Introduction
The question about the source of a very high activity of metal sites produced by cation exchange in a silicalite environment of framework oxygens is the important area of the research in zeolite community. One of the main points of interest is the Cu
+ centre in Cu
+ZSM-5, known as a good catalyst for decomposition of nitrogen oxides [
1], classified among the most dangerous pollutants. Another interesting property of Cu
+ in ZSM-5 is the possibility of bonding N
2 at room temperature [
2]. In spite of numerous efforts, however, little is still known about the nature of the Cu
+ site. Experimental difficulties (e.g. in XRD) stem from low content of framework aluminum and hence very low concentration of copper sites.
Cu
+ sites are produced from Cu
2+ exchanged cations by the process of self-reduction during dehydration and a number of hypotheses have been proposed as to the structure and coordination of both copper forms [
3,
4]. In addition, the interaction with NO molecule also triggers out reduction-oxidation processes leading to the modification of the site structure and properties. Experimental techniques can reach only selected properties: while Cu
2+ is EPR visible centre, the reduced form cannot be followed by this tool; on the contrary, the complex with NO is EPR silent for the Cu
2+ form. XRD which is useful for Al rich zeolites has several limitations in the case of high siliceous zeolites because of low cation content and low symmetry of zeolite framework [
5]. Nevertheless, the consensus has already been met that there exist several positions of Cu cation depending on the oxidation state and the interaction with an adsorbate [
