1 Introduction
Indirect nuclear magnetic resonance (NMR) spin-spin coupling constants (SSCCs)
nJ(A
, B) are sensitive to the geometrical features of a molecule and, therefore, their magnitude provides a direct insight into the geometry and electronic structure of a molecule [
1,
2,
3,
4,
5]. Most celebrated in this connection is the Karplus relationship, which relates in a simple trigonometric way vicinal SSCCs of the type
3J(HCCH) to the dihedral angle
τ(HCCH) [
6,
7,
8]. The Karplus relationship (often called Karplus equation) has been extended beyond the field of proton,proton coupling constants and is extensively used to determine dihedral angles and by this conformational features of molecules, in particular biomolecules, from measured SSCCs [
9,
10,
11].
In previous work, we described a generalization of the Karplus relationship in the case of pseu-dorotating ring molecules [
12]. In the case of a free (or nearly free) pseudorotor, it is difficult to establish a Karplus relationship because only average SSCCs < J > can be measured, which do not seem to provide any insight into conformational features of a molecule. In this situation, however, reliable quantum chemical calculations of individual constants J [
13,
14] in dependence of the pseudorotational mode of a ring can be used to compliment the limited information obtained from NMR experiments to derive a Karplus relationship for the ring molecule [
12]. Our investi-gation of cyclopentane led to a number of interesting conclusions concerning the description of pseudorotating molecules with the help of SSCCs [
12].
1) The establishment of a Karplus relationship using the dihedral angles of the ring is problematic because often one dihedral angle can be associated with two different SSCCs. However, a useful extension of the Karplus relationship to pseudorotating rings is achieved by expressing SSCC as a function of the puckering coordinates of the ring molecule.
2) For cyclopentane, SSCCs were calculated employing both CCSD theory [
15,
16,
17] and coupled perturbed DFT (CPDFT) [
13] with the B3LYP functional. Both approaches led to reasonable SSCCs. However, for a given basis set of moderate size CPDFT turned out to be superior in terms of both cost and accuracy [
12].
3) All SSCCs of cyclopentane could be expressed as simple analytical functions of the ring puck-ering coordinates of a five-membered ring, namely the pseudorotational phase angle φ and the puckering amplitude q. Average values < J > were calculated by integrating functions J(φ) over φ. Results were compared with measured < J > values.
4) It was shown that by a combination of measured and calculated < J > values, important conformational properties of a ring molecule can be determined.
The work carried out for cyclopentane represents a first step of determining J-hypersurfaces spanned in terms of the most important conformational coordinates of a ring compound. Once a J-hypersurface is known, it can be used to investigate the conformational behavior of biochemically interesting molecules such as ribose, 2’-deoxyribose, proline, etc.
In this work, we extend the investigation of pseudorotating ring molecules to tetrahydrofuran (THF). THF is the appropriate test case in connection with an NMR-spectroscopic investigation of ribose. Hence, we will determine the the pseudorotational potential of THF, then calculate all NMR SSCCs of THF for selected molecular forms located along its pseudorotational path. These SSCCs will be the basis to derive Karplus relationships of the form J = J(q,φ), which reflects the conformational behavior of THF.
Because of its importance as a model for ribofuranose, THF has been extensively investigated by various experimental techniques ranging from far-infrared spectroscopy [
18,
19] and microwave spectroscopy [
20,
21] to electron diffraction [
22], X-ray [
23], and neutron diffraction techniques [
24]. In addition, there are several quantum chemical investigations [
25,
26] and molecular mechanics (MM) studies [
27,
28], which have focused on the conformational features of THF. However, relative little is known about the SSCCs of THF either from experiment [
29,
30] or theory [
31,
32,
33] so that the current investigation fills a gap in the description of this important molecule.
2 Computational Methods
In the following, the theory of ring puckering coordinates, the quantum chemical methods used, and the derivation of the Karplus relationships used in this work are shortly described.
Description of THF in terms of Ring Puckering Coordinates. As shown by Cremer and Pople (CP) [
34], the conformational space of any puckered N-membered ring can be spanned by N-3 puckering coordinates, which split up in pairs of pseudorotational coordinates q and
φ, and (for even-membered rings) an additional puckering amplitude describing ring inversion. Hence, for THF (N = 5) there is just one pseudorotational mode defined by the pseudorotational phase angle
φdescribing the mode of ring puckering and the puckering amplitude q describing the degree of ring puckering [
25,
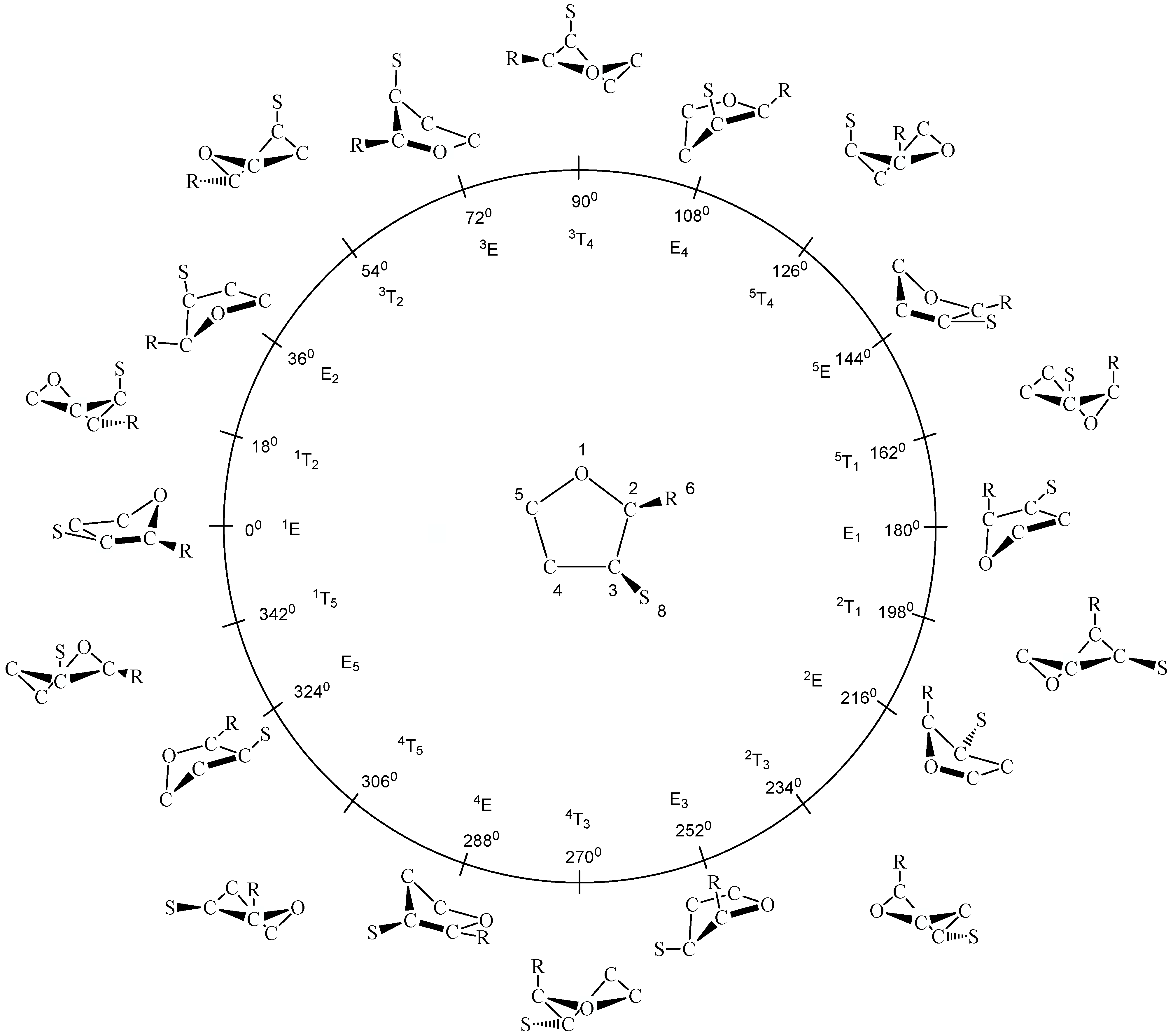
34]. An infinite number of ring conformations is located along the pseudorotational path, of which in the case of THF a subset of 20 forms is easy to recognize because it comprises the ten envelope (E) forms (
φ = (0 + k*360)/10 for k = 0,1,2,...,9 ) and the ten twist (T) forms (
φ = (18 + k*360)/10 for k = 0,1,2,...,9) of a five-membered ring (see
Figure 1). Since planar THF possesses
C2v symmetry, the conformational energy surface (CES) must comply with this symmetry, which means that puckered THF forms with
φ = 0
± ∆
ψ and
φ = 180
± ∆
ψ are identical for unsubstituted THF (see
Figure 1). There are just three unique E (
φ = 0, 36, 72°), three unique T forms (
φ = 18, 54, 90°), and the planar form (q = 0,
φ not specified), which have to be considered in an investigation of THF. Of course, this minimum set of conformations can be augmented, if necessary, by setting the phase angle to other values than those of the T and E forms.
According to the CP theory of ring puckering, the out-of-plane coordinates
zj of atom j=1,...,5 of any five-membered ring conformation are given by [
34,
35,
36,
37,
38,
39,
40,
41]
where the coordinates
zj are normalized according to
The full set of 3N-6 independent Cartesian coordinates of any puckered N-membered ring can be determined once the N-3 puckering coordinates (N = 5: q and
φ), N-3 bond angles (N = 5: two internal ring angles of the five-membered ring), and N (5) bond lengths are specified. First, the
zj coordinates are calculated according to Eq. (1) (or similar formulas for N > 5) [
34,
35]; then the N bond lengths and N-3 bond angles are projected onto the mean plane of the ring. Finally, the projected ring is partitioned into segments, for which the coordinates
xj, yj are calculated according to a procedure described by Cremer [
37].
The use of the ring puckering coordinates has the advantage [
40,
41] that the geometry of puckered forms of THF with a given value of
φ (or q and
φ) can be optimized even if these forms do not occupy a stationary point of the CES. This would not be possible when using Cartesian or internal coordinates [
37]. Another advantage results from the fact that any calculated property of THF (in particular any SSCC) can be expressed as a function of the puckering coordinates.
In
Figure 1, the planar form, ten E and ten T forms of THF are shown in the conformational space spanned by q and
φ. Hence, the planar form is located at the center for q = 0 and the puckered E and T forms along a pseudorotation itinerary (given by a circle). The numbering of the ring atoms (see
Figure 1) defines the conformation at
φ = 0° and all subsequent conformations. Often the different E and T forms are identified according to a notation based on suitable reference planes taken from the E (atoms 2, 3, 4, 5) and the T form (atoms 1, 2, 5) of cyclopentane. Ring atoms, which lie above the reference plane are written as superscripts and precede the conformational symbol E or T, while ring atoms , which lie below the reference plane are written as subscripts and follow the symbol E and T [
42]. Since experimentalists often prefer this notation instead of using puckering coordinates, it is also given in
Figure 1. However, in the discussion, we will exclusively use the pseudorotational angle to identify a given ring form since this notation can be applied to any ring form in the pseudorotational space.
Quantum Chemical Methods and Basis Sets. Energies, geometries, frequencies, enthalpies, and density distributions of seven THF forms were calculated by using second-order many-body perturbation theory [MBPT(2)] with the Møller-Plesset (MP) perturbation operator [
43] and Dunning’s cc-pVTZ basis set [
44]. This level of theory leads to a very accurate description in the case of cyclopentane as we showed recently [
12]. Since the actual target of the current project is the investigation of larger ring systems such as ribose and 2′-deoxyribose sugars, for which MBPT(2)/cc-pVTZ calculations become too expensive in view of the large number of conformations involved, the usefulness of a more economic method was also tested in this work. For this purpose, a second set of calculations was carried out using density functional theory (DFT) [
45,
46] with the hybrid functional B3LYP [
47,
48,
49] and Pople’s 6-31G(d,p) basis set [
50]. For the B3LYP frequency calculations, an ultrafine pruned (99,590) grid was used.
Geometries were calculated with the help of analytical gradients for ring puckering coordinates developed by Cremer [
37]. These were also used to calculate vibrational frequencies and to determine zero-point energies (ZPE) and enthalpies H(298) at 298 K. When calculating the vibrational contributions to enthalpies, large amplitude vibrations corresponding to the ring puckering modes were treated separately similar to the way internal rotations are treated [
51].
The electron density distribution, atomic charges, and the degree of hybridization was investigated by calculating MP2 response densities [
52,
53] and applying Weinhold’s natural bond orbital (NBO) analysis [
54,
55,
56].
