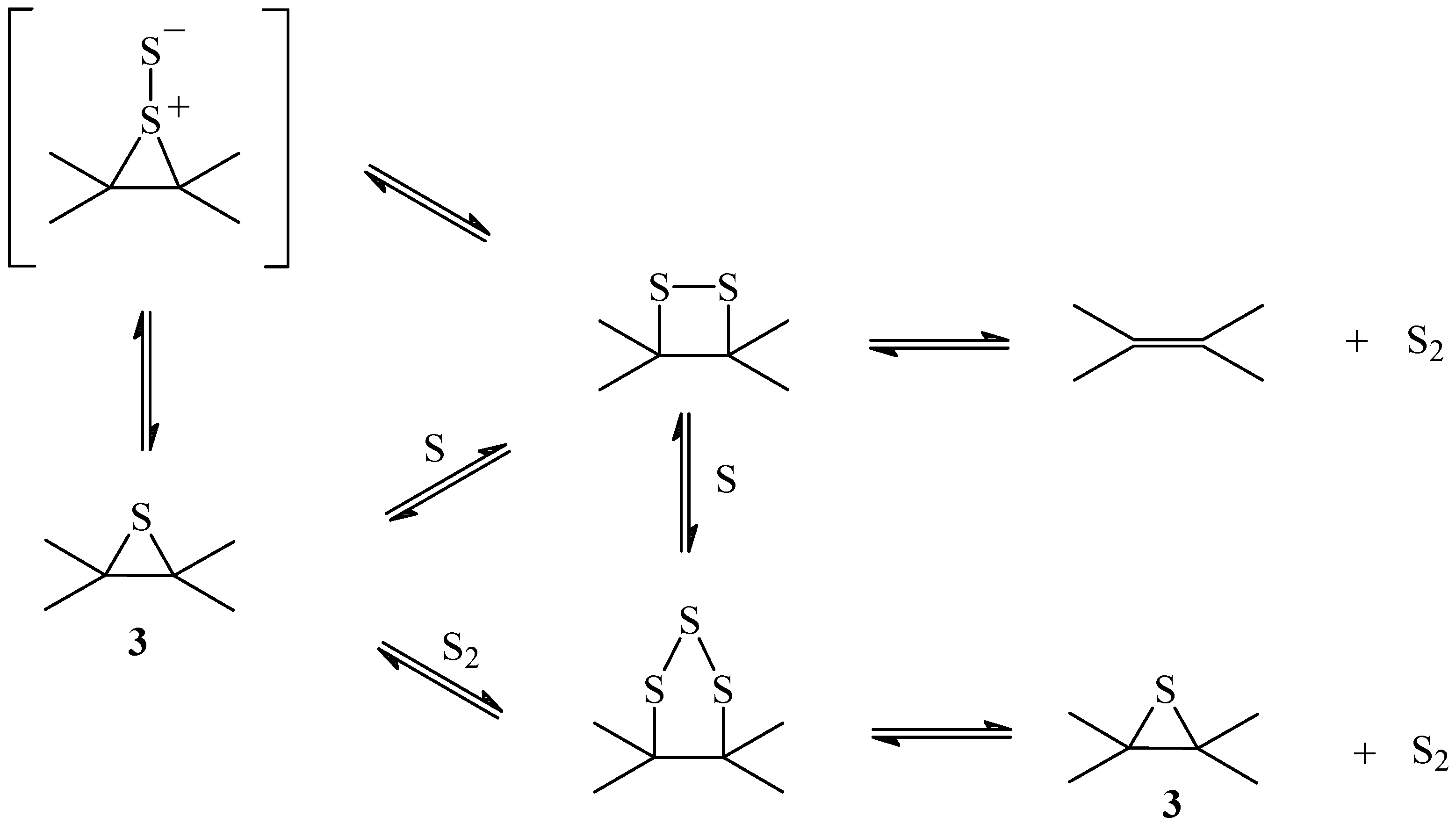
Scheme 1.
