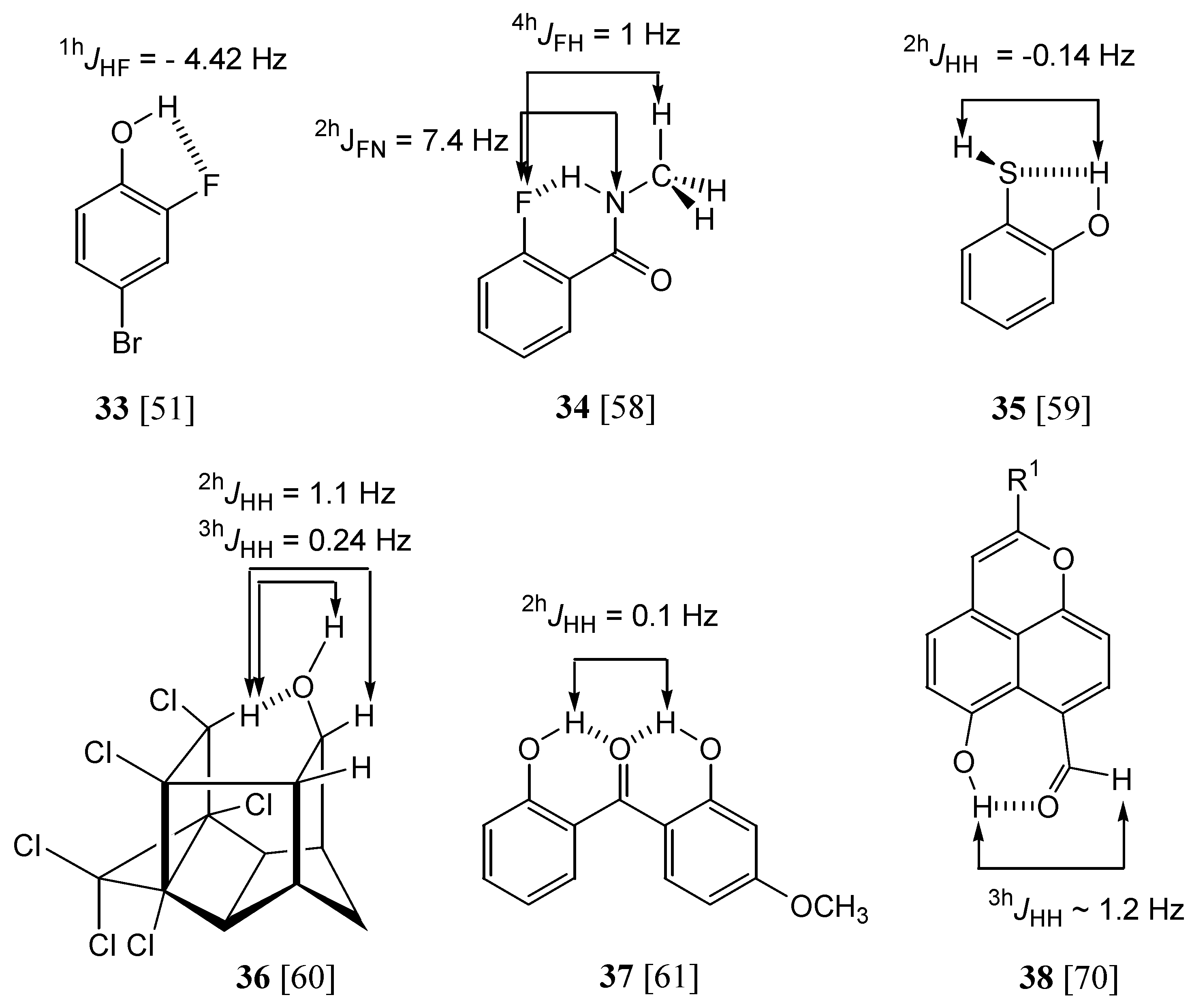
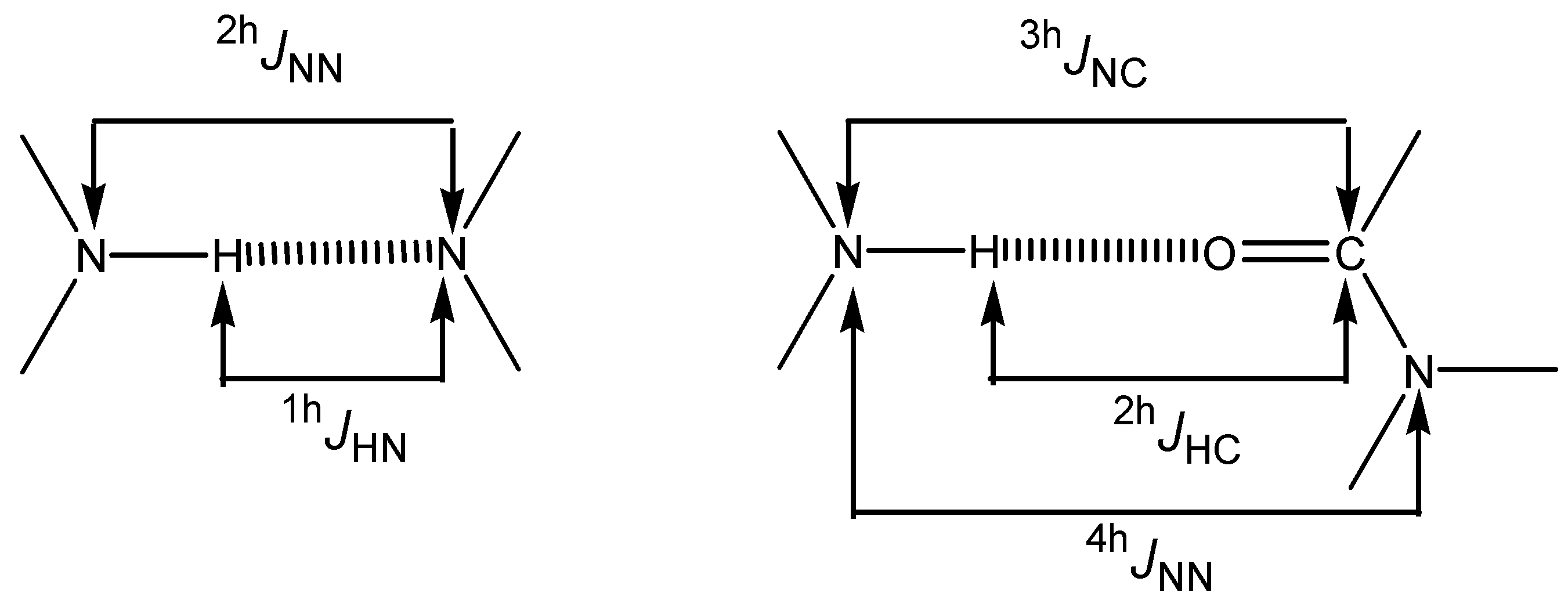
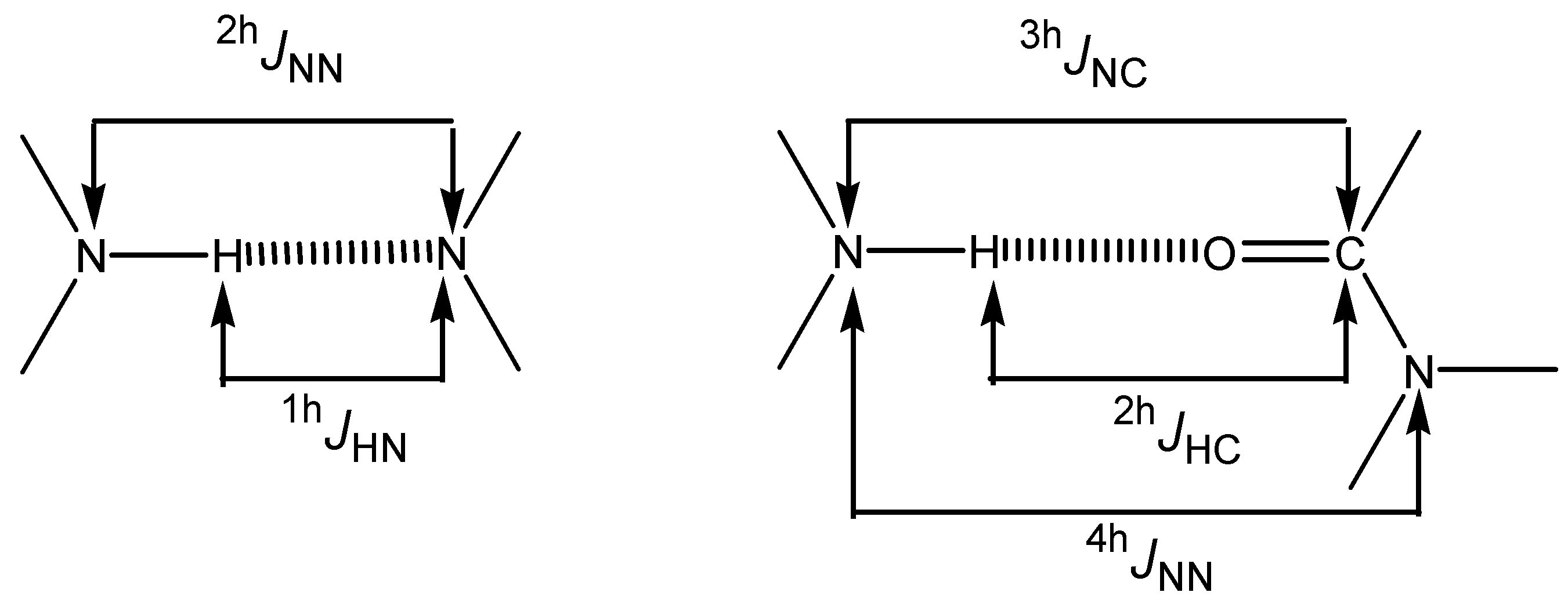
Hydrogen bonded molecules: couplings through a hydrogen bond
The hydrogen-bond-induced changes of the intramolecular spin-spin coupling constants will not be discussed systematically (for instance,
1JNH before and after the NH group was involved in an HB). Any hydrogen bond (HB) of the A–H···B type can adopt three limit situations (we will use IMHB for intramolecular hydrogen bonds): classical, shared and proton transfer. In cases were the proton is shared between A and B the distinction between
1JAH and
1hJB···H disappears; finally, when the proton is completely transferred, the situation again becomes "classical", with
1JBH and
1hJA···H.