Calculation of the CES of THF. The CES function V(q
, φ) of any puckered five-membered ring can be given as a Fourier expansion in the pseudorotational phase angle
φ and a power series in the puckering amplitude q [
25,
12]:
where
![Ijms 04 00158 i004]()
and
![Ijms 04 00158 i005]()
are expressed as
In view of the
C2v-symmetry of planar THF, the CES function V(q
, φ) can be simplified and truncated after the quartic term according to Eq. (5):
where the constant V
00 is set equal to the energy of the planar ring. Coefficients V
02,
· · ·, V
44 were determined by optimizing the geometry of the six puckered ring forms with
φ = 0, 18, 36, 54, 72, and 90°.
Similarly to V, each property P of THF can be expressed as a function of the puckering coordinates. Global properties P of the molecule lead to functions P(φ), which always comply with the molecular symmetry whereas local properties can lead Fourier expansions in φ of lower symmetry.
In the latter case, the property in question has to be determined for all 20 THF forms given in
Figure 1 (and others not considered there) to obtain accurate functions P(
φ). Nevertheless, the symmetry of the molecule helps again to reduce the number of necessary calculations. For example, calculation of the E form at
φ = 0 leads to the length of the equatorial bond C2H6, but provides also the length of this bond at
φ = 180 where it occupies an axial position and becomes identical to the length of the axial bond C2H7 at
φ = 0. Use of
Figure 1 helps to identify these relationships.
Calculation of the SSCCs of THF. SSCCs were determined by coupled perturbed density functional theory (CPDFT) as recently described and implemented by Cremer and co-workers [
13]. In this approach, Fermi contact (FC), paramagnetic spin-orbital (PSO), diamagnetic spin-orbit (DSO), and spin-dipole (SP) contribution to the total SSCC are consistently determined at the CPDFT level of theory (see also Ref. 57). Previous investigations showed that reliable SSCC values are obtained with the B3LYP functional [
12,
13,
57] and TZ+P or QZ+P basis sets [
58]. In this work, a (9s,5p,1d/5s,1p)[6s,4p,1d/3s,1p] basis set was employed (basis II of ref 58).
Using CPDFT/B3LYP all SSCCs of the type J(13C,13C), J(17O,13C) J(13C,1H), J(17O,1H) and J(1H,1H) were calculated. In the following, we will simplify the notation of SSCCs by using symbols such as 3J(HCCH, cis) where C and H denote 13C and 1H, the major coupling path H-C-C-H of the 3-bond SSCC is given, and cis identifies the positions of the H atoms on the same side of the ring.
SSCCs were calculate for both MBPT(2) and B3LYP geometries. However, because the current work will provide reference data for a B3LYP investigation of ribofuranose, for which MBPT(2)/cc-pVTZ geometry optimizations become too expensive, we will exclusively discuss the SSCCs calculated for the B3LYP/6-31G(d,p) geometries of THF.
For the analysis of the FC term, the
s-density at the nucleus
K was calculated according to
where
δ(
rK) is the Dirac delta function and
φl is the localized bond orbital. The product
gs(
K,L) =
gs(
K)
gs(
L) was related to the magnitude of the FC term.
Calculation of the Karplus Relationships J(q
, φ)
or J(
φ)
. The SSCC
nJ was expanded as a function of the puckering coordinates q and
φ according to Eq. (7).
where the term A
0(q) converges to the SSCC of the planar ring for q
→ 0. A
0, B
k and C
k can be expanded as power series in the puckering amplitude q.
Note that for B
k and C
k the constant terms with
l = 0 vanish because
For pseudorotating ring molecules, only average SSCCs can be measured. For the purpose of calculating average SSCCs, one has to determine both the CES function V and the SSCCs as a function of puckering coordinates q and
φ. Averaging of the SSCC can be simplified by considering just the dependence of
nJ on the pseudorotational phase angle
φ:
where
c and
s are truncation limits, which can differ. Then, the average SSCC <
nJ > is given by
The conformational probability distribution
g(
φ) is given as a Boltzmann distribution:
The potential V is determined according to Eq. (5) and used to calculate average SSCCs according to Eq.s (10) and (11).
For the quantum chemical calculations, the program systems COLOGNE2002 [
59] and Gaussian98 [
60] were used.
3 The Flexible Pseudorotor THF
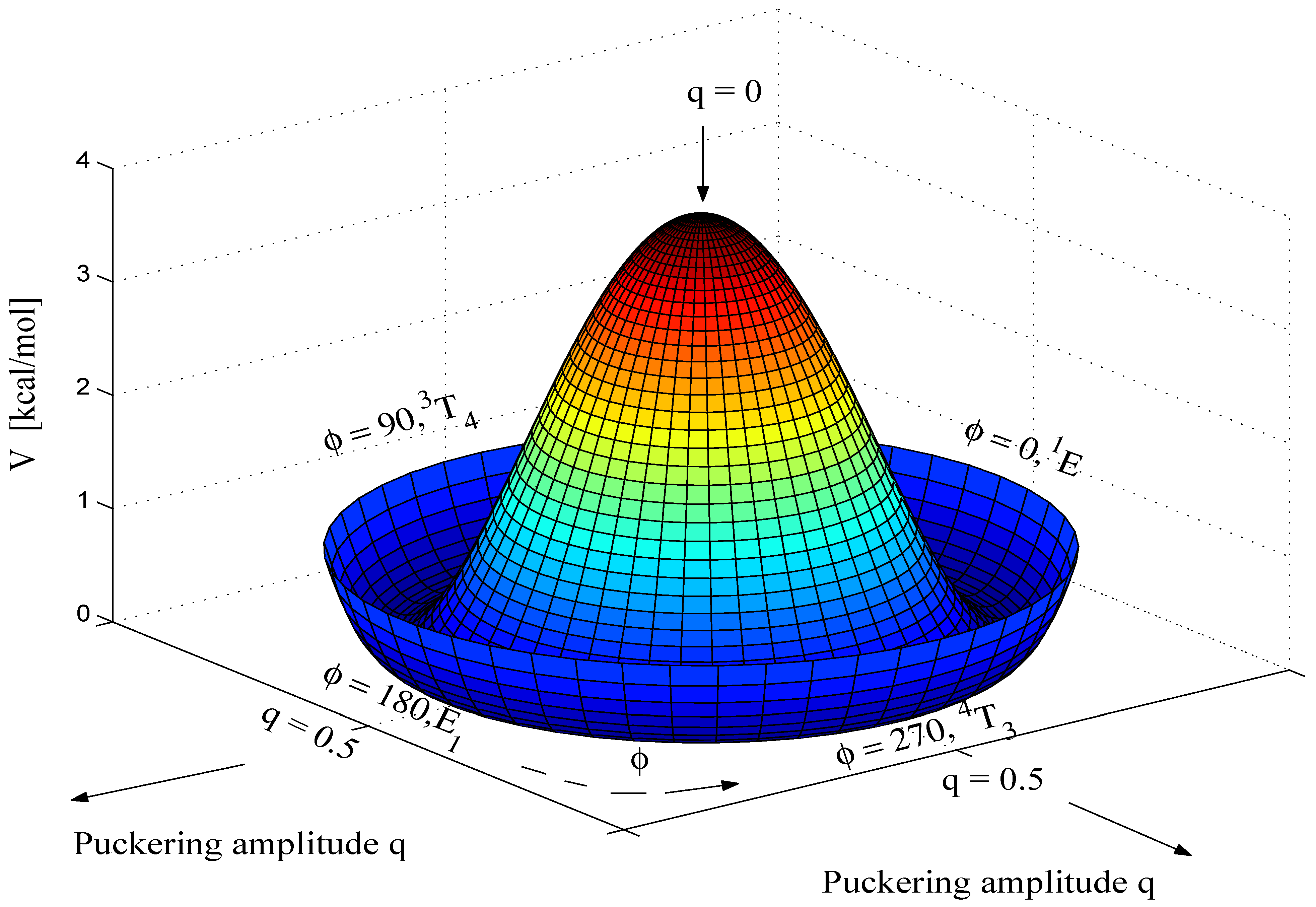
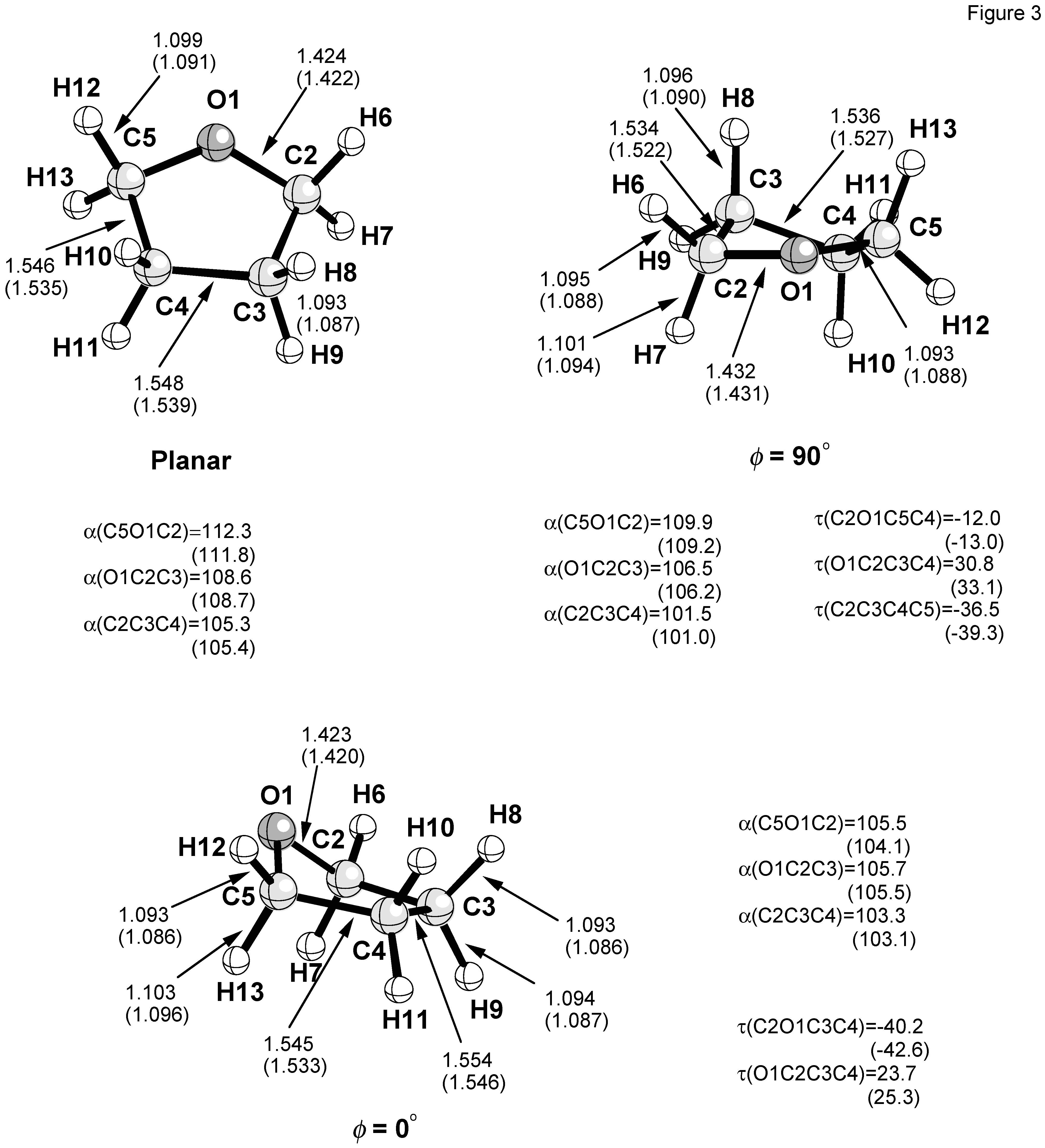
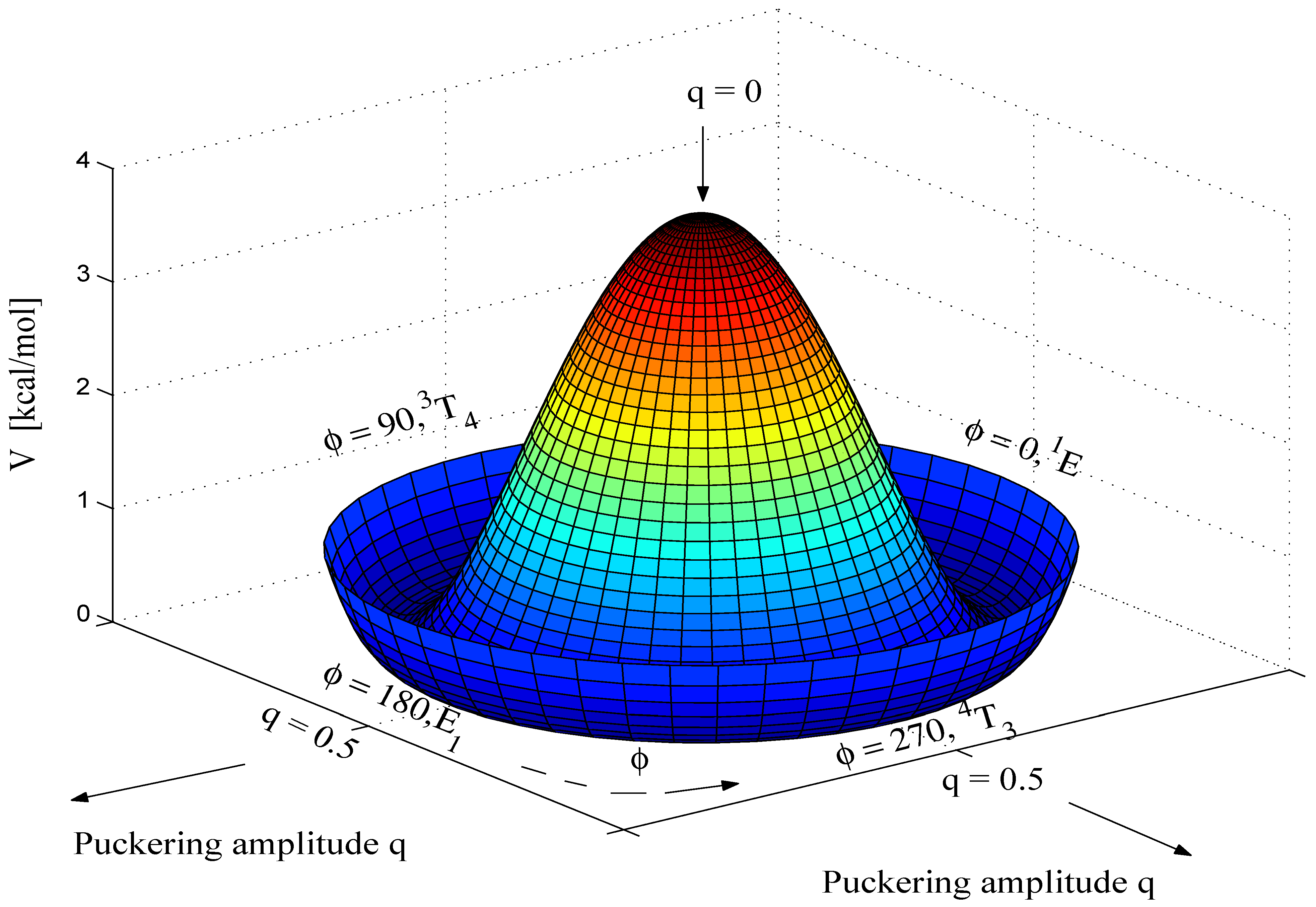
Calculated energies and puckering coordinates obtained in this work are summarized in
Table 1. A three-dimensional perspective drawing of the calculated CES V(q
, φ) is given in
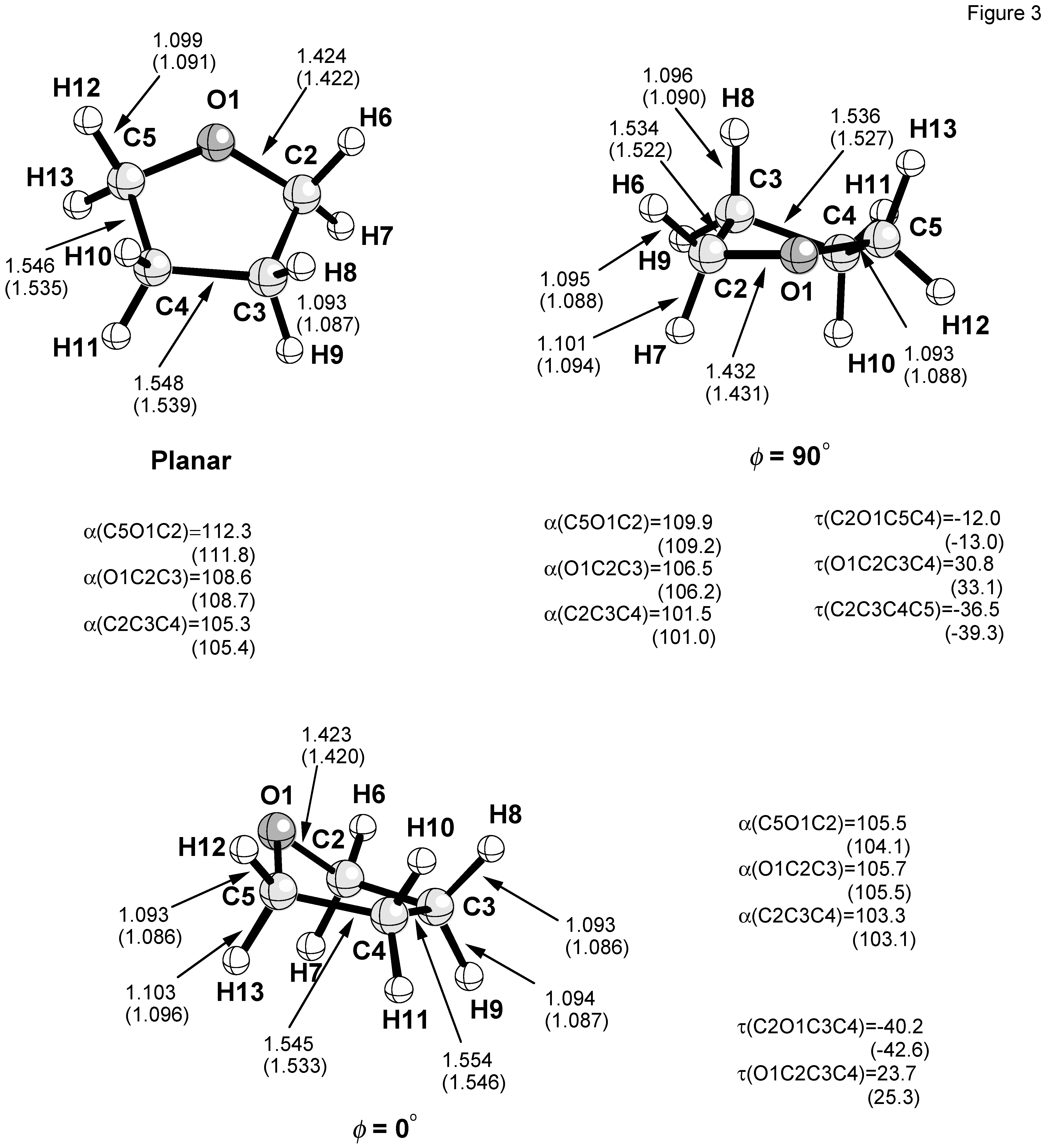
Figure 2. The geometries of the planar form, the E form with
φ = 0°, and the T form
φ = 90° are shown in
Figure 3.
The gross features of the CES of THF resemble those of the CES of cyclopentane (see
Figure 2). Incorporation of a hetero atom into the ring makes THF a slightly hindered pseudorotor (energy barrier 0.14 kcal/mol,
Table 1). Both MBPT(2) and B3LYP predict a twofold pseudorotational potential that locates the T forms with
C2-symmetry (
φ = 90°, 270°) at the the global minima of the CES. The E forms at
φ = 0° and 180° represent first order saddle points of the pseudorotational mode while the planar form occupies a local maximum at the center of the q,
φ-coordinate system (
Figure 2). This is consistent with all previous
ab initio and MM calculations [
25,
26,
27,
28] and with most, but not all experimental studies. An electron diffraction study of Bartell and co-workers [
22] also predicts the
C2-symmetrical T forms to represent the equilibrium conformation of THF.
Results based on far-infrared spectroscopy [
18,
19] are in linewith this. However, microwave studies by Engerholm and co-workers [