Mechanism of the primary reaction
Scheme 1.
Mechanism of the primary reaction
The oxidized intermediates
1 and/or
2 decompose to produce CO
2 and atomic sulfur. According to this mechanism the non-oxidized sulfur intermediate is an episulfide
3 formed from the reaction of the atomic sulfur with the nearest double bond. It is known that atomic sulfur reacts with olefins producing episulfides and mercaptans [
25,
26].
The formation of the episulfide
3 is probably concerted with the formation of CO
2. However, the mechanism of formation of CO
2 is not known and in
Scheme 1 the fragment if the residual carbon matrix was not included. The fact that the reaction is reversible [
13] imposes the condition that the sulfur should be at the proper place to react with CO
2 to re-form SO
2. In fact, the reversibility of the reaction imposes the condition that the atomic sulfur formed from the oxidized intermediate C(SO) should not go freely into the gas phase, but should remain as the intermediate on the carbon matrix in order to regenerate SO
2 from CO
2. For this reason, monoatomic sulfur extrusion from the episulfide should not be considered [
27].
Since sulfur does not accumulate [
13] and episulfides inserted in a carbon matrix are very unreactive and thermally stable [
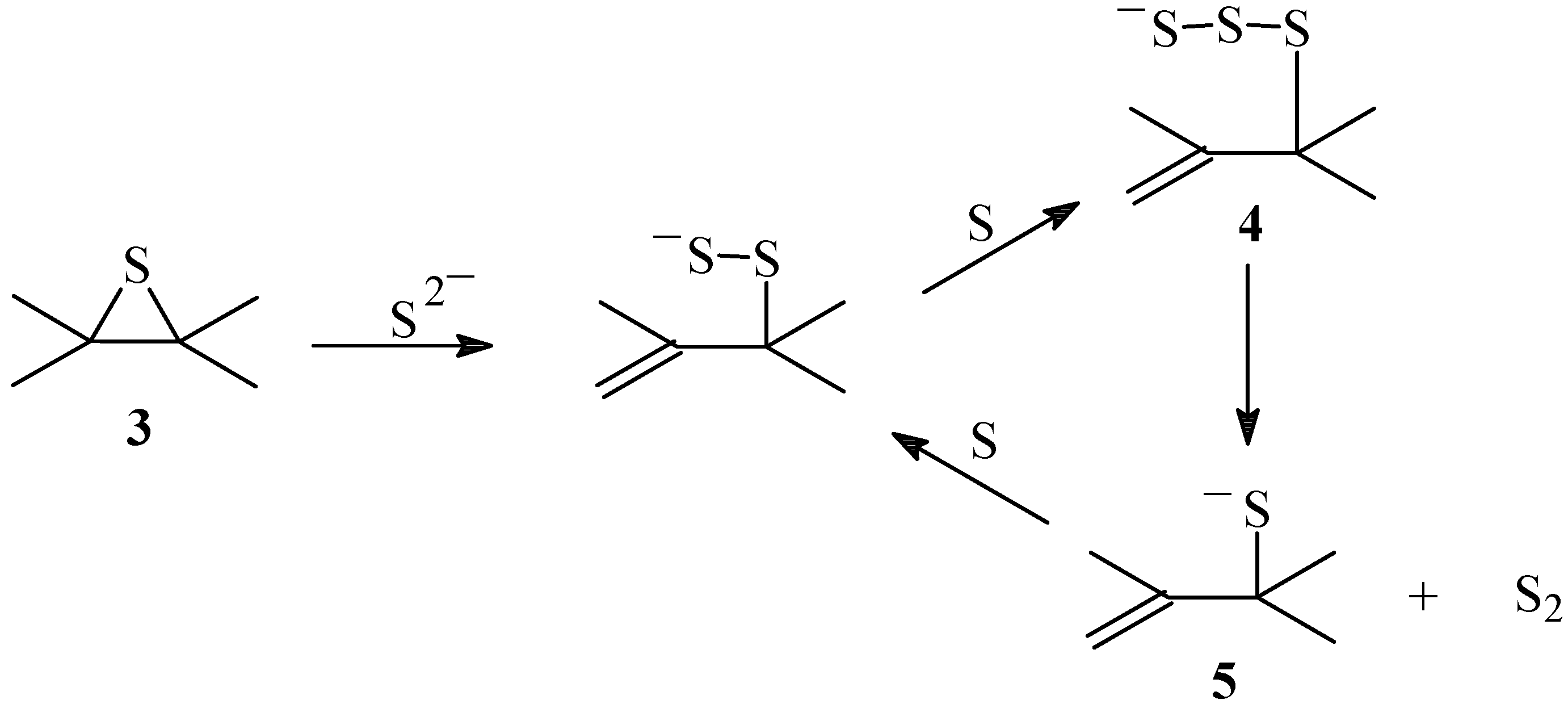
4], this reaction, that should occur during the pre-steady-state period, must be followed by consecutive reactions of insertion of atomic sulfur to form intermediate(s) able to liberate free sulfur when the steady-state has been reached (