Traditional AH···B HBs have normal A···B distances, A–H slightly elongated (relative to the monomer); similarly the ion-pair, B+–H···A, have A···B distances comparable to the preceding case, B+–H slightly elongated (relative to the isolated B+–H cation). The proton-shared structure is characterized by a short A···B distance and an elongation of the A···H and B···H distances.
The contribution of the Bartlett, Perera and Del Bene's group to this topic is so significant that we have decided to divide this section in three parts: a) Other groups [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24]; b) Bartlett-Perera-Del Bene [
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36]; c) Data analysis [
37,
38,
39,
40,
41].
a) Other groups. Three publications by Pecul, Sadlej and Leszczynski are noteworthy for the quality of the calculations. In the first one, Pecul and Sadlej [
9] described their calculation on water dimer. In the first part they use the conventional case of water monomer (
Table 1) to compare different approaches to the calculation of spin-spin coupling constants (they were dominated by the Fermi contact and paramagnetic spin-orbital terms).
Table 1.
The nuclear spin-spin coupling constants (Hz) in the water monomer
Table 1.
The nuclear spin-spin coupling constants (Hz) in the water monomer
| | | 1JOH | 2JHH |
|---|
| Experimental | | | |
| | cyclohexane | –78.7 | — |
| | nitromethane | –80.6 | –7.3 |
| Calculated | | | |
| | MCSCF (RAS4/HIII) | –77.2 | –12.6 |
| | MCSCF (CAS/HIIIa) | –75.2 | –9.3 |
| | EOM-CCSDa | –74.9 | –10.8 |
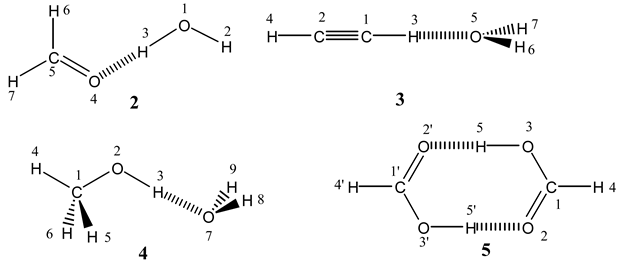
These results show that MCSCF (included in the DALTON program) when used with a basis set of very high quality are comparable to EOM-CCSD calculations. For the water dimer 1, at the RAS level, they found 2hJO1-O2 = 1.6, 1hJO1-H5 = 4.4, 3hJO1-H6 = 0.5, 3hJO2-H3 = 0.2, 2hJH3-H5 = 0.3 and 4hJH3-H6 = –0.08 Hz.
Pecul, Leszczynski and Sadlej published two papers on scalar coupling constants in hydrogen bonded systems. In the first one [
10], they studied X-H···O complexes [CH
2O–H
2O
2, C
2H
2–H
2O
3, CH
3OH–H
2O
4 and (HCOOH)
2 5]. The calculations were carried out at the MCSCF level and the main conclusions are:
1hJOH correlates with the strength of the HB (expressed in terms of the interaction energy); the intermolecular
1hJOH couplings are determined primarily by the FC term (these couplings are similar to those through covalent bonds, what distinguishes them is their magnitude and, more surprisingly, their sign); the
2hJXY couplings through the X-H···Y HB are also dominated by the FC term. In the formic acid dimer
5, the
2hJOO coupling attains 7.1 Hz.
The second paper [
11] deals with the important case of N-H···O=C (this last atom is designed C' in most publications following the common use in peptides) and N-H···N=C systems. Thus, formamide dimer (Fa-Fa)
6 and formamide-formamidine dimer (Fa-Fi)
7 have been studied at the MCSCF level. Complex
7 should present a large
2hJNN coupling of 8.3 Hz. Another interesting conclusion is that
2hJCH couplings are positive for N–H···N=C bonds and negative for N–H···O=C bonds. The authors have examined the variation of the couplings with the ON distance, for instance, there is a fast exponential decay of
2hJNN.