20] carried out in the late 60ies and recently repeated and extended by Meyer and co-workers [
21] suggest a fourfold pseudorotational potential (V
4l coefficients dominate the CES function (5) rather than the V
2l coefficients) with equivalent minima at
φ = 0
±52.5° and 180
± 52.5°, which are close to the T forms at
φ = 54, 126, 234, and 306°.
A careful analysis of calculated energies and geometries (see below) provides now clue for a fourfold pseudorotational potential. In particular, the analysis of the electronic effects determining the stability of the various THF forms along the pseudorotation path clearly suggests the T forms at
φ = 90 and 270° to be more stable than any other T form. The argument given by Meyer and co-workers [
21] that
ab initio theory is not sufficiently accurate to provide a reliable description of the pseudorotational potential of THF has to be rejected on the following grounds. The quantum chemical description of conformational processes does not require high-level correlation corrected
ab initio methods because the bonding pattern of the molecule (and by this also the pair correlation effects) remains largely unchanged during internal rotation, pseudorotation, etc. Although MBPT(2) covers just pair correlation effects [
43], it is known to provide an accurate account of energies and geometries along the pseudorotational path of a free (or slightly hindered) pseudorotor if carried out with a cc-pVTZ basis set [
12,
35]. The consistency of the MBPT(2) description along the pseudorotation itinerary excludes the existence of
C1-symmetrical minima as suggested by the microwave results.
It is noteworthy in this connection that both the spectroscopic [
18,
19,
20,
21] and the electron diffraction study [
22] had to fit experimental parameters such as diffraction intensities, frequency splittings (due to tunneling) or rotational constants to a constrained model of THF based on assumed geometrical parameters and simplifications of its conformational flexibility. A more direct way of determining the equilibrium conformation of THF was provided by X-ray diffraction experiments at 103 K of Luger and Buschmann [
23] and the high resolution neutron powder diffraction experiments at 5 K of David and Ibberson [
24]. Both investigations identify the T forms at
φ = 90° and 270° as the equilibrium forms of THF in line with the twofold pseudorotational potential calculated in this work.
The enthalpy barriers ∆H along the pseudorotational path obtained from MBPT(2) and B3LYP calculations are 0.22 and 0.25 kcal/mol (
Table 1), respectively, corresponding to 77 and 87 cm
−1, which are in good agreement with experimental estimates from a far-infrared measurement (< 0.5 kcal/mol) [
18], and from microwave experiments (0.21 kcal/mol or 73 cm
−1 [
20]; 0.13 kcal/mol or 45 cm
−1 [
21]). The barrier to planarity, ∆E(inv) (∆H(inv)), is calculated to be 3.36 (2.94) kcal/mol at B3LYP and 4.46 (4.15) kcal/mol at MBPT(2) (see
Table 1). Both ∆H(inv) values compare well with experimental estimates of 3.49 kcal/mol (microwave spectroscopy [
20]) and 3.86 kcal/mol (far-infrared experiment [
19]). B3LYP seems to slightly underestimate, MBPT(2) to slightly overestimate the barrier to planarity as was also observed in the case of cyclopentane [
12].
All E and T forms are predicted to have the same puckering amplitude q (B3LYP: 0.371
± 0.001; MBPT(2), 0.396
± 0.002 Å;
Table 1), which is typical of a free or slightly hindered pseudorotor [
12,
25]. Agreement with the puckering amplitude q
g = 0.38
± 0.02 Å obtained in the electron diffraction experiment [
22] is satisfactory considering vibrational effects and the fact that the latter investigation used a highly constrained geometrical model (e.g., equal CC and CO bond lengths) of THF. David and Ibberson determined a q value of 0.343 Å for the T forms (
φ = 90°, 270°) on the basis of the neutron diffraction data [
24] while the X-ray diffraction study of Luger and Buschmann [
23] led to a q value of 0.348 Å. Considering again vibrational effects and the fact that the interactions between THF molecules in the unit cell imply a slight flattening of the five-membered ring, agreement with theory is reasonable.
Previous computational investigations of THF were carried out with MM methods (∆E = 1.4 kcal/mol, q = 0.370 - 0.399 Å) [
27,
28], at the Hartree-Fock level of theory (∆E(inv) = 2.30 - 3.41 kcal/mol; q = 0.314 - 0.370 Å) [
25,
26], or at the MBPT(2) level of theory (∆E(inv) = 4.77 kcal/mol; q = 0.395 - 0.398 Å) [
26]. Calculated pseudorotational barriers ranged from 0.39 to 0.65 kcal/mol [
25,
26]. None of these investigations provided an accurate account of the CES of THF because either force fields of moderate accuracy, uncorrelated wave functions, too small basis sets, just constrained geometry optimizations, or ∆E rather than ∆H (neglect of vibrational effects) were used because of computational limitations.
Each geometrical parameter P of THF was expanded in this work using the general Eq. (12):
In
Table 2, the Fourier expansion coefficients
![Ijms 04 00158 i016]()
and
![Ijms 04 00158 i017]()
of the internal coordinates of THF are listed. In view of the low energy barriers to pseudorotation, the constant term
![Ijms 04 00158 i018]()
of the Fourier expansion is close to the average value < P > obtained when using Eq. (12) in connection with (10) and considering the correct Boltzmann statistics (11) for the pseudorotational itinerary (compare with
Table 3).
In
Table 3, calculated average values < P > of the the most important geometrical parameters of THF are compared with available experimental values. Comparison of these data reveals that both MBPT(2) and B3LYP provide a reasonable account of the geometry of THF.