Scheme 2).
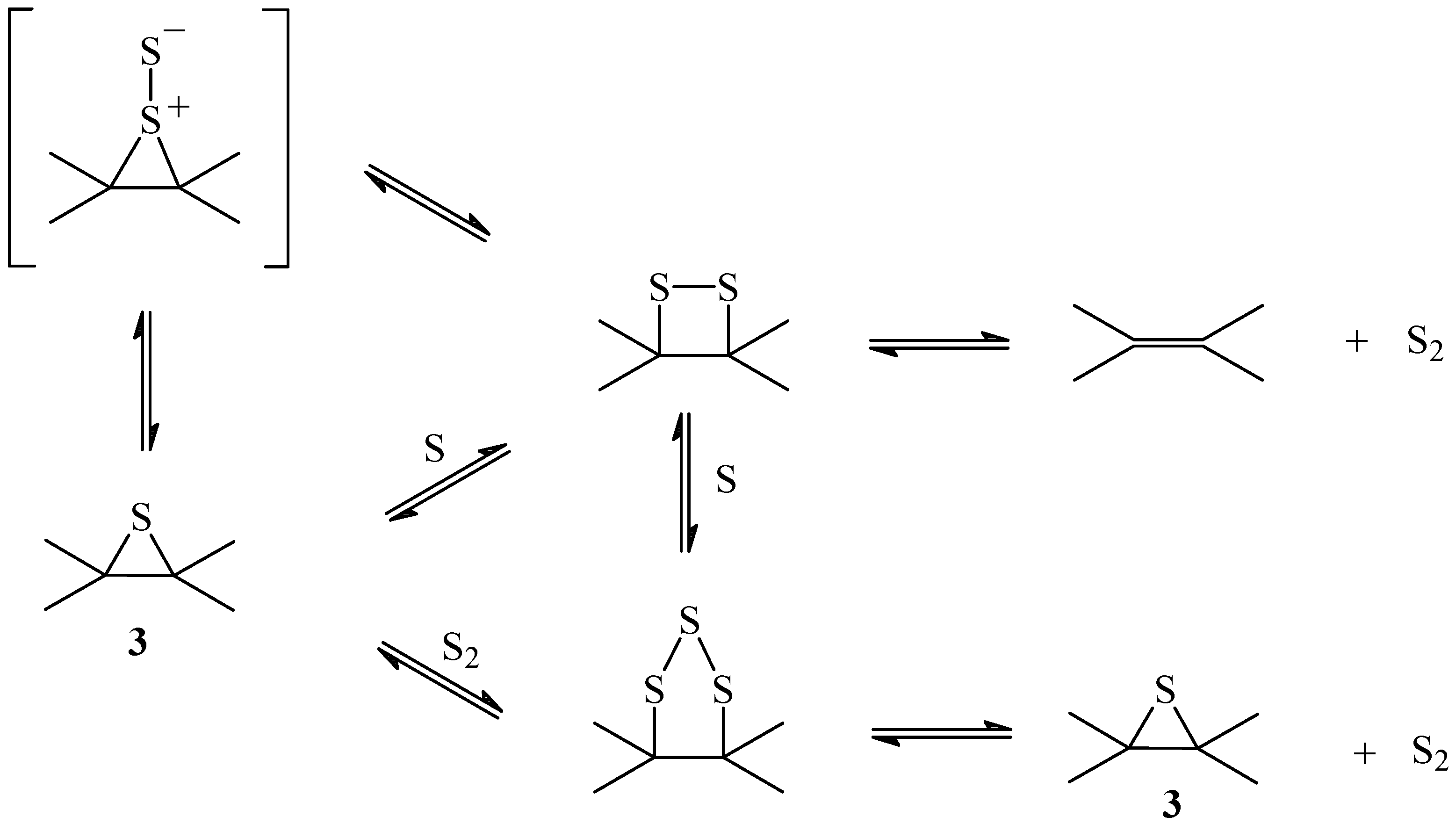
Scheme 2.
Mechanism of the sulfur transport
Scheme 2.
Mechanism of the sulfur transport
The insertion of a second sulfur atom in the episulfide produces a disulfide that can either decompose, to produce a diatomic sulfur S
2 and the original double bond, or accept a third atom forming a trisulfide. Anthracene endodisulfide decomposes to form anthracene and singlet diatomic sulfur
1S
2 [
28]. Singlet diatomic sulfur is very reactive and can react with dienes to form episulfides and disulfides.
Formation of episulfides can be understood if
1S
2 reacts with the olefin to form a thiosulfoxide intermediate followed by desulfurization. The reverse reaction would produce a disulfide from the episulfide through the thiosulfoxide intermediate (
Scheme 2). However, transport of sulfur through the disulfide to release S
2 would render the reaction irreversible and could only occur after the concentration of episulfide had reached the steady state. Formation of a trisulfide from the disulfide or the episulfide allows the extrusion of a more stable form of sulfur such as
1S
2 [
29,
30,
31,
32] that would regenerate the sulfide, establishing an equilibrium sulfide-disulfide-trisulfide that would function as a capture-release cycle of sulfur. Trisulfides can result from the insertion of S