Scheurer and Brüschweiler [
12] have studied the
N-methylacetamide dimer
8 as a model of the trans-hydrogen
3hJNC' coupling involved in polypeptide N–H···O=C' hydrogen bonds (trans here does not mean a
trans disposition but that the coupling occurs through the HB). Using the DFT-based methodology of Malkin, Malkina and Salahub (SOS-DFTP), they explored the double dependence of
3hJNC' with the NO distance and the out-of-plane tilt angle and they showed that there exists a linear correlation between
3hJNC' and the isotropic H
N chemical shift. They computed also the
2hJNN couplings in guanine/cytosine (5.36 Hz), uracil/adenine (7.65 Hz) and thymine/adenine pairs (6.45 Hz).
A second paper by Czernek and Brüschweiler [
13] is related to the experimental observation of
2hJ(
31P-
1H) and
3hJ(
31P-
15N) in protein-nucleotide complexes (see Appendix,
Table 7). Using the same methodological approach, they calculated two complex fragments
9 and
10 with different N–H···O=P geometries.
Fragment
9 is a model of Gly15 together with Mg[PO
4(CH
3)] and fragment
10 is a model of Ala18 together with [PO
4(CH
3)
2]
–. They devised a two-dimensional surface of the form:
Each value of nhJPX (for a pair of N···O distance and P–O···H angle) depends on a series of parameters that have been calculated. For instance, a0 = –9628.6 Hz, a1 = 2.7232 Å–1, a2 = 2.4761, αoffset = 3.1853 rad for 3hJPN. They proposed to use equation (2) in a similar way to the Karplus equation.
It will be interesting to know whether the type of equation known as the Karplus equation is followed in supramolecular complexes. For instance, if the dependence of
3JFF in compound
11 has a counterpart in
4hJFF in complex
12.
Barfield's contribution is important having published four papers between 1999 and 2002 [
14,
15,
16,
17,
18]. In his first paper, published with Grzesiek, two DNA triplets
13 (T
·A-T) and
14 (C
+·G–C) were studied theoretically (DFT-FTP//UB3PW91/6-311G**) to be compared with their experimental results (
Table 7 of the Appendix) [
14,
15]. In these systems, two trans-hydrogen bond
2hJNN and
1hJHN scalar couplings were calculated. Interesting correlations were found between the chemical shift of the proton of the HB and
1hJHN (positive),
2hJNN (positive) and
1JNH (negative), both between calculated values (Eqs. 3 and 4) and between experimental values (Eq. 5).
Barfield, O'Leary
et al. measured (see
Table 7, Appendix)
2hJHH couplings in 1,3 and 1,4-diols of favorable conformation and computed (DFT/FTP) these couplings for different geometries of the fragment O–H···O–H with acceptable results [
16,
17]. In his last publication, Barfield addressed the problem of proteins, and specifically, the notorious
3hJNC' coupling [
18]. He used as a model, formamide dimer, but instead of a cyclic dimer, like
6, he used a more realistic open-dimer
15.
He found that
3hJNC' depends on
rOH, θ
1, θ
2 and the torsion angle ρ about the C=O bond (H···O=C'–N). Several equations were devised in this very important publication. Those relating
3hJNC' with cos
2ρ bear a clear similarity with the Karplus relationship.
The authors of one of the most widely used methods for calculating coupling constants, Malkin and Malkina, collaborated with Limbach in the study of the experimental findings of these last authors (reported in
Table 7). Thanks to experiments at very low temperatures (between 110 and 150 K) using freons as solvents, Golubev, Limbach and their coworkers measured
1H-
19F coupling constants in fluoride ion/hydrogen fluoride complexes (see
Scheme 1) [
19].