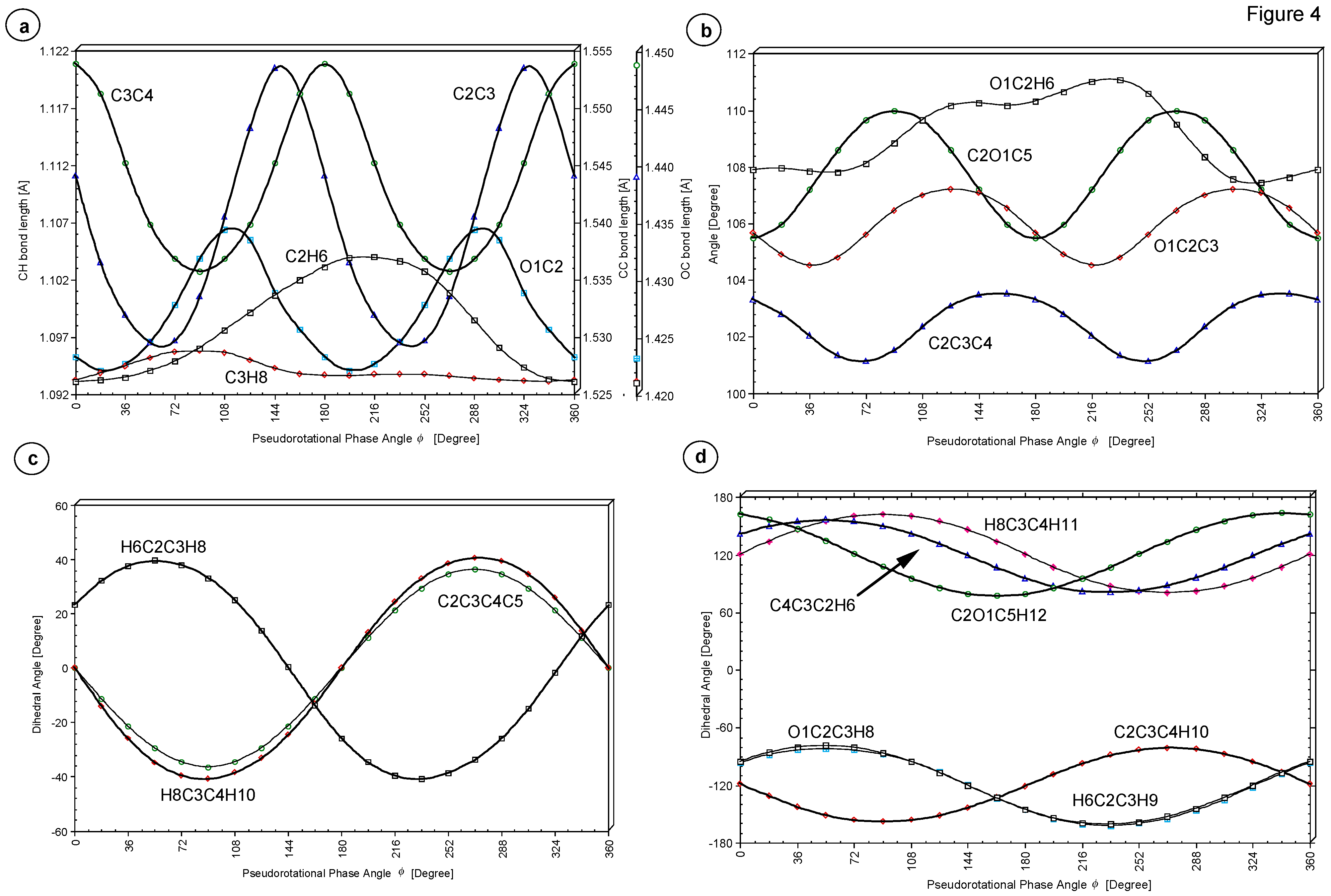
For the purpose of rationalizing the relative stability of the various THF forms along the pseudorotation itinerary, we next consider changes in the internal coordinates caused by a variation of
φ. In
Figure 4, P(
φ) is given for some of the internal coordinates of THF (see also
Table 2).
The known rotational barriers of ethane (2.9 kcal/mol) [
61,
62] and methanol (1.1 kcal/mol) [
63,
64] suggest that bond eclipsing in the H
2C3C4H
2 unit for
φ = 0° and 180° causes a destabilization of the two E forms accompanied by a lengthening of the C3C4 bond to 1.554 Å (see,
Figure 4a). For the two T forms at
φ = 90° and 270° bond staggering in the H
2C3C4H
2 unit is maximized (see
Figure 4c) thus yielding a shorter C3C4 bond (1.536 Å) and larger stability of THF. For the H
2C2C3H
2 (H
2C4C5H
2) unit bond staggering is maximized for
φ = 54° and 234° (
φ = 128° and 306°) (compare with bond length C2C3 in
Figure 4a). Any model of THF, which exaggerates this effect by using a too large barrier to rotation in methanol, gets the four
C1-symmetrical T forms too stable (see above).
In addition to bond eclipsing/bond staggering effects, there are also anomeric interactions [
12,
65] involving an O electron lone pair and bonds C2H6 and C2C3. In the case of bond C2C3, they enhance changes in its length to ∆ = 0.024 Å (variation from 1.530 Å at
φ = 54 to 1.554 Å at
φ = 144°, see,
Figure 4a) while for bond C2H6 ∆ is about 0.01 Å. At
φ = 198 (18)°, bond C2H6 (C2H7) is perfectly positioned to support a delocalization of one of the lone pair electrons at O into its
σ*(CH) orbital. This leads to a weakening of the CH bond but a strengthening of the CO bond (see
Figure 4a). The anomeric effect implies that the
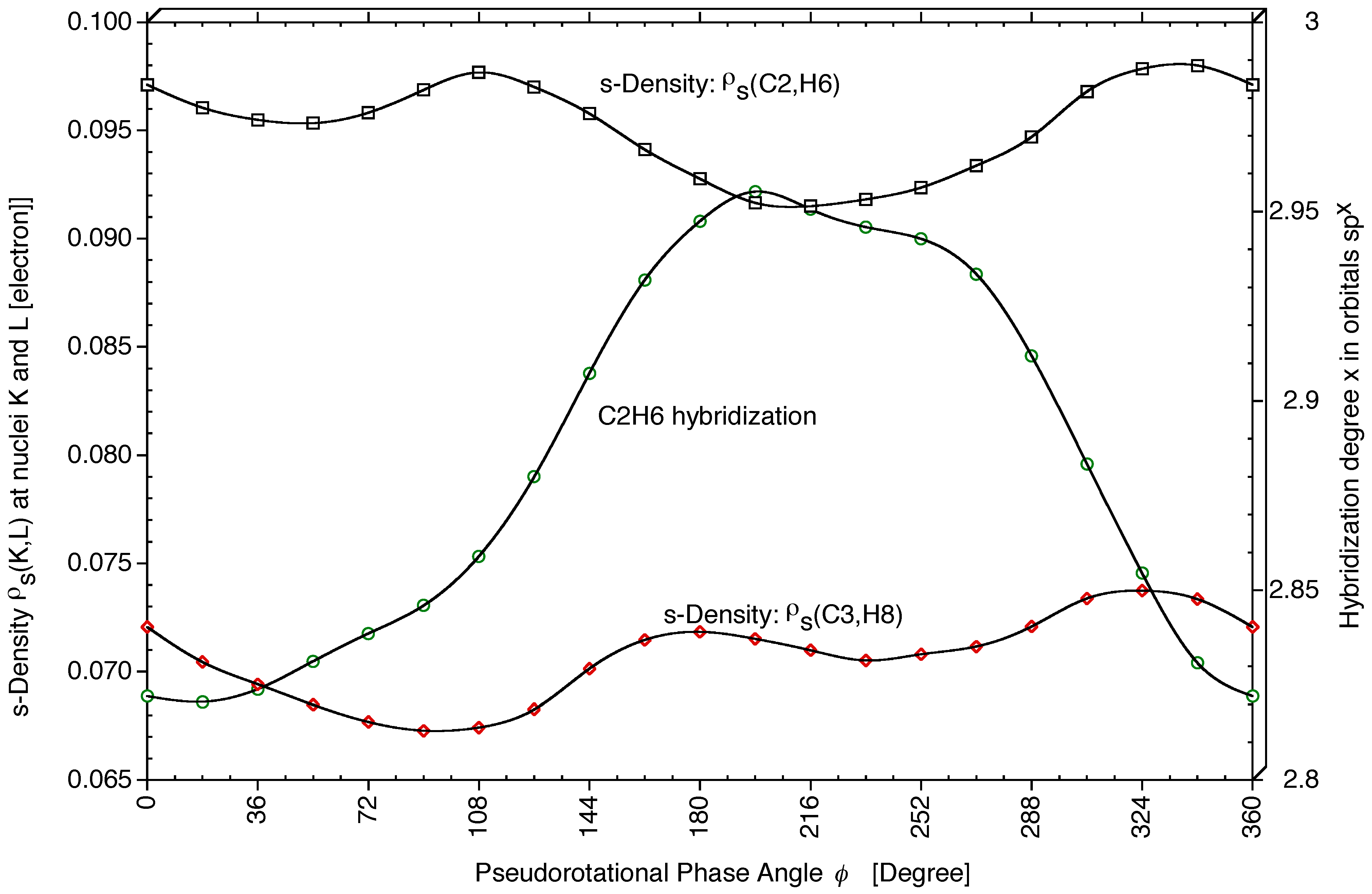
p-character of the CH bond orbital increases. Hybride orbital calculations for THF employing the NBO analysis [
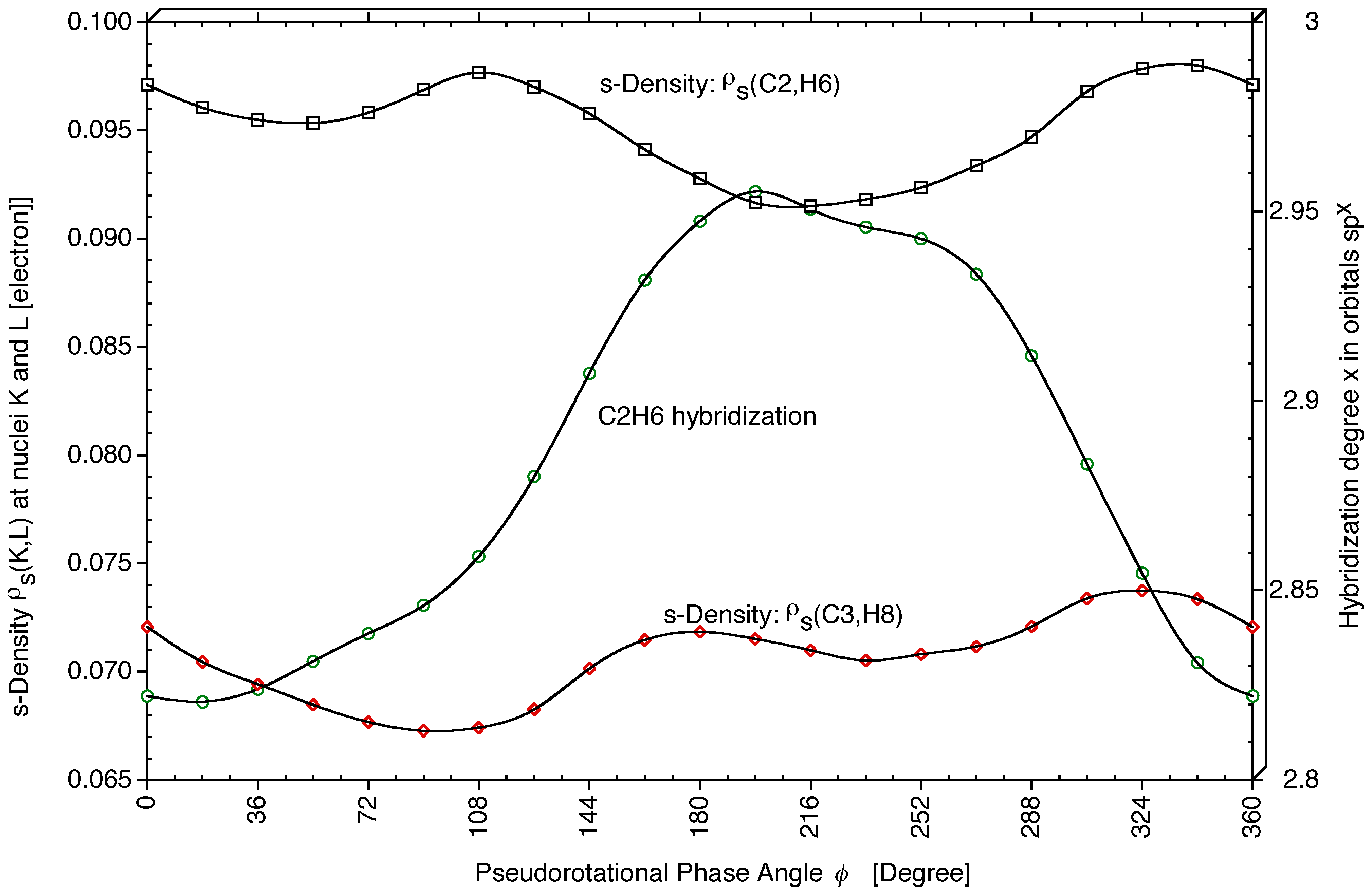
54,
55,
56] confirm this, although effects are small: For C2H6, hybridization changes from sp
2.82 (
φ = 18°) to sp
2.96 (
φ = 198°)(see
Figure 5), which corresponds to a slight decrease in the
s-character of the CH hybrid orbital from 26.2% to 25.2%. These changes are interesting in view of the changes calculated for the SSCCs
1J(CH) (see next section).