2 into a previously formed disulfide, followed by the extrusion of sulfur [
31].
Releasing sulfur from the trisulfide as S
2 is reasonable because at the temperature of the reaction the main species is S
2. At the boiling point, gaseous sulfur consists mainly of species S
8 and S
6, which dissociate into S
2 when the temperature is raised. At 730
oC and 1 torr, S
2 is almost 99% pure and dissociation to monoatomic sulfur requires a temperature above 1500
oC [
33,
34].
For the dioxathiolane intermediate
1 to accumulate, according to
Scheme 1, the rate determining step should be the decomposition of the episulfide
3. The ratio non-oxidized sulfur C(S): oxidized sulfur C(SO) should be one. The XPS spectrum (
Figure 2a) showed that the ratio is 1.2, indicating that the most stable intermediate is the episulfide and that the formation of S
2 from the disulfide is not important (
Scheme 2).
In the presence of salts
Since the mechanism of reduction of SO
2 on different carbons proved to be the same with respect to the stoichiometry of the reaction and the formation of secondary products [
12], a more detailed study of the catalysis by salts on activated carbon can provide some important pieces of evidence on the mechanism of the catalytic pathway. It is considered that the addition of salts on a carbon surface forms species that act as catalytic active sites of the reaction [
35,
36] and whose concentration depends on the amount of salt deposited.