6,
7,
8]. The most probable coordination for Cu
2+ is the fourfold bonded, approximately planar square structure, while Cu
+ prefers lower coordination between 2 and 3 exhibiting lower symmetry. Cu
+ are supposed to be compensated by a single framework aluminum atom while two framework aluminums are required to compensate a bare Cu
2+ [
9]. In consequence many prospective sites hosting exchanged Cu cations in ZSM-5 framework have been proposed. It has already been suggested, e.g. from photoluminescence studies that among many possibilities accessible for Cu
+ in ZSM-5 two positions: α and β are prevailing while one of them (α) is suggested to be catalytically active [
10]. In view of the limited experimental information available on Cu binding sites molecular modelling of their properties is burdened with high degree of arbitrariness and uncertainty and always needs additional support and justification by parallel experimental measurements.
IR spectroscopy provides another insight into the nature of copper sites in ZSM-5: it is the best probe of modifications undergone by the adsorbed molecule and, indirectly, gives some information on the influence of the presence of exchanged cation and the adsorbed molecule onto the site structure and framework properties. As already known, such information may be extracted both from the analysis of NO stretch and of T-O-T skeletal vibration [
11,
12,
13,
14,
15]. In this paper, on the basis of our theoretical investigations related to experimentally available information, we discuss the problem of speciation of Cu ions in MFI framework and its migration in the course of a catalytic process.