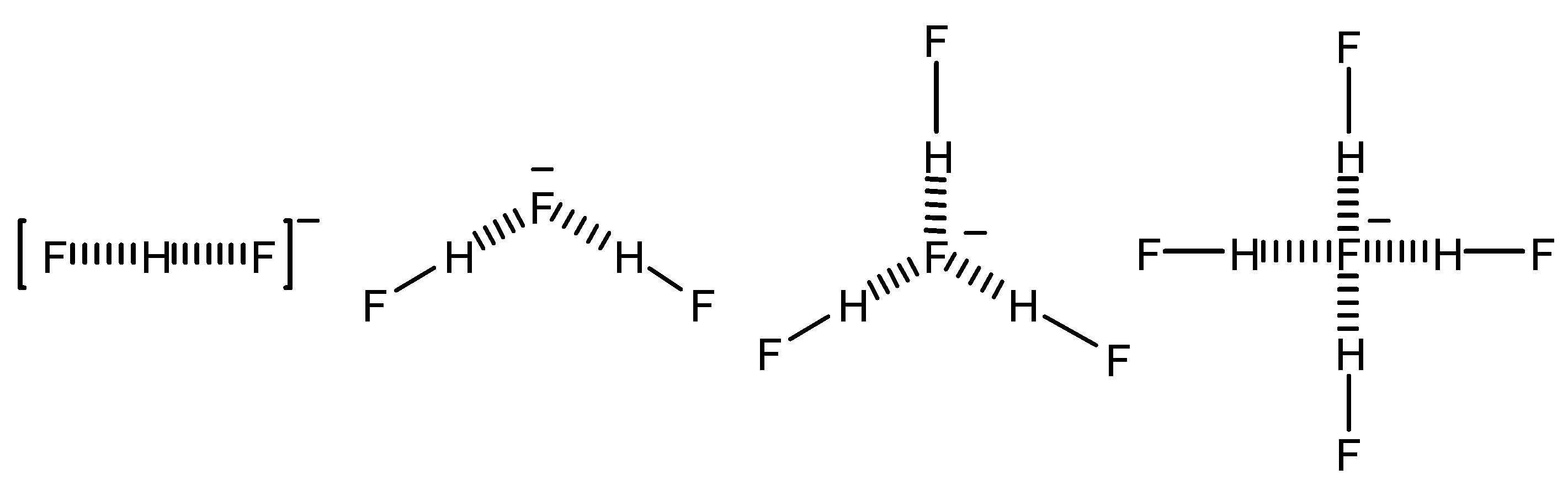
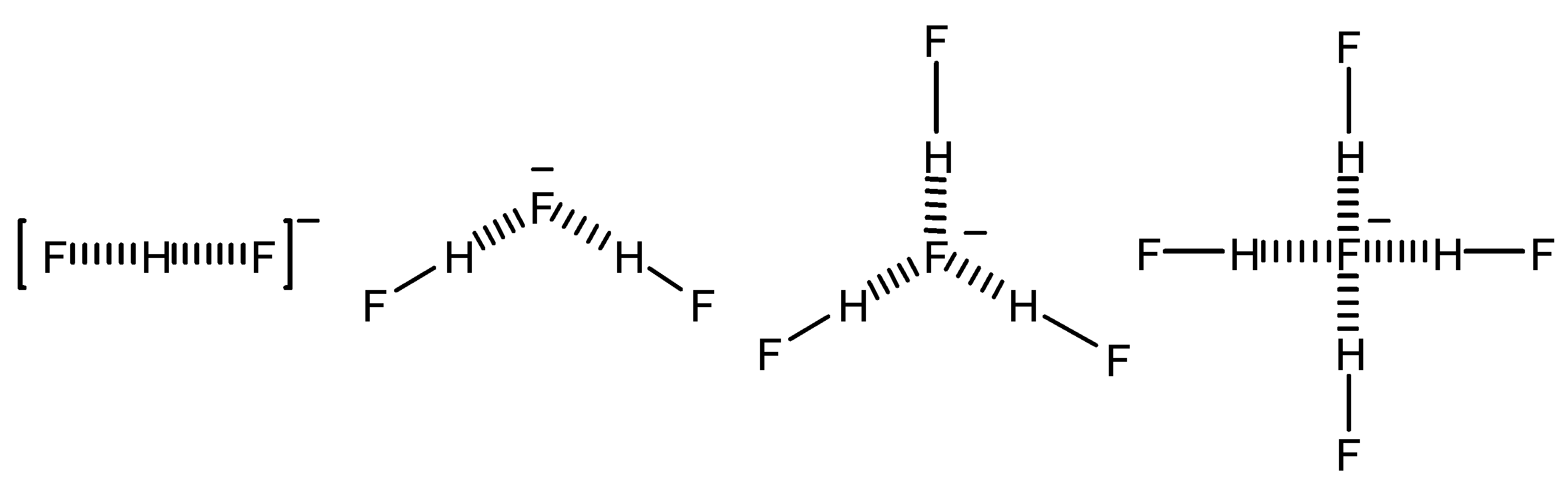
Scheme 1.
Species formed by mixing HF and tetrabutylammonium fluoride
Scheme 1.
Species formed by mixing HF and tetrabutylammonium fluoride
They measured or estimated all the possible coupling constants that were also calculated at the MCLR-CAS level [
19]. This collection of data is still remarkable for the wealth of information it contains being completely apart from peptides, nucleic acids or their combination.
The second publication is of a more theoretical nature [
20]. The authors used the famous Steiner-Limbach diagrams, where geometrical (such as q
2) and NMR properties (such as δ
1H of the H in the HB,
nJ and
nhJ) were represented against q
1:
The cases of [F(HF)
n]
–,
n = 1-4, discussed above, of [C≡N…N≡;C]
–, formamidine dimer
16 were calculated. For instance, we report below the calculated value (in Hz) of
2hJNN of
16.