Nitrates of sodium, potassium or calcium melt and decompose during the thermal pre-treatment [
37] producing the corresponding oxide that could be reduced to metallic form in the presence of carbon, and, upon the reaction of the carbon with SO
2, could generate sulfides which are thermodynamically more stable. At the temperature range used in this work for the reduction reaction, they remained as solids.
Activated carbon impregnated with sulfides and polysulfides of sodium and potassium increased the reactivity by about four times, as shown in
Table 4. No difference between Na
+ and K
+ salts was observed, and mono and polysulfides showed the same reactivity, independent of the number of sulfur atoms in the polysulfide. Potassium polysulfides K
2S
n (n = 2-4) decompose at the experimental temperature, and the fact that the rate of carbon conversion R
o was the same for the series of potassium polysulfides indicates that they decomposed to formed the same catalytically-active species.
Table 4.
Effect of sulfides and polysulfides on the reaction of activated carbon with SO2 at 700 oC a
Table 4.
Effect of sulfides and polysulfides on the reaction of activated carbon with SO2 at 700 oC a
| Salt | None | NaNO3 | Na2S | K2Sn, n = 1-4 |
|---|
| 109 Ro, mol m-2.s-1.atm-1 | 1.08 | 3.80 | 4.34 | 4.07 |
| 103 mol S/g carbon | 2.2 | 6.2b | 5.4b | |
Sodium nitrate also decomposed during the pre-treatment at 900 oC, producing Na2O and NOx along with a small amount of CO2 from the carbon oxidation. No nitrogen was observed in the XPS spectrum, indicating a total decomposition of the nitrate.