Results
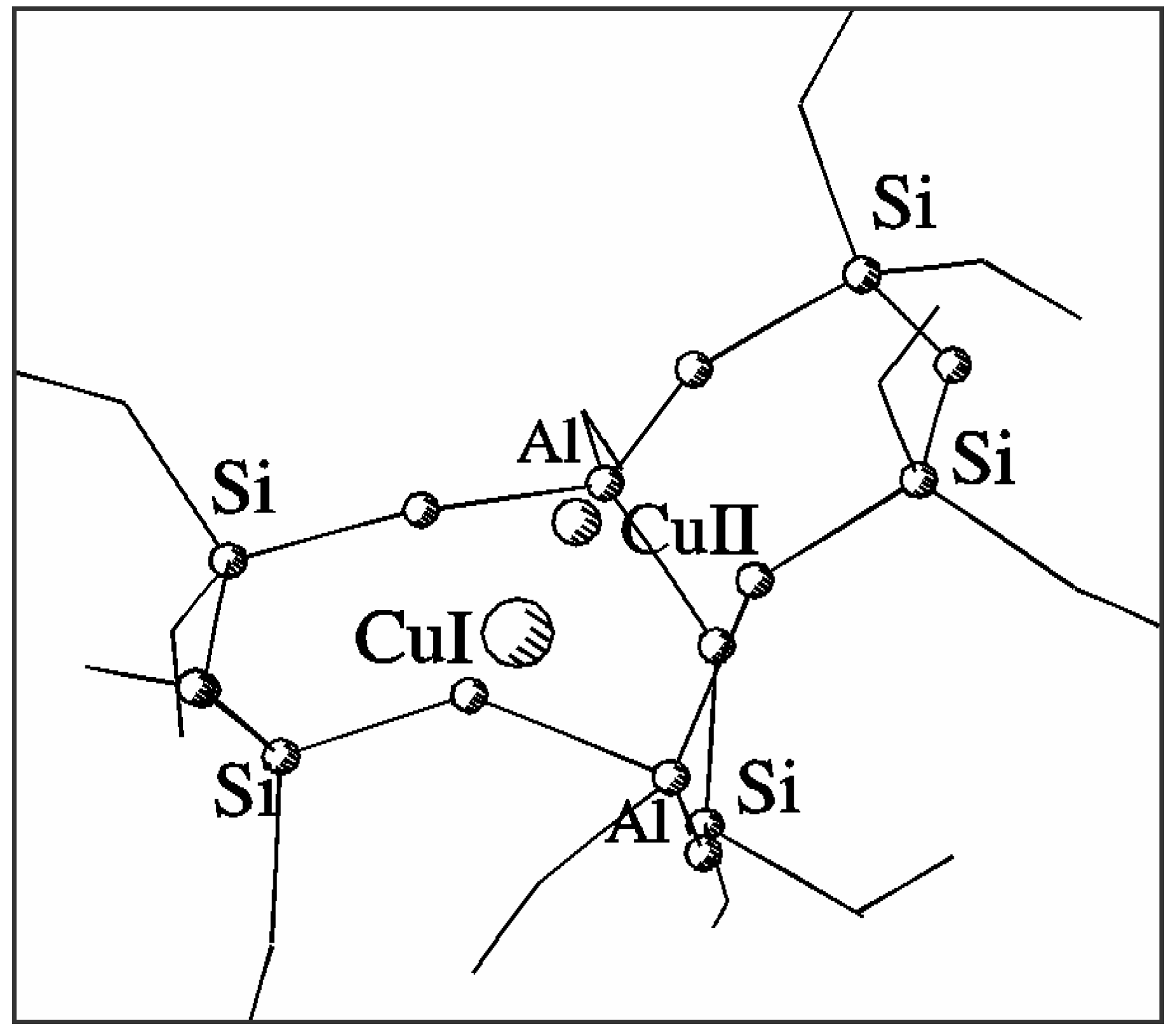
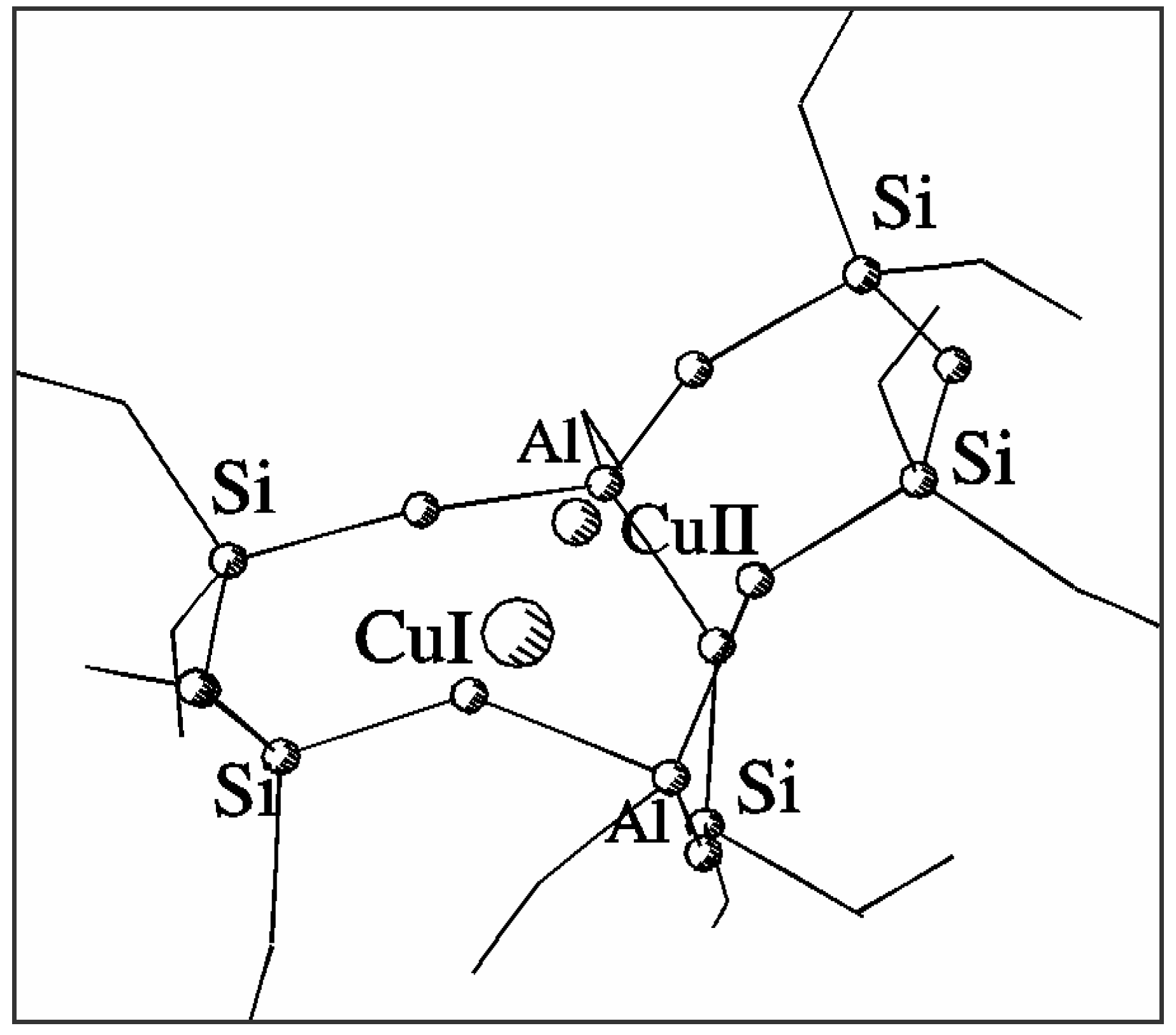
Fig. 1 shows the optimised positions of Cu
2+ and Cu
+ in the common basket model of the α site in ZSM-5. Column 2 in
Table 1 gives the distances between the appropriate cation and nearest framework oxygens (below 2.5 Å). It can be easily verified that divalent copper prefers planar square coordination in the centre of 6T ring of the 7T model while monovalent copper shifts towards the centre of the 5T part of the model forming three bonds with bridging Al-O-Si oxygens in a five-ring. Nevertheless, both forms of the cation remain bonded in the central area of the model. The shift between the positions of Cu
2+ and Cu
+ sites in our model amounts to 1.22 Å. This result directly confirms the assumption of different environment for both forms of copper cation and its migration in the zeolite framework upon reduction-oxidation. The position of monovalent copper differs significantly from the one obtained by Nachtigallova et al. [
21], where the difference between the positions of Cu
2+ and Cu
+ was not so distinct. Thus, we have recalculated this model and we have found the other geometry to be the local minimum slightly higher in energy. In addition, geometrical parameters of both sites agree perfectly with those obtained previously for smaller, single-ring models [
15,
16]. Therefore we regard the 7T basket model as the smooth extension of our previous single-ring models.
Figure 1.
Structure and coordination of Cu2+ and Cu+ sites in a common 7T basket.
Figure 1.
Structure and coordination of Cu2+ and Cu+ sites in a common 7T basket.