Although not reporting coupling constants, a third paper by Limbach is worth quoting because it describes the influence of an electric field on the geometry and properties of A–H···B complexes [
21].
Bagno has examined the case of formamide dimer
15 in relation with the case of ubiquitin (see
Table 7) [
22]. He has built up the coupling surface of
3hJNC' in function of the N···O distance and the dihedral angle. His conclusions, based on FTP/DFT calculations are similar to those of Barfield [
18], which were obtained two years later.
Finally, we have calculated the coupling constants of structures
17-
19 in an attempt to determine whether the coupling of 1.5 Hz between
15N and
31P (not directly bonded) is transmitted through the skeleton (
4JNP) or through the HB (
3hJNP) [
23]. Only the Fermi contact term was calculated using the FTP/B3LYP/aug-cc-pVTZ approach. The conclusion is that the main transmission pathway is through the covalent bonds.
Bryce and Wasylishen have modeled the
2hJNN in nucleic acid base pairs using methylene-imine dimer
20 in function of hydrogen bond geometry (
rNN and θ) [
24]. The variation of
2hJNN as a function of
rNN is fit to an exponential decay [
rNN = 3.36 – 0.37 ln (
2hJNN)]. The influence of θ is less pronounced.
b) Bartlett, Perera, Del Bene's contribution [
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36].
Remember that all papers by this group are based on high-level EOM-CCSD calculations. Perera and Bartlett [
25] initiated their systematic exploration of scalar couplings through hydrogen-bonds inspired by Limbach-Malkina's paper on [FHF]
– and [F(HF)
n]
– (
n = 1-4) [
20]. These authors criticize the use of DFT methods when dealing with fluorine atoms "The MCLR results reported for [FHF]
– by Limbach et al. are unrealistic". The estimated value for
2hJFF is > 200 Hz, the MCLR calculated value is –133 Hz and the EOM-CCSD value is + 225 Hz. We will see in the following discussion that
19F NMR properties are amongst the most difficult to calculate.
In the second publication of the same authors with Del Bene [
26], they examined the case of the "low-barrier" hydrogen bonds, a topic of general interest for its biological implications, trying to find out whether predicted NMR coupling constants across hydrogen bonds can be used as fingerprints for specifying hydrogen bond types. They calculated a certain number of
17O–
17O,
15N–
15N and
15N–
17O systems (
Table 2). Amongst their main conclusions are that for these nuclei, the FC term dominates, that the relationship between
2hJOO (Hz) and
dOO (Å) shows an exponential decay, and that
2hJNN for
dNN = 2.9 Å is 7.2 Hz close to the experimental findings (see
Table 7).
Table 2.
NMR total and FC coupling constants (Hz) for A–H–B systems
Table 2.
NMR total and FC coupling constants (Hz) for A–H–B systems
| Complex | AB | HB typea | dAB (Å) | Fermi-contact (Hz) | Jtotal (Hz) |
|---|
| O2H5+ | OO | PS | 2.38 | 39.92 | 39.54 |
| O2H3– | OO | PS | 2.44 | 16.28 | 17.96 |
| O2H4 | OO | T | 2.91 | 1.47 | 1.29 |
| +H2OH:NCH | ON | PS | 2.47 | 34.07 | 34.12 |
| HOH:NC– | ON | T | 2.82 | 6.62 | 6.55 |
| CNH:OH2 | NO | T | 2.84 | 8.57 | 8.51 |
| HOH:NCH | ON | T | 3.13 | 1.14 | 1.16 |
| +HCNH:NCH | NN | PS | 2.52 | 32.46 | 32.52 |
| CNH:NC– | NN | PS | 2.58 | 21.47 | 21.52 |
| CNH:NCH | NN | T | 3.00 | 5.60 | 5.62 |
Then, Del Bene and Jordan examined the case of the complex between hydrogen chloride and ammonia that Del Bene had already studied in what concerns its geometry, energy and vibrational aspects [
27]. To the Cl–H···NH
3 complex they applied an external electric field along the HB to modify the character of the HB, an approach already used by Limbach and ourselves [
21]. When the field increases, the system moves from traditional, to shared, to ion-pair. We report their most significant results in
Table 3. They criticized the use by Limbach and ourselves of the q coordinates concluding that they are only useful for symmetric systems (which is not correct), that the FC term dominates and showing that
2hJ15N-35Cl is negative.
Table 3.
Equilibrium Cl···N distances, proton chemical shifts and 2hJ15N-35Cl coupling constants as functions of external field strength
Table 3.