Upon reaction with SO
2, the sulfur content of the activated carbon increased in the presence of NaNO
3 and Na
2S to a constant value of 2.2 x 10
-3 mol S/g carbon, while the sodium content after the reaction with SO
2 did not change (
Table 4). Considering the sulfur content due to the non-catalyzed reaction, the sulfur excess in the presence of salt with respect to sodium was in the ratio Na : S = 3.0 ± 0.3 (
Table 4). A sodium polysulfide Na
2S
6 with such a ratio would be very unlikely because hexasulfides are very unstable at 700
oC [
38].
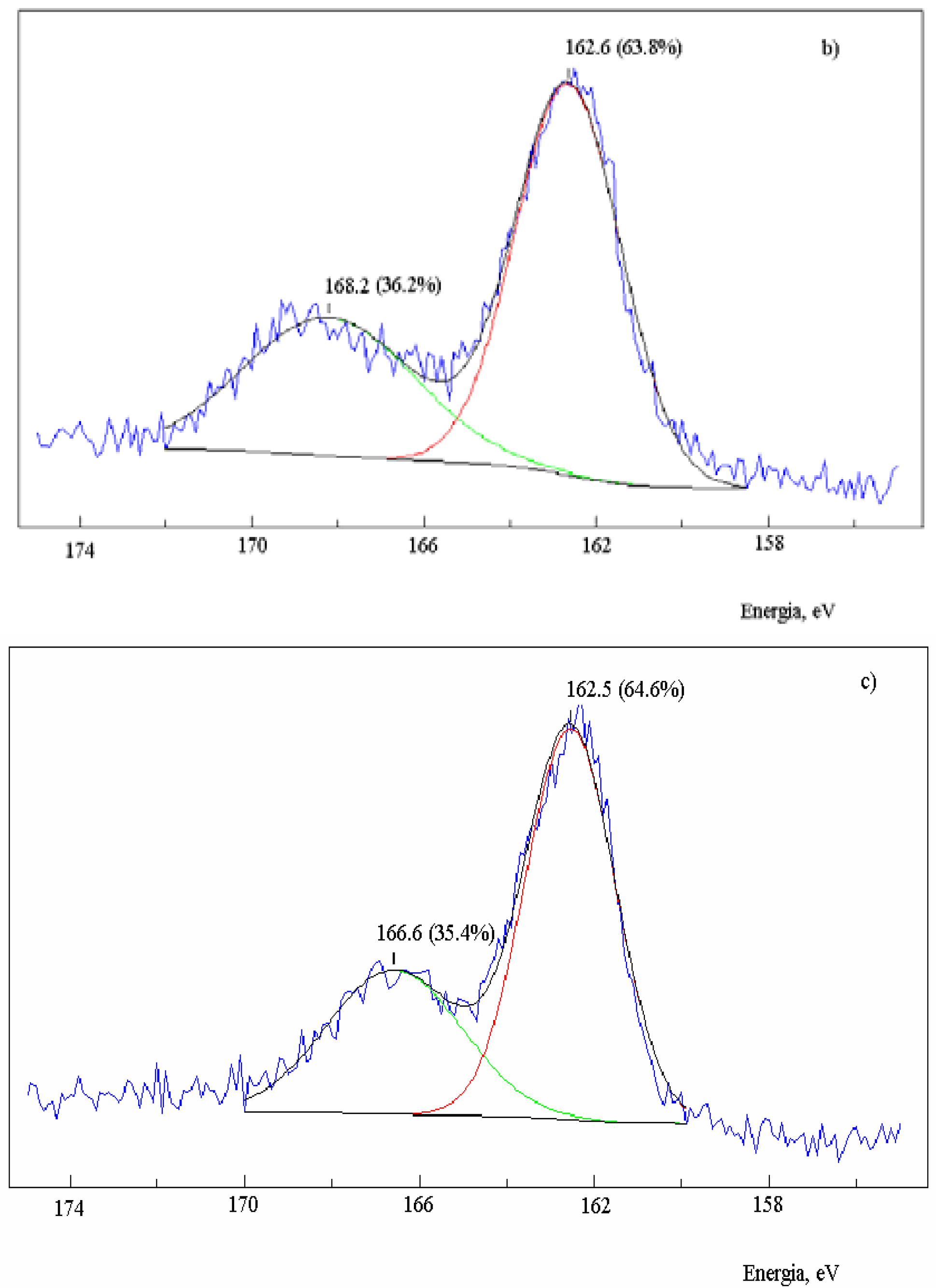
The residual carbon impregnated with sodium nitrate produced an XPS spectrum (
Figure 2b) with a band of oxidized sulfur centered at 168.2 eV (36.2%) [
39]. On the other hand, the band of non-oxidized sulfur presented the same energy as it did in the absence of salt, but increased to 63.8% with respect to the oxidized sulfur. The same result was observed for the residual carbon containing Na
2S (
Figure 2c), also with an increase of the non-oxidized sulfur band to 64.6%. Therefore, there was an increase of the chemically bound non-oxidized sulfur in the presence of salt, and the ratio C(S):C(SO) increased to 1.8.
These results are consistent with the assumption that once the episulfide
3 is formed (
Scheme 1), the sulfide anion from the salt produces a disulfide anion upon the reaction with
3 (
Scheme 3). Sulfur is known to react with even trace amounts of S
2- to give polysulfide ions [
40].