The C2O1C5 bond angle changes by 4.5 ° from 105.5° to 110.0° where the smallest angle is found for the E forms at
φ = 0° and 180° and the largest angle for the T forms at
φ = 90° and 270° (see
Figure 4b). This also supports the stability of the latter conformations.
4 Analysis of NMR Spin-Spin Coupling Constants
There are 26 different NMR SSCCs for THF: two
nJ(
17O-
13C), two
nJ(
17O-
1H), four
nJ(
13C-
13C), eight
nJ(
13C-
1H), and ten
nJ(
1H-
1H) SSCCs. Because of the small natural abundance of
17O and its large quadrupole moment,
17O SSCCs are difficult to measure and only a relatively small number of reliable SSCCs involving
17O is available [
66]. Nevertheless, it is still interesting to investigate the conformational dependence of SSCCs involving
17O and test their use as descriptors of the electronic structure of THF. In this work, we provide the full set of all 26 SSCCs of THF.
For cyclopentane [
12], we showed that
3J(HCCH) expressed as a function of the associated dihedral angle
τ(HCCH) can only be described by two rather than one Karplus relationship because one
τ(HCCH) value can lead to two significantly different
3J(HCCH,cis) values. For THF, even six Karplus relationships are needed to represent all
3J(HCCH,cis) values for fragments H6C2C3H8 and H8C3C4H10 as a function of the associated dihedral angle
τ(HCCH) in an accurate way. These problems vanish when SSCCs J are expressed as functions of the ring puckering coordinates according to Eq.s (7) or (9).
In
Table 4, the Fourier expansion coefficients of Eq. (9) are given for all 26 SSCCs of THF. The calculated average SSCCs <
nJ > based on B3LYP and MBPT(2) geometries are listed in
Table 5 and compared with measured J values and the corresponding SSCCs of cyclopentane [
12]. Karplus relationships (9) are shown for selected SSCCs in
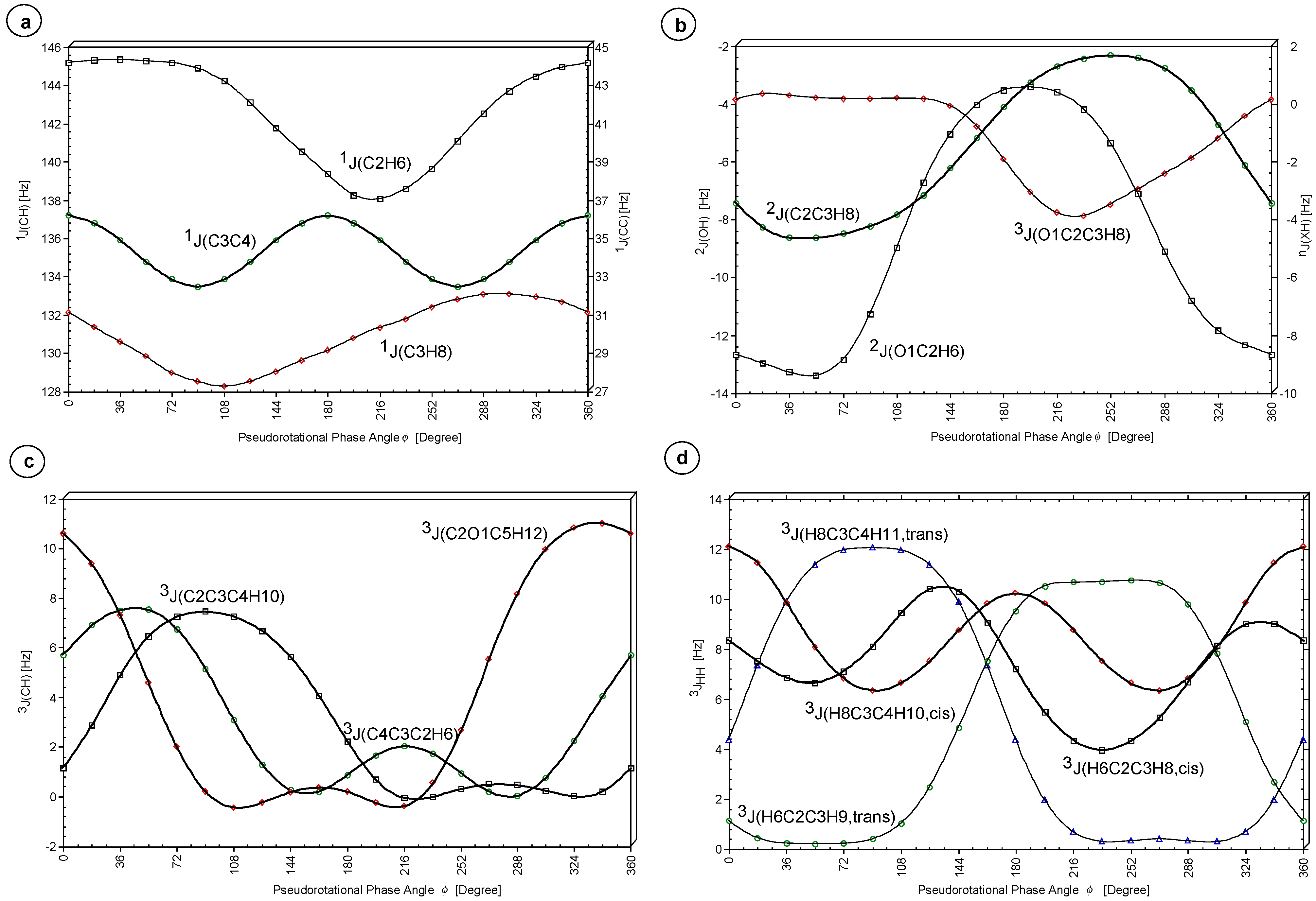
Figure 6.
In the following, we will discuss the SSCCs of THF (
Table 4 and
Table 5) separately where the emphasis will be on those SSCCs that can be used for the determination of the conformation of THF and its derivatives. The SSCCs of THF are dominated by the FC contribution whereas the PSO, DSO, and SD contributions make small but significant contributions (≤ 3 Hz, e.g. for the one-bond SSCCs). Hence, the calculation of these terms is absolutely necessary to obtain reliable J values, however the dependence of J on the puckering coordinates can be rationalized by concentrating on the FC term only.
One-Bond Coupling Constant 1J(OC). This SSCC is dominated by the FC term, which changes significantly with φ. However, the changes in PSO+DSO+SD are opposite to that of the FC contribution so that the total SSCC changes by only 0.4 Hz from 26.1 (φ = 18, 198°) to 26.6 Hz (φ = 126, 306°). Hence, the total SSCC is of little use for the conformational analysis.
One-Bond Coupling Constants 1J(CC). The position of the electronegative O atom has little effect on
1J(CC) constants. The calculated SSCC value is 34.0 Hz for <
1J(C2C3) > and 34.2 Hz for <
1J(C3C4) >, respectively, which are comparable to the measured values for propane (34.6) [
30], cyclohexane(32.7) [
30] and the calculated value of <
1J(CC) > for cyclopentane (34.0 Hz [
12]). The variation of
1J(C3C4) (∆ = 3.7 Hz) is much larger than the changes in
1J(C2C3) (∆ = 1.1 Hz), which is opposite to the changes in the corresponding bond lengths (see discussion in previous section). The changes in the one-bond SSCCs can however be directly explained considering the products
gs(C,C) of the densities at the nuclei (see Eq. 6). This will be large if exchange repulsion (bond eclipsing) increases. A large density at the site of the nuclei guarantees a stronger transmission of spin polarization from one nucleus to the other.
In this respect, exchange repulsion effects increase both the FC term of the SSCCs and the bond length, i.e. the latter do not directly influence the one-bond SSCC.
One-Bond Coupling Constants 1J(CH). In contrast to
1J(CC),
1J(CH) exhibits a strong dependence on the position of the O atom. The average SSCC values are 142.6 Hz for
1J(C2H6) and 130.9 Hz for
1J(C3H8), respectively, which are in reasonable agreement with measured values of 144.6 for
1J(C2H6) and 132.2 for
1J(C3H8) (see
Table 5 and
Figure 6a). The difference of 12 Hz between <
1J(C2H6) > and <
1J(C3H8) > is a result of the inductive effect of the O atom [
30]. Analysis of the
s-density reveals that at C2 (bond C2H6: ≃ 0.096 electron) it is significantly larger than the s-density at C3 (bond C3H8: ≃ 0.070 electron; see
Figure 5).
1J(C2H6) changes by 7.3 Hz from 145.4 (
φ = 36°) to 138.1 Hz (
φ = 216°). This decrease is caused by a change in the position of bond C2H6 relative to the mean plane of the ring. Calculating the ring substituent orientations according to Cremer [
36] reveals that C2H6 is in the equatorial position for
φ = 36
± 18° and in the axial position for
φ = 216
± 72°. In the latter situation, the Perlin effect (the magnetic analogue of the anomeric effect) [
5,
67] decreases
1J(C2H6) and enlarges the difference
1J(CH
eq) -
1J(CH
ax). The Perlin effect is well-known for the
1J(CH) constants of heterocyclohexanes [
68] and, obviously, plays also a role in five-membered rings with hetero atoms.