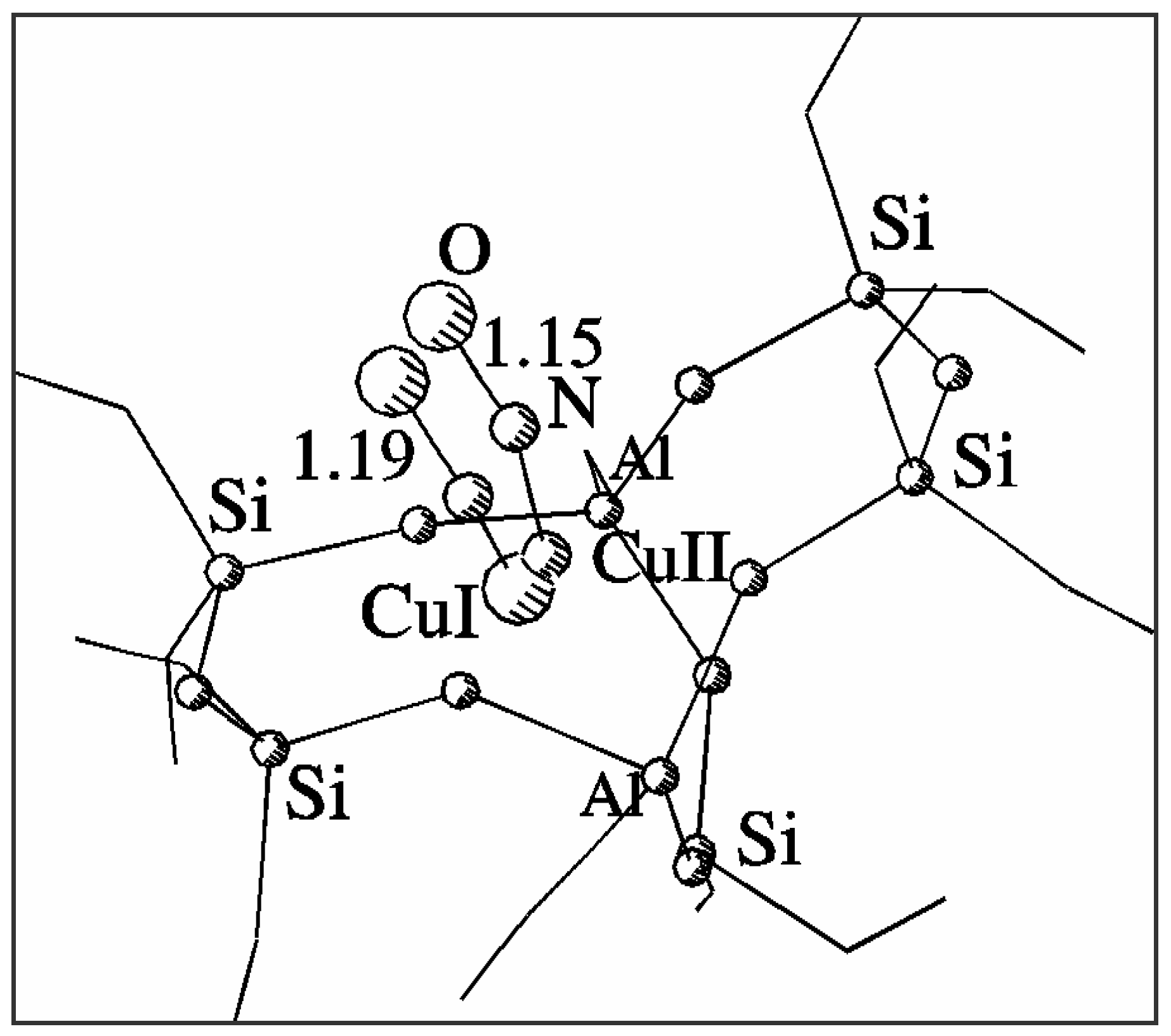
Figure 2.
Geometry of NO adsorption on Cu2+ and Cu+ sites in a common 7T basket model of CuZSM-5.
Figure 2.
Geometry of NO adsorption on Cu2+ and Cu+ sites in a common 7T basket model of CuZSM-5.
Thus in the next step the extended 7T basket model was used to study properties of adsorption complexes of nitrogen oxide with exchanged copper cations.