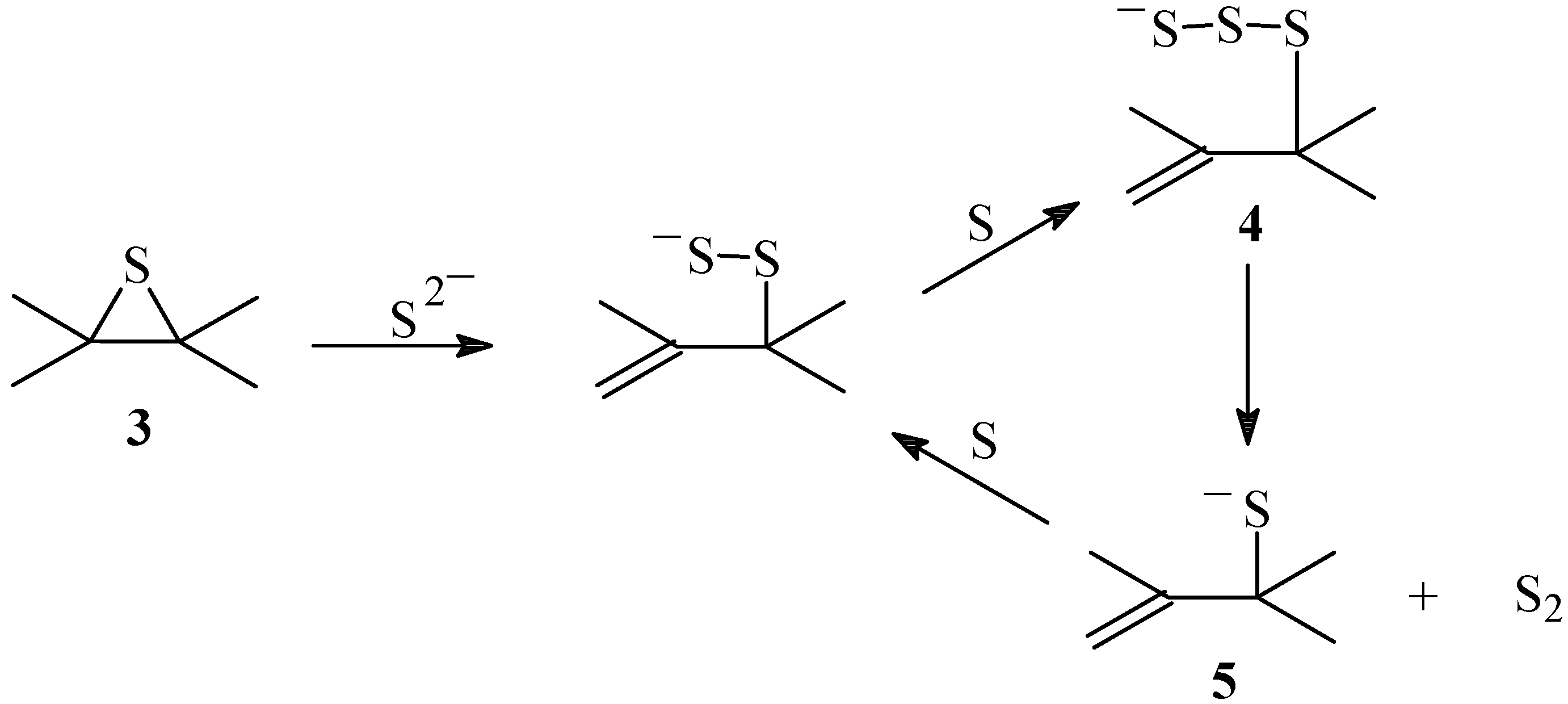
Scheme 3.
Mechanism of the sulfur transport catalyzed by salts
Scheme 3.
Mechanism of the sulfur transport catalyzed by salts
Reaction of the disulfide anion with the sulfur arising from the decomposition of the dioxathiolane intermediate
1 would form the trisulfide anion
4 that decomposes and releases singlet diatomic sulfur
1S
2, thus transporting the sulfur and generating the powerful nucleophile thiolate
5 that is part of the catalytic cycle [
41,
42]. A catalytic capture-release cycle of sulfur is then established which is similar to the non-catalyzed reaction. These results support the assumption that the transport of sulfur occurs through a trisulfide intermediate. The reaction of organometallic trisulfides in the presence of triphenylphosphine produced
1S
2 through a trisulfide intermediate that extruded S
2 to form a sulfide [
30,
32].
Finally, from the XPS spectra and sulfur content, it can be concluded that in the absence of salt the main component of the non-oxidized sulfur intermediate is the episulfide, and therefore its decomposition must be rate determining of the sulfur transport. In the presence of salt on the other hand, since the trisulfide is the main component, the sulfur transport being determined by the decomposition of the trisulfide and consequently the product distribution in the presence of salt does not change.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}