Equilibrium Cl···N distances, proton chemical shifts and 2hJ15N-35Cl coupling constants as functions of external field strength
| Field (a.u.) | dCl···N (Å) | δ (ppm) | 2hJ15N-35Cl (Hz) |
|---|
| 0.0000 | 3.080 | 10.0 | –5.7 |
| 0.0010 | 3.056 | 10.8 | –6.1 |
| 0.0025 | 3.019 | 12.0 | –6.9 |
| 0.0040 | 2.975 | 13.6 | –8.0 |
| 0.0055 | 2.832 | 20.9 | –11.8 |
| 0.0100 | 2.896 | 19.1 | –9.1 |
| 0.0150 | 3.004 | 16.4 | –6.7 |
Contrary to most authors, they prefer graphical representations to statistical models. Their graphs clearly showed that their seven data belong to two families, the first set corresponds to 0.0000-0.0040 a.u. fields and the second one to 0.0055-0.0150 a.u. fields. These sets can be adjusted to linear correlations, although they probably correspond to the two branches of an exponential [
21] (eqs. 8-13).
First set (traditional Cl–H···N HB):
dN···Cl = (3.082±0.002) – (26.2±0.9) field, n = 4, r2 = 0.998 (8)
δ1H = (8.93±0.11) + (893±47) field, n = 4, r2 = 0.994 (9)
2hJCl···N = – (5.60±0.11) – (576±49) field, n = 4, r2 = 0.986 (10)
Second set (ion-pair Cl–···H–N+ HB):
dN···Cl = (2.73±0.02) + (18±2) field, n = 3, r2 = 0.987 (11)
δ1H = (23.6±0.4) – (475±40) field, n = 3, r2 = 0.993
2hJCl···N = – (14.6±0.3) + (536±35) field, n = 3, r2 = 0.996 (13)
The proton shared situation is very unstable [
21] and should be found in the 0.0040-0.0055 au region.
The next contribution by Del Bene and Bartlett was devoted to the
2hJNN coupling present in some
15N–H···
15N fragment [
28] (see
Table 7). Here again, the FC term dominates, other terms being negligible. Their results are reported in
Table 4 (note that is
2hJNN positive).
Table 4.
Equilibrium N···N distances, symmetry and 2hJ15N-15N coupling constants
Table 4.
Equilibrium N···N distances, symmetry and 2hJ15N-15N coupling constants
| Complex | symmetry | dN···N (Å) | 2hJ15N-15N (Hz) |
|---|
| pyrrole:NCH (21) | C2v | 3.16 | 3.0 |
| CNH:NCH (22) | C∞v | 3.00 | 5.5 |
| CNH:NH3 | C3v | 2.85 | 8.7 |
| CNH:NCLi | C∞v | 2.83 | 9.6 |
| CNH:pyridine (23) | C2v | 2.79 | 10.7 |
Reference [
29] reports the calculations carried out on the problem of a long range coupling constant present in iminophosphorane-substituted proton-sponges
24 (see
Table 7). Using as a model structure
25, the results reported in
Table 5 were found (only the FC term was computed).
Table 5.
N···N and P···P distances and computed 4hJ31P-31P coupling constants in model 25
Table 5.
N···N and P···P distances and computed 4hJ31P-31P coupling constants in model 25
| N···N distance (Å) | P···P distance (Å) | 4hJ31P-31P (Hz) |
|---|
| 2.53 | 4.79 | 14.3 |
| 2.83 | 5.09 | 8.1 |
| 3.03 | 5.29 | 5.9 |
| 3.33 | 5.59 | 3.9 |
| 3.53 | 5.79 | 3.0 |
These data can be analyzed statistically leading to eqs. (14) and (15):
Del Bene, Parera and Bartlett used in another contribution the reduced coupling constants
K to correct some erroneous statements about the relative importance of scalar couplings [
30]; for the N–H
+–N and O–H
+–O complexes
2hKX···X >
1KX–H >
1hKX···X. Jordan, Del Bene
et al. devoted another paper to the N–H···N system [
31]. They used the pyrrole:hydrogen cyanide (
21) and the hydrogen isocyanide:hydrogen cyanide (
22) systems and, as perturbation, an electric field or chemical substitution (fluorine and Be
2+ on the pyrrole, Li, Na, S
– and O
– instead of H in NCH). Some of their results are reported in
Table 6.
Table 6.
Chemical shifts of the hydrogen-bonded proton and 2hJ15N-15N coupling constants as a function of field strength
Table 6.
Chemical shifts of the hydrogen-bonded proton and 2hJ15N-15N coupling constants as a function of field strength
| Field (a.u | δ1H (ppm) | 2hJ15N-15N (Hz) |
| 0.0000 | 1.9 | 6.4 |
| 0.0040 | 2.4 | 7.3 |
| 0.0100 | 3.2 | 8.8 |
| 0.0150 | 4.1 | 10.3 |
| 0.0200 | 5.2 | 12.3 |
| 0.0225 | 6.0 | 13.7 |
| 0.0250 | 7.1 | 15.5 |
In all cases, proton-shared and ion-pair N
a–H–N
b HBs are unlikely to be formed in neutral complexes. These authors introduced an important representation to understand the nature and properties of hydrogen-bonded systems, plotting N
a–H (Å) against N
b–H (Å) and representing the square of the ground-state vibrational wave function superimposed on the potential energy surface.