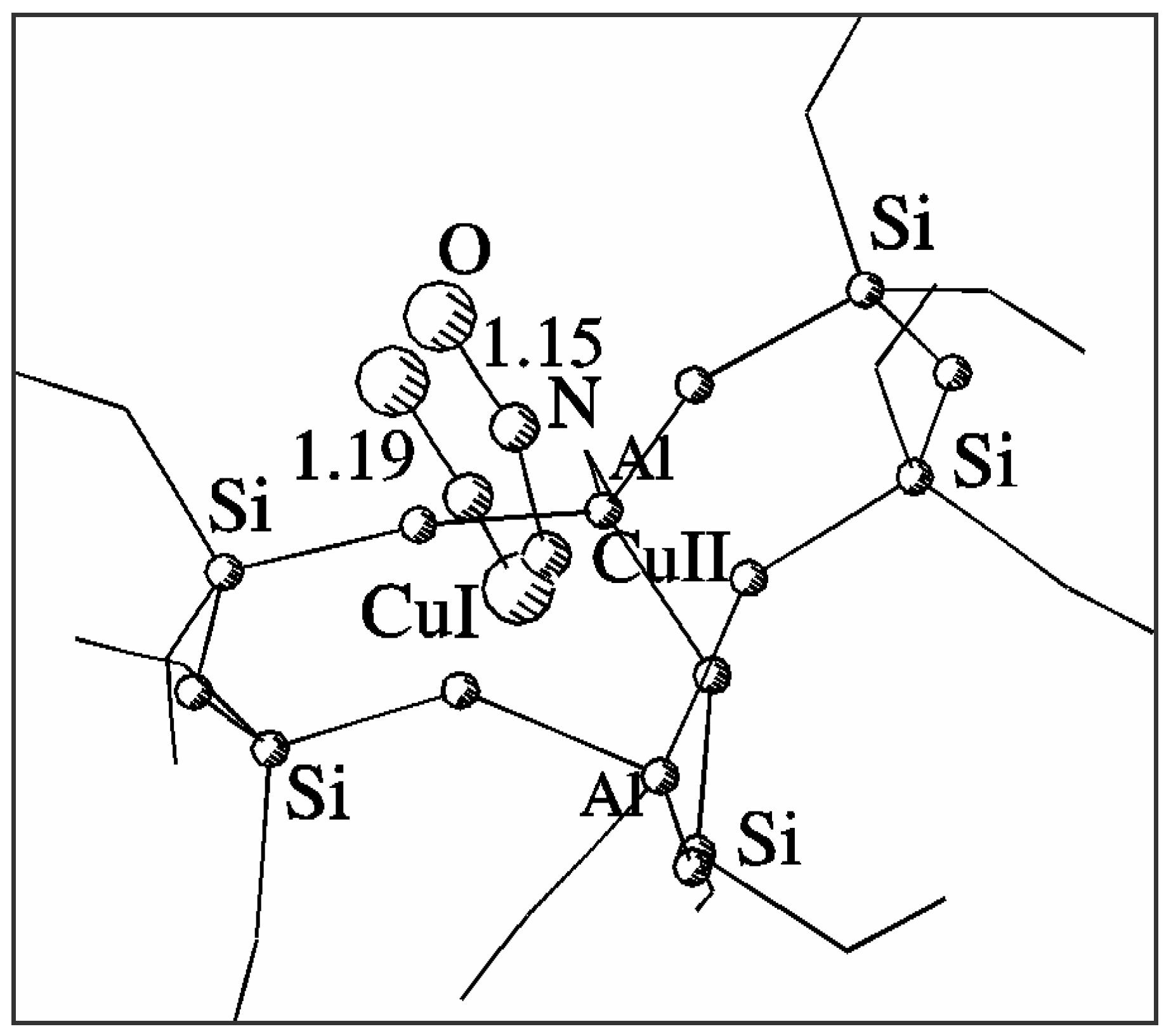
Fig. 2 shows superimposed optimised geometries of NO adduct with Cu
2+ and Cu
+ sites in ZSM-5. The NO molecule is strongly bonded by both Cu
2+ and Cu
+ sites. NO adsorption modifies the position of the cation and its interaction with the framework, which is particularly effective for Cu
2+. Column 3 in
Table 1 lists the distances between the appropriate cation interacting with nitrogen oxide and nearest framework oxygens. The cation becomes withdrawn by the interaction from its original position and changes its coordination to the threefold one close to the centre of the 5T part of the model. The interaction of Cu
+ with NO is weaker and the modification of its coordination is much smaller than that of Cu
2+, although the shift of Cu
+ to Cu
2+ position is also observed. The distance between the positions of the two copper cations decreases from 1.22 Å to 0.36 Å when interacting with NO, which indicates additional copper migration after adsorption. These detailed properties of the sites and their adsorption complexes with NO could not be obtained with the use of simplified single-ring models even if they were verified to provide good description of selected geometrical properties of the sites.
Table 1.
Bond distances between copper cations and bridging oxygens below 2.5 Å, (index 5T denotes oxygen in 5T ring, otherwise in 6T ring).
Table 1.
Bond distances between copper cations and bridging oxygens below 2.5 Å, (index 5T denotes oxygen in 5T ring, otherwise in 6T ring).
| | RCu-O [Å] |
|---|
| Centre in M7 | Site | Site with NO |
|---|
| Cu2+ | 2.03, 2.03, 2.05, 2.05 | 2.02, 2.03, 2.105T |
| Cu+ | 1.93, 2.01, 2.035T | 2.02, 2.06, 2.065T |
The two preceding paragraphs showed that very bride scope of valuable information could be extracted from the proposed extended model. On the other side, the model was by no means perfect and burdened with high degree of arbitrariness similarly to all molecular models of extended systems. Thus it was highly desirable to confirm its validity by testing against existing experiment in the similar manner to that utilised for the first time in the case of small models [
15]. There we used the mean value of deviation between cartesian coordinates of superimposed structures with and without guest moiety (Cu or NO) as the measure of framework distortion after cation exchange or NO sorption and correlated this parameter with known shifts in skeletal vibration. This seemed to be the only way of theoretical interpretation of such shifts because skeletal vibration frequencies could not be obtained from direct vibrational analysis since this is physically extended motion and could be seriously perturbed by cluster model calculations and contamination with artificial boundary conditions. Here the basket model has been again validated via indirect interpretation of shifts in T-O-T skeletal frequencies caused by cation exchange and NO sorption, measured by infrared spectroscopy [
11,
12,
13,
14,
15]. Calculated parameters characterising geometrical framework distortion were very similar to those determined for small models and changed consequently with the shift of skeletal vibration. In this work we propose additional way to approach the problem of interpreting shifts in skeletal vibration. T-O-T vibration may be viewed as a combination of T-O stretching frequencies and thus its shift should correlate with the average strength of T-O bonds in the vicinity of perturbing cation. In the M7 model the positions of both forms of copper cation and their complexes with NO could be precisely determined and therefore we could additionally correlate shifts in T-O-T frequency with calculated changes of Mayer bond orders of T-O pairs for all Al-O-Si bridges in the model.
Inspection of
Table 2 shows that red shift in T-O-T frequency corresponds perfectly to global weakening of these T-O bonds, where the effect of Cu
2+ is the strongest.
Table 2 shows also that the interaction with NO substantially weakens the Cu
2+ - MFI framework interaction and makes the T-O bonds stronger what makes the perturbing effect of Cu
2+ on the framework comparable to that of Cu
+. It is consistent with previous discussion of geometrical changes caused by adsorption where we have already shown that Cu
2+ cation is withdrawn by the interaction with NO from its original position and changes its coordination to the threefold one. The interaction of Cu
+ with NO is weaker and the modification of its coordination is much smaller than that of Cu
2+, although the shift of Cu
+ towards Cu
2+ position is also observed. In this way predictions concerning the nature of a cation-framework and cation-sorbed molecule interaction, inferred from geometrical analysis of the models, acquires experimental support.
Table 2.
The frequency shift from the position of unperturbed ring (Δν=1020cm
-1-ν) of IR band of T-O-T vibration for Cu
2+ and Cu
+ ions and their complexes with NO [
11,
14], and calculated changes in sums of T-O bond orders.
Table 2.
The frequency shift from the position of unperturbed ring (Δν=1020cm-1-ν) of IR band of T-O-T vibration for Cu2+ and Cu+ ions and their complexes with NO [11,14], and calculated changes in sums of T-O bond orders.
| | Cu2+ZSM-5 | Cu+ZSM-5 | Cu2+-NO | Cu+-NO |
|---|
| Δν cm-1 | 83 | 43 | 58 | a) |
| Δ(Σb.o.)T-O | -0.68 | -0.43 | -0.43 | -0.40 |
Table 3 displays the properties of Cu-NO adsorption complexes obtained from electronic structure calculations for the basket model. Indeed, the adsorption energy of nitrogen oxide on Cu
2+ZSM-5 is much higher than that on Cu
+ZSM-5 even if the bond length and bond angle is similar in each case. Distinct difference, however, shows up in the N-O bond distance. It becomes strongly elongated after sorption of NO on Cu
+ZSM-5 while the influence of Cu
2+ site leads to slight shortening of the bond. This indicates that the nature of NO bonding with the active site in the two cases should be different. In order to discuss more precisely the features of the substrate – active site bonding additional electronic factors should be considered.
Table 3.
Calculated properties of adsorption complexes between NO molecule and CuZSM-5