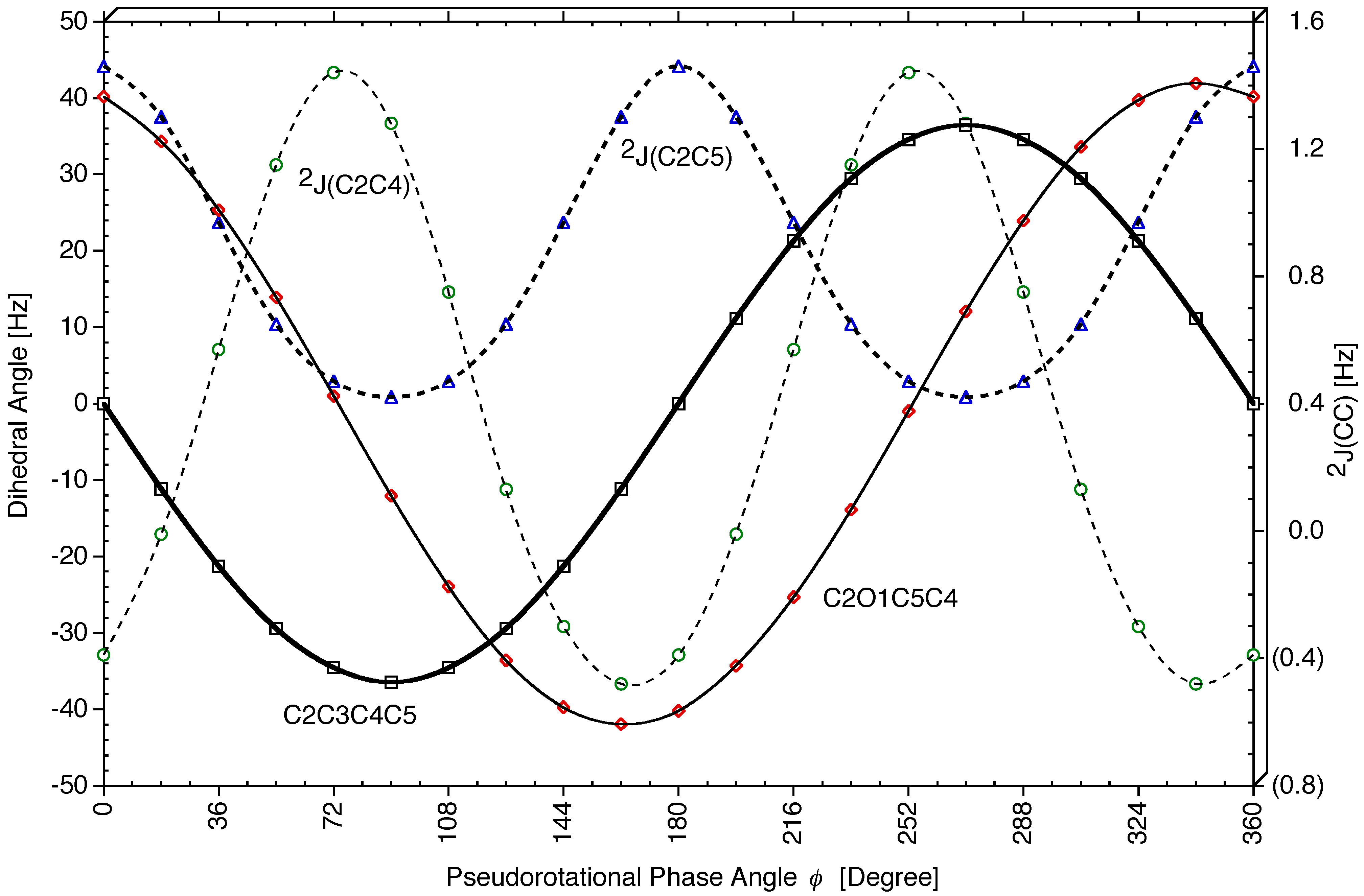
Geminal Coupling Constants 2J(OCC) and 2J(CCC). The three
2J(X,C) constants possess positive average values: 0.3 Hz for <
2J(O1C2C3)>, 0.5 Hz for <
2J(C2C3C4)>, and 0.8 Hz for <
2J(C2O1C5)>. For the purpose of explaining the
φ-dependence of these SSCCs, one has to consider that there are two coupling paths as reflected by notations such as
2+3J(XCC,XCCC) with X = C or O. The two-bond contribution of
2J(CXC) and
2J(XCC) are almost constants during pseudorotation. Hence, the total coupling constants depend on the changes in the three-bond contributions, which in turn correlate with the dihedral angle of the corresponding fragments XCCC and CXCC in the sense that a larger deviation of the dihedral angle from 0° leads to a smaller value of the coupling constant (see
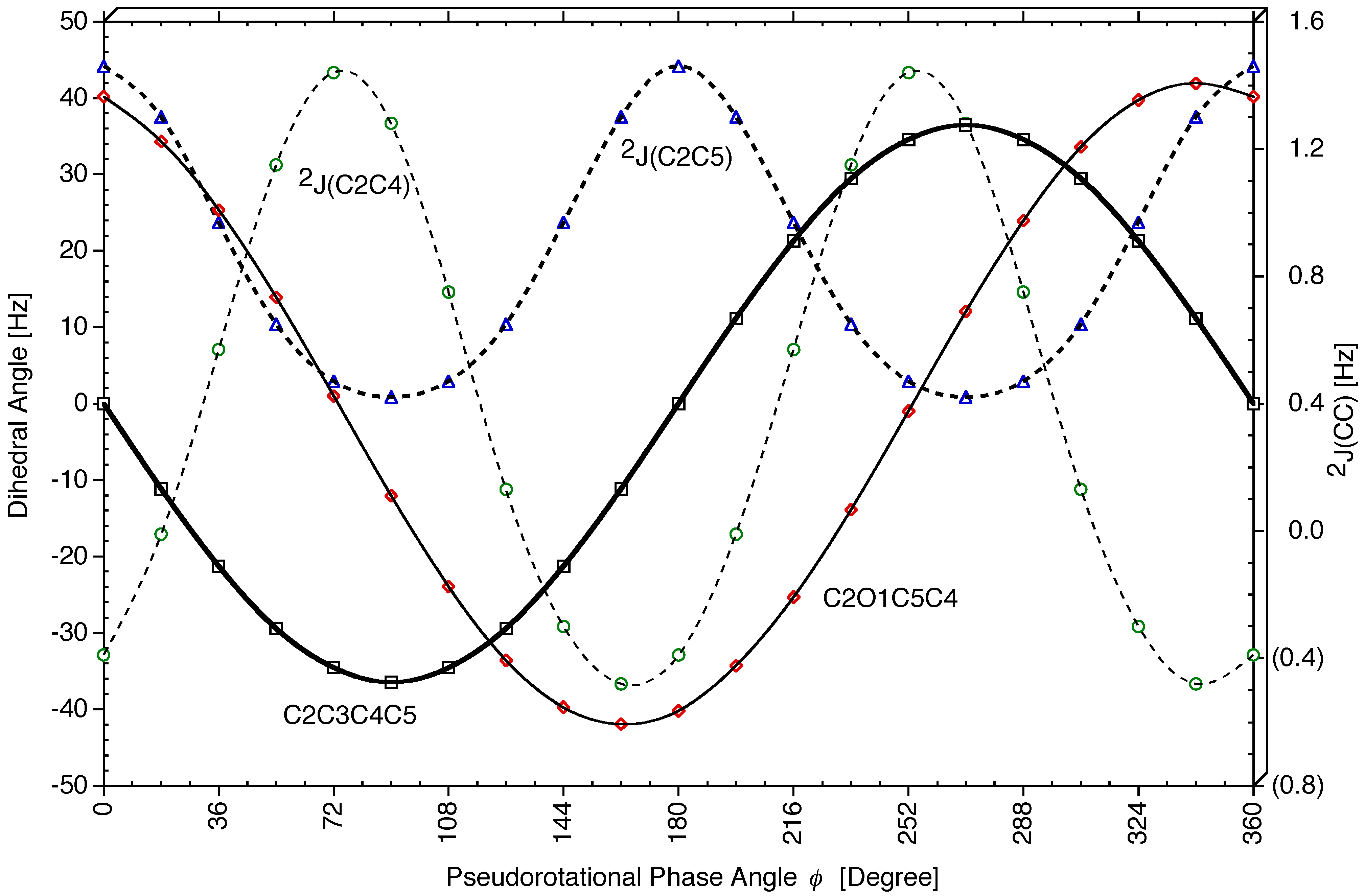
Figure 7).
Geminal Coupling Constants 2J(OCH) and 2J(CCH). The calculated value of the SSCC <
2J(C3C4H10) > is -2.8 Hz and agrees well with <
2J(CCH) > for cyclpopentane (-2.6 Hz,
Table 1). The inductive effect of an O atom leads to a positive contribution of 1 - 2 Hz depending on the position of O and the relative orientation of CO and CH bond [
30]. This is also reflected by <
2J(C3C2H6) > = -0.7 Hz and <
2J(C2C32H8) > = -1.5 Hz. Of the four
2J(XCH) SSCCs,
2J(C2C3H8) and
2J(O1C2H6) are most sensitive to the pseudorotational phase angle
φ (see
Figure 6b).
2J(C2C3H8) changes by 6.3 Hz from -4.6 (
φ = 36°, 54°) to 1.7 Hz (
φ = 252°) where its value depends on the relative orientation of the CO bond as reflected by
τ(O1C2C3H8) (the larger
τ, the more positive
2J(C2C3H8); see
Figure 4d and
Figure 6b). Similar effects have been reported [
5,
67] and frequently utilized to study the conformation of important biological compounds [
68].
2J(O1C2H6) changes by 10 Hz from -13.4 Hz (
φ = 54°) to -3.4 Hz (
φ = 198°). The
s-character of the hybrid orbital of bond C2H6 influences
2J(O1C2H6),
1J(C2H6), and
2J(H6C2H7). An increase in the
s-character of sp
x(C2H6) leads to a more negative
2J(O1C2H6) SSCC (see
Figure 5 and
Figure 6b). If
17O SSCCs could be measured,
2J(O1C2H6) would be an useful conformational probe.
Geminal Coupling Constants 2J(HCH). The dependence of
2J(HCH) on the hybridisation degree of the C atom and the impact of an electronegative atom are well known [
1,
4,
5,
30]. The calculated values of <
2J(H6C2H7) > (-8.7 Hz,
Table 5) and <
2J(H8C2H9) > (-12.0 Hz) reflect these trends. <
2J(H8C2H9) > agrees well with the calculated <
2J(HCH) > value for cyclopentane (-12.4 Hz,
Table 5).
Vicinal Coupling Constants 3J(OCCH), 3J(CCCH). All
3J(XCCH) couplings depend strongly on the pseudorotational phase angle
φ (see
Figure 6c). The values of <
3J(C
,H) >, are all positive (
3J(C4C3C2H6): 2.9;
3J(C2C3C4H10): 3.0;
3J(C2O1C5H12): 4.0 Hz,
Table 5) and comparable to the corresponding cyclopentane value of 3.9 Hz (
Table 5). The O atom as an external substituent of the CCH unit decreases the SSCC, however O in the ether position increases the SSCC [
30]. The value of <
3J(OCCH) > is -1.2 Hz whereas
3J(OCCH) varies by 4.3 Hz from -3.9 (
φ = 234°) to 0.4 Hz (
φ = 36°) and thus reflects the corresponding changes in
τ(O1C2C3H8) (
Figure 4d). Variations in the vicinal SSCCs by 7.5 Hz (
3J(C2C3C4H10)), 11.5 Hz (
3J(C2O1C5H12)), and 7.6 Hz (
3J(C4C3C2H6),
Figure 6c) can be explained by considering both the
s-character of CH (or CC) bonds and the dihedral angle
τ(CCCH) (see also Ref. 12).
Vicinal Coupling Constants 3J(HCCH). The SSCC
3J(H8C3C4H10,cis) possesses a similar dependence on
φ than the corresponding SSCC in cyclopentane [
12], adopting large values for
τ(H8C3C4H10) = 0° (12.1 and 10.2 Hz at
φ = 0
◦ and 180°;
Figure 6d) and smaller values for
τ(H8C3C4H10) = 40.7° (6.4 Hz for
φ =90° and 270°). The difference ∆ = 1.86 Hz between the
endo-3J(H8C3C4H10,cis) (
φ = 180°) and the
exo-3J(H8C3C4H10,cis) value (
φ = 0°) is another example for the
Barfield transmission effect [
70,
71], which has been studied by various authors and which has been explained in different ways [
69,
70,
71]. Noteworthy in this connection is that differences in SSCC due to the Barfield transmission effect are automatically covered when the SSCCs are expanded as functions of the puckering coordinates (see
Figure 6d).
An accurate description of the variation in
3J(H8C3C4H10,cis) during pseudorotation requires cos3
φ and cos4
φ terms (standard deviation 0.01 Hz; see
Table 4). The average value of SSCC
3J(H8C3C4H10,cis) is calculated to be 8.51 Hz, which is in good agreement with the measured value of 8.65 Hz [
29]. For cyclopentane the corresponding SSCC is 0.8 Hz larger (see
Table 5).
The Karplus relationship
3J(H6C2C3H8,cis) =
3J(
φ) is shifted by 216
◦ relative to that for
3J(H8C3C4H10,cis) (see
Figure 6d). It has a similar, however somewhat asymetrical shape (caused by a similar asymmetry in
τ(H6C2C3C8)) where J varies by 6.4 Hz from 4 Hz (
φ = 234°) to 10.4 Hz (
φ = 126°). Again, the electronegative O atom leads to a lower <
3J(H6C2C3H8
,cis) > value (7.33 Hz,
Table 5), which is in reasonable agreement with the experimental value of 7.94 Hz [
29]. The Barfield transmission effect is now smaller as reflected by an
exo-
endo difference of 1.33 Hz (
endo,
φ = 324°, 8.99 Hz;
exo,
φ = 144°, 10.32 Hz).
As in the case of cyclopentane, the dependence of
3J(HCCH,trans) on pseudorotation is considerably different from that observed for
3J(HCCH,cis) (
Figure 6d). Utilizing the
φ-dependence of
3J(H8C3C4H11), the conformational space can be partitioned into several regions:
3J(H8C3C4H11) adopts values
≥ 10 Hz for 36° ≤
φ ≤ 144°; it becomes ≤ 2 Hz for 198° ≤
φ ≤ 342°, and takes intermediate values in the remaining regions. The Karplus relationships for
3J(HCCH,trans) are given in
Table 4. The values of <
3J(HCCH
,trans) > are 5.50 for
3J(H8C3C4H11) and 5.35 Hz for
3J(H6C2C3H9), which are 0.7 - 0.8 Hz smaller than the measured values of 6.25 and 6.14 Hz [
29], respectively. We note that as in the case of cyclopentane the average cis-SSCCs are better described than the average trans-SSCCs. Since the latter are largely insensitive to ring puckering, this deviation is due to shortcomings of the method (lack of higher order correlation effects). We note in this connection that <
3J(HCCH
,trans) > is also calculated somewhat too small for alkanes and alkenes irrespective of the basis set.
Long Range Coupling Constants 4J(HCCCH). The four long-range SSCCs (two cis- + two trans-
4J(HCCCH) constants, see
Table 4 and
Table 5) change only slightly (≤ 1.5 Hz) during pseudorotation. Average values are -0.23 Hz for <
4J(H6C2O1C5H12) >, -0.16 Hz for <
4J(H6C2C3C4H19) >, -0.55 Hz for
4J(H6C2O1H13), and -0.61 Hz for
4J(H6C2C3C4H11).
5 A General Karplus Relationship for Puckered Rings
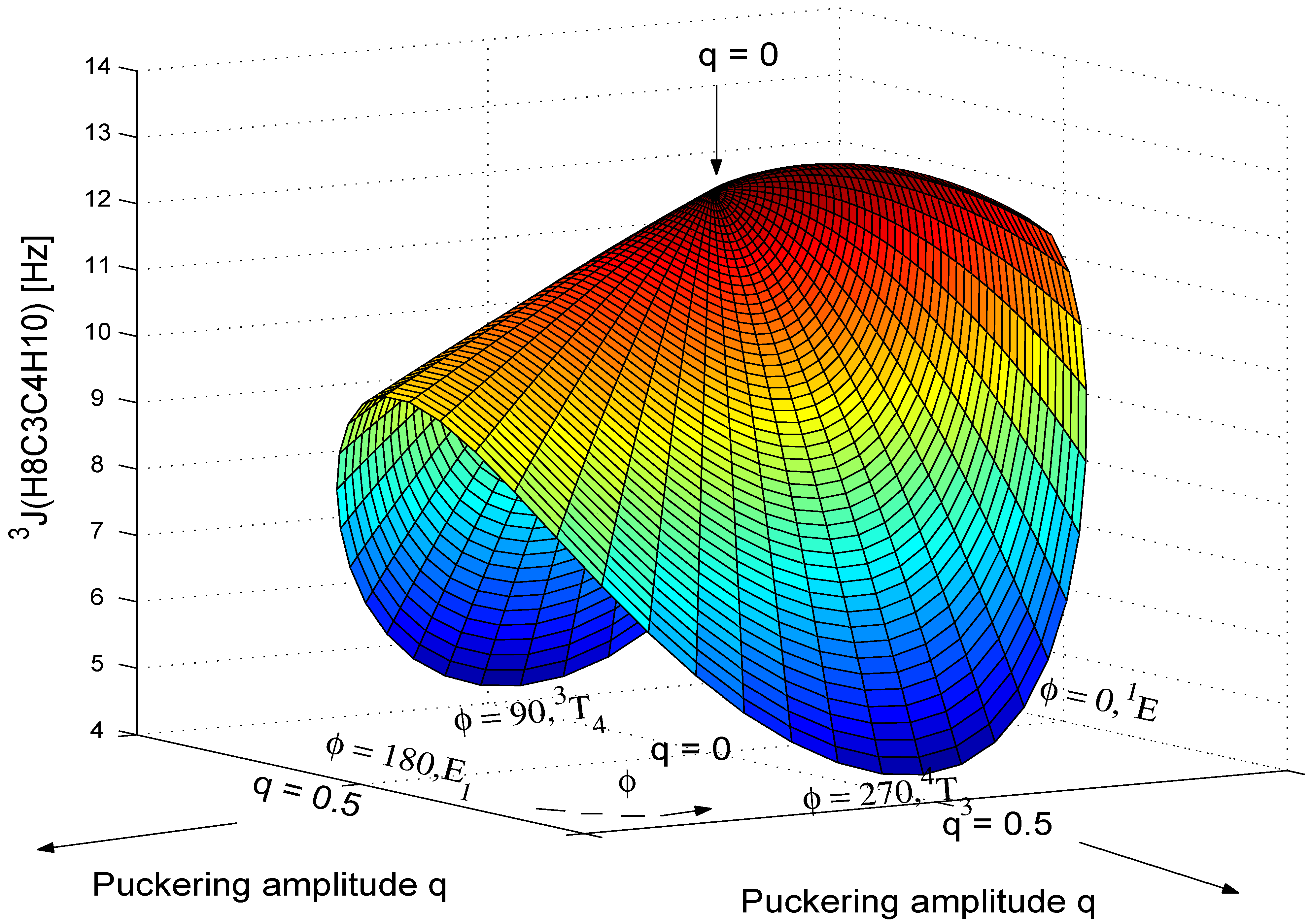
In this section, we discuss the more general Karplus relationship J(q,φ) given in Eq. (7), which leads to a three-dimensional (3D) surface for the SSCC. If function (7) is known, it can be used to describe the conformation of THF and its derivatives more accurately in terms of the mode and the degree of ring puckering.
In the case of THF, the puckering amplitude is largely constant along the pseudorotational path (
Table 1) so that one can discuss the dependence of J on q and
φ separately. For the purpose, of evaluating the q-dependence of the SSCCs, we repeated geometry optimizations at the B3LYP/6-31G(d,p) level of theory for fixed values of q, varying q in the range from 0.33 to 0.41 Å. Subsequent calculation of the SSCCs for the new set of geometries made it possible to derive the coefficients in Eq.s (8a), (8b), and (8c) truncating the power series in q after the quadratic term (see below). Insertion into Eq. (7) led to the general Karplus relationship J(q,
φ). As a representative example, we discuss here results for SSCC
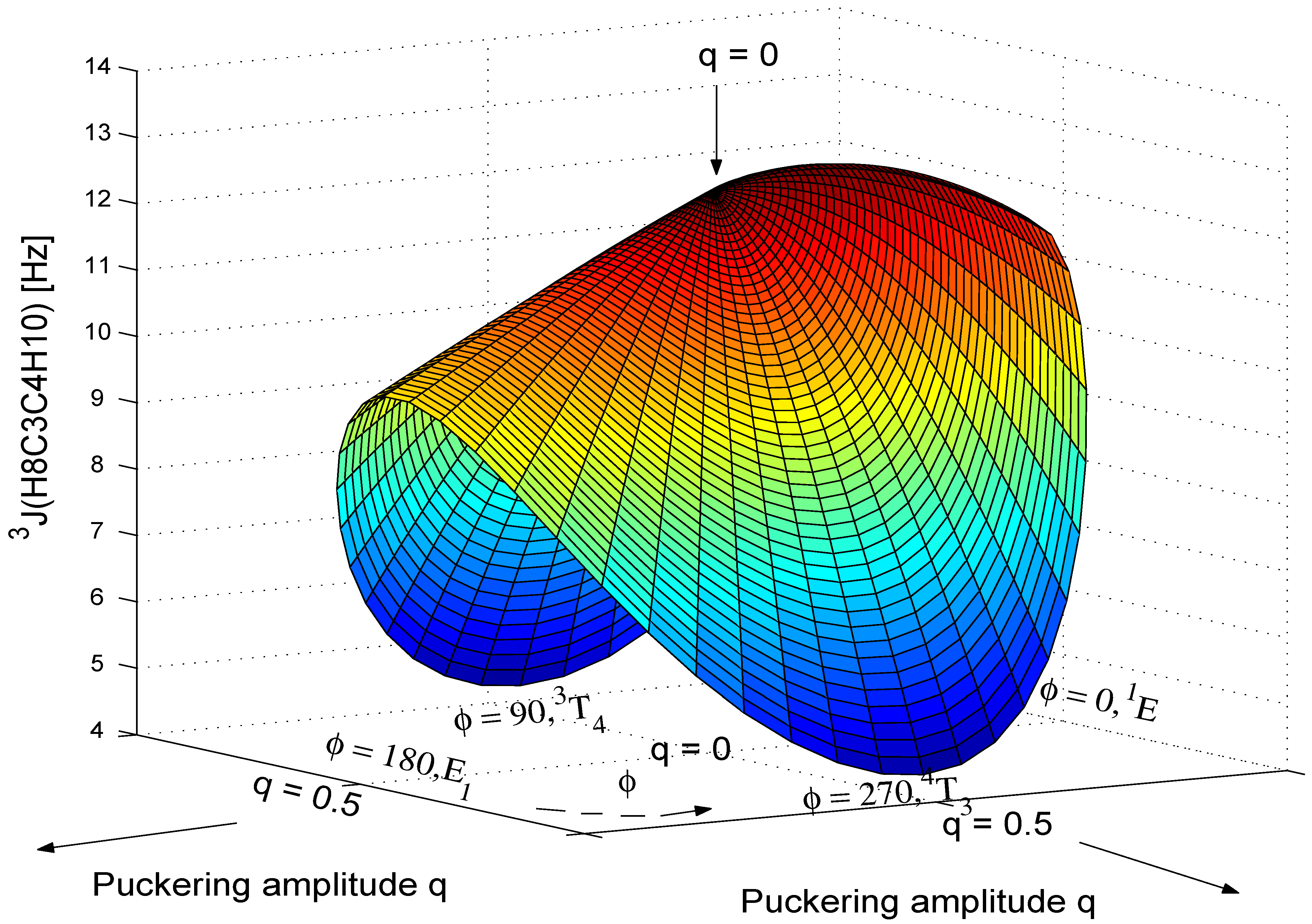
3J(H8C3C4H10,cis).
The Fourier expansion coefficients B
k and C
k are listed in
Table 6 for each q value chosen. The calculated 3D-surface of the
3J(H8C3C4H10,cis) SSCC is shown in
Figure 8.
As stated in section 2, the Fourier coefficient A
0(q) approaches for q
→ 0 the value of the SSCC
3J(H8C3C4H10,cis) for the planar form. All other coefficients become zero for q = 0. The experience obtained in this work suggests that a truncation after the quadratic term is sufficient to give a reliable account of the dependence of the power series coefficients A
0l, B
kl and C
kl on q.
Figure 8 shows that
3J(H8C3C4H10,cis) decreases rapidly with increasing q where the decrease for the
C2-symmetrical T forms is stronger than for the
Cs-symmetrical E forms. It can also be seen that in the latter case the SSCC is always larger for
φ = 0° than for
φ = 180° and that the difference becomes smaller for q approaching 0 while
3J(H8C3C4H10,cis) converges to 12.48 Hz, the SSCC of the planar form. The value of <
3J(H8C3C4H10
,cis) > also exhibits a significant dependence on q, which in turn can be used for the determination of the puckering amplitude q as discussed in Ref. 12.
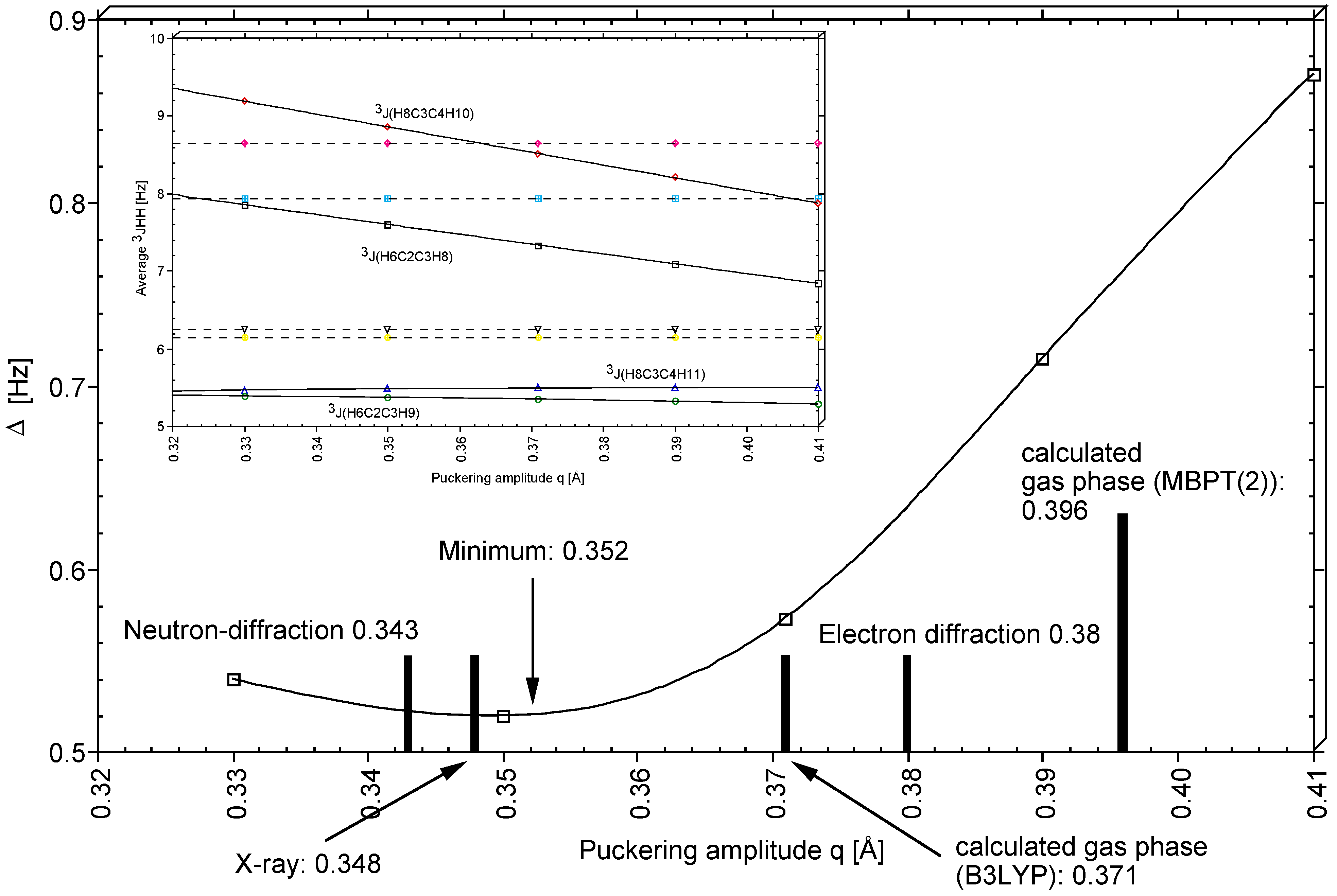
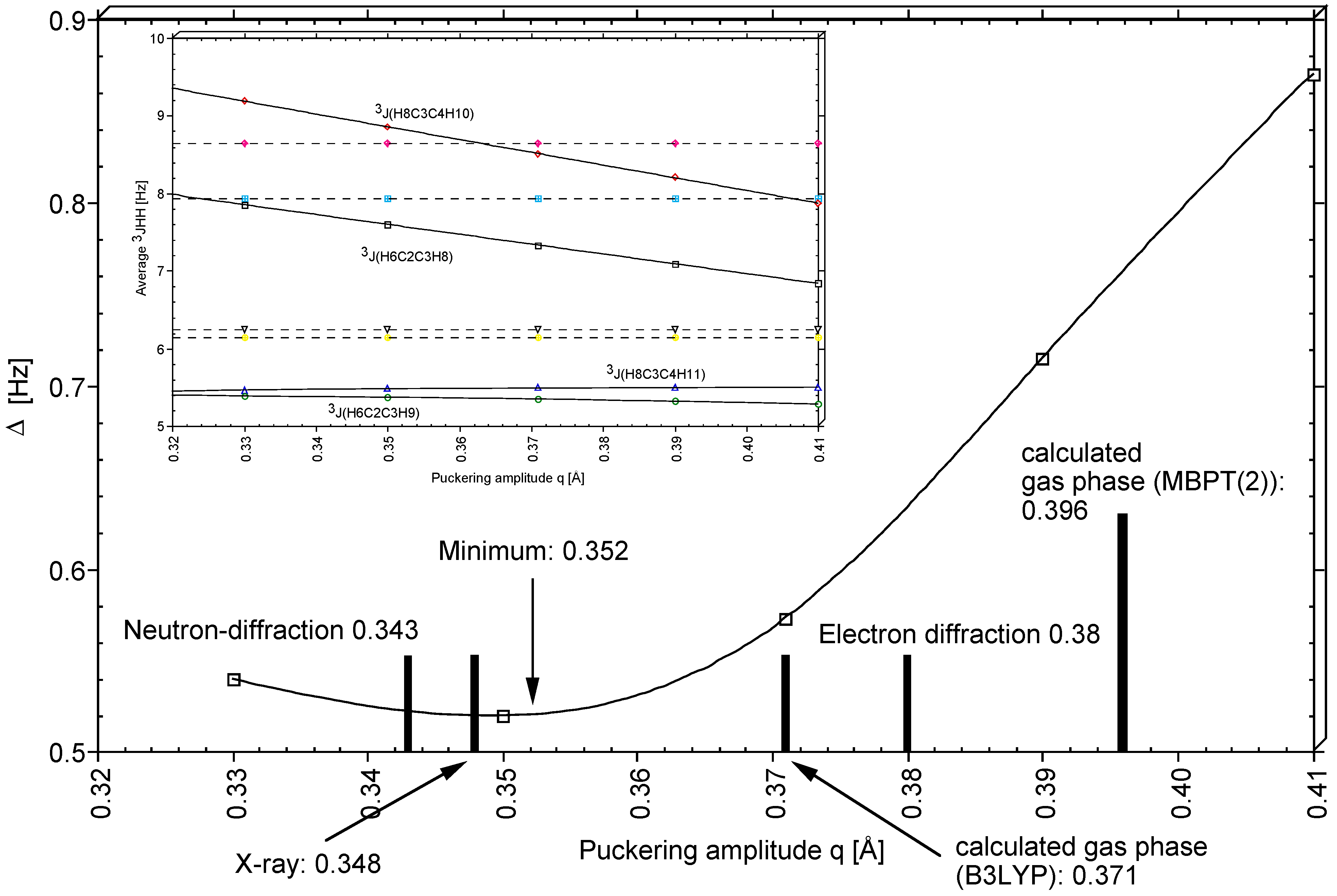
For the vicinal proton,proton SSCCs of THF, general Karplus relationship J(q,
φ) were determined and used in connection with the corresponding experimental values to determine in a least square sense that puckering amplitude q of THF that leads to the best agreement between measured and calculated averaged SSCCs. As shown in
Figure 9, a q-value of 0.352 Å was obtained in this way (predominantly determined by
3J(HCCH,cis), see
Figure 9), which is clearly smaller than the gas phase value of 0.396 Å (
Table 1). The difference of 0.044 Å reflects vibrational and environmental effects on the puckering amplitude q where one has to consider that the vicinal proton,proton SSCCs of THF were measured in CHCl
3 [
29]. While the large amplitude vibration along the pseudorotational path does not change q very much, the second large amplitude vibration perpendicular to the mean plane of the ring decreases q because of the relatively low barrier to inversion (4 kcal/mol,
Table 1). Molecular interactions in condensed phases seem to flatten the ring as was also observed in the X-ray [
23] and the neutron diffraction study [
24] of THF. We note that the q-values obtained in the solid state (0.348 and 0.343 Å;
Table 3) are close to the q-value determined with the help of the vicinal SSCCs. Hence, a realistic puckering amplitude of THF in solution should be close to 0.35 Å.
6 Conclusions
The following conclusions can be drawn from this work.
1) MBPT(2)/cc-pVTZ provides a reliable description of the conformational behavior of THF. THF is a slightly hindered pseudorotor with a constant puckering amplitude of 0.396 Å; the global minima correspond to the C2-symmetrical T forms at φ = 90 and 270°. The pseudorotational and inversion barriers ∆H(298) are 0.22 and 4.15 kcal/mol, respectively, which agree well with the available spectroscopic data.
2) A fourfold pseudorotational potential for THF as suggested by microwave spectroscopic investigations [
20,
21] is not confirmed by the quantum chemical calculations. Analysis of the electronic structure of THF along the pseudorotational path clearly indicates that the T forms at
φ = 90 and 270° are most stable while the E forms at
φ = 0 and 180° occupy the first order saddle points of the CES. We note that the microwave spectroscopic results strongly depend on the constrained THF model used in the investigation and suggest that these investigations should utilize THF models based on the accurate geometries provided in this work.
3) B3LYP/6-31G(d,p) calculations are almost as reliable as MBPT(2)/cc-pVTZ results in the case of THF. This result is in so far important as it provides a basis to investigate pseudorotation of ribose and other biochemically interesting ring compounds at the DFT level of theory. In view of the coupling of this mode with the internal rotation of the substituents of ribose such an investigation would become too expensive at the MBPT(2)/cc-pVTZ level of theory.
4) CPDFT/B3LYP calculations with the [6s,4p,1d/3s,1p] basis set provide SSCCs in good agreement with the available experimental values for THF and the known SSCCs for cyclopentane. The influence of the O atom on SSCCs involving
13C and
1H is in line with observations made for acyclic O-containing compounds [
30].
5) Karplus relationships
nJ(
φ) and
nJ(q,
φ) are established for all 26 SSCCs of THF. Since all internal coordinates of THF are described in the same way, the analysis of the SSCCs in terms of geometrical parameters is straightforward. Also, SSCCs <
nJ > averaged over the pseudorotational mode so that they become directly comparable to measured SSCCs, have been calculated. Magnitude and sign of the <
nJ > values agree well with the corresponding values found for cyclopentane [
12] provided the influence of the O atom is considered.
6) In
Figure 6, those SSCCs are identified that strongly vary during pseudorotation. It has been shown that in particular the vicinal SSCCs, if accurately measured, make it possible to exactly determine the conformation of the THF molecule in solution. A puckering amplitude of 0.352 Å was determined in this way.
Work is in progress to develop a procedure that utilizes the Karplus relationships established in this work to determine the conformation of substituted THF compounds with the help of measured SSCCs. This method and the results summarized in this article provide an excellent platform for analyzing the conformational behavior of biochemically interesting molecules such as ribose, 2′-deoxyribose, proline, etc.




 and
and  are expressed as
are expressed as










 and
and  of the internal coordinates of THF are listed. In view of the low energy barriers to pseudorotation, the constant term
of the internal coordinates of THF are listed. In view of the low energy barriers to pseudorotation, the constant term  of the Fourier expansion is close to the average value < P > obtained when using Eq. (12) in connection with (10) and considering the correct Boltzmann statistics (11) for the pseudorotational itinerary (compare with Table 3).
of the Fourier expansion is close to the average value < P > obtained when using Eq. (12) in connection with (10) and considering the correct Boltzmann statistics (11) for the pseudorotational itinerary (compare with Table 3).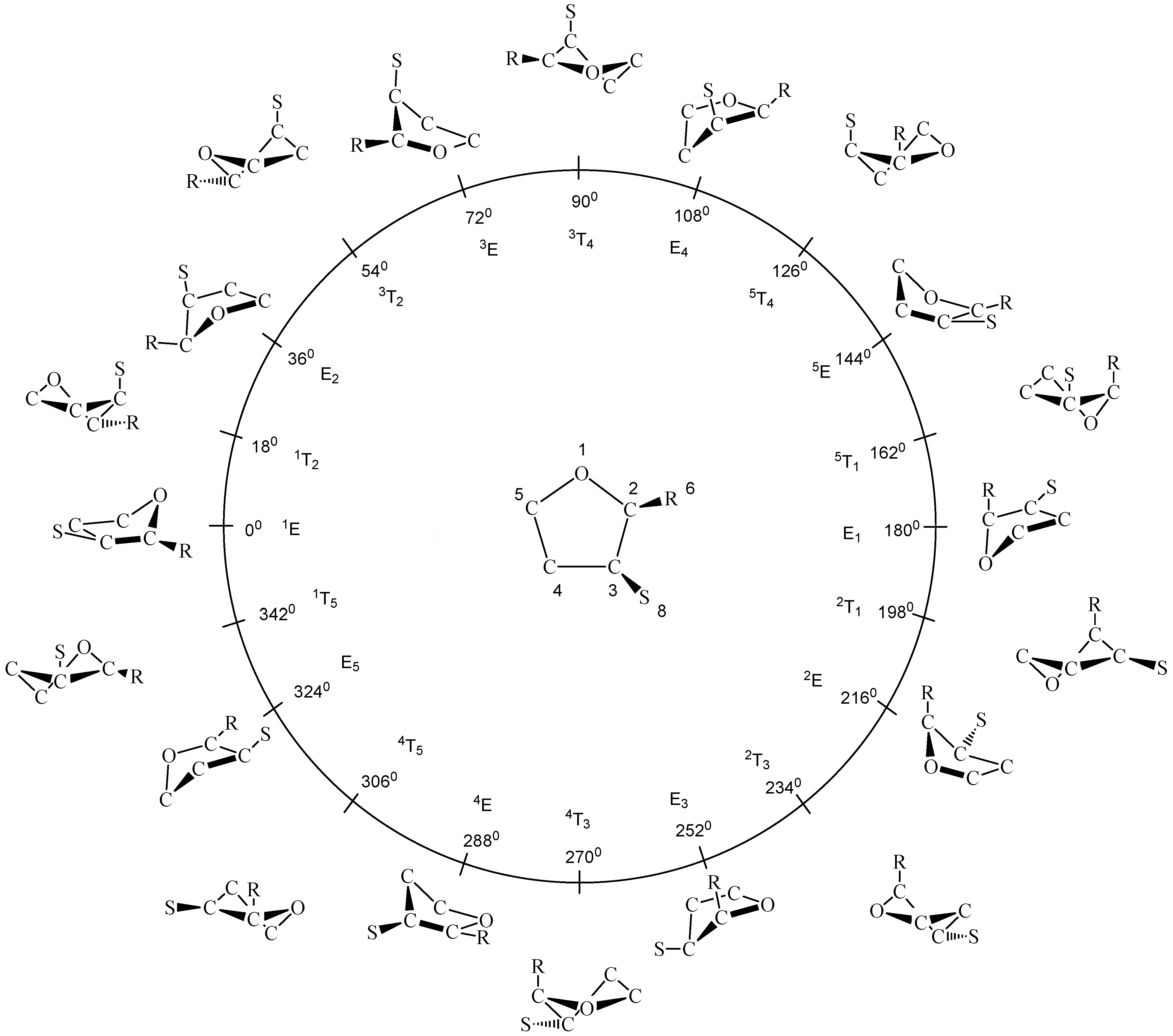



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}