The next problem they tackled concerns the hydrogen chloride:pyridine complex (
26) [
32]. Pyridine is a very good HBA because the positive charge can be delocalized over the ring. In this example, increasing the field, they succeeded to attain the proton-shared and afterwards the ion-pair structures. They carried out the calculations using an isotropic external electric field (analogous to the Onsager's model), optimizing the structure for each field (under some restrictions) and then calculating the coupling constants with the field turned off (turning on the external field during the calculations increases the absolute value of
2hJ35Cl···15N). Their calculations are in agreement with Limbach's results on temperature effects o HBs: decreasing the temperature corresponds to an increase of the field and the traditional system evolves to proton-shared and then to ion pair.
Del Bene, Jordan, Parera and Bartlett [
33] returned to the problem of
2hJFF in [FHF]
– and [FDF]
– that was their first publication on scalar couplings across HBs [
25]. They examined the possible effects of zero-point vibrational effects and of vibrational averaging, to conclude that both have essentially no effect on
2hJFF. They confirmed that, although the FC term dominates, the PSO term is also important. Using Limbach's estimation of 220 Hz for
2hJFF in [FHF]
– for comparison, they reported for [FHF]
– 212.7 Hz and for [FDF]
– 223.1 Hz. This is an important result that allows a new entrance to isotope effects.
Two papers [
34,
35] can be consulted to get an overview of the preceding series of papers. One dealing with
15N,
15N spin-spin coupling constants across N–H–N and N–H
+–N hydrogen bonds and entitled "Can coupling constants provide reliable estimates of N–N distances in biomolecules?" (with a positive answer both for the distance and the angular variations) and the other with "what a difference a decade makes: progress in
ab initio studies of the hydrogen bond".
The last paper until now reports calculations of
3hJ15N-31P spin-spin coupling constants across N-H···O-P hydrogen bonded complexes [
36]. This work is similar in subject but different in methodology to that of Czernek and Brüschweiler [
12]. Instead of complex structures
9 and
10, equation-of-motion coupled cluster singles and doubles (EOM-CCSD) calculations were performed in model cationic and anionic complexes including NH
4+:OPH, NH
4+:OPH
3, NH
3:
–O
2PH
2, NFH
2:
–O
2PH
2, and NF
2H:
–O
2PH
2. Three-bond coupling constants can be appreciable when the phosphorous is P(V), but are negligible with P(III).
3hJN-P values in complexes with cyclic or open structures are less than 1 Hz, a consequence of the nonlinear arrangement of N, H, O, and P atoms. For complexes with these structures,
3hJN-P may not be experimentally measurable. In contrast, complexes in which the N, H, O, and P atoms are collinear or nearly collinear have larger values of
3hJN-P, even though the N-P distances are longer than N-P distances in cyclic and open structures. In linear complexes,
3hJN-P is dominated by the Fermi-contact term, which is distance dependent. Therefore, N-P (and hydrogen-bonding N-O) distances in these complexes can be determined from experimentally measured
15N-
31P coupling constants.
c) Data analysis.
Peralta, Contreras
et al. analyzed using NBOs (Natural Bonding Orbitals) the C-H···O interaction in NCH···OH
2 and NCH···O=CH
2 27 and their effect on nuclear magnetic shielding constants [
37]. In a subsequent paper, they expanded the same methodology to the dissection of fluorine–fluorine couplings (see below,
31). They proposed a partition of the Fermi contact term as a sum of contributions of core orbitals, non-bonding electron pairs and bonding orbitals. As expected the coupling constant through the space are governed by the non-bonding electron pair term in all the cases studied [
38].
Weinhold, the creator of NBO analysis, applied his method first to an example of covalent bonds (
3JHH couplings in ethane) [
39] and then to
1hJNH and
2hJNN in DNA A-T base pair [
3]. The contributions of the orbitals involved in the HB to the coupling constants are able to explain the differences observed between the
2hJNN and
1hJNH [
3]. Arnold, Oldfield
et al. used DFT theory to calculate and analyze through-space
19F-
19F coupling constants [
40] and found an exponential relationship between the coupling constant value and the internuclear distance. The same authors compared the
nhJNC' in proteins with the electron density properties obtained using Bader’s Theory of Atoms in Molecules (AIM) [
41]. They found an exponential relationship between the mutual penetration of the electronic clouds and the coupling constant values.





















{kind=link}
{kind=link}
{kind=link}
{kind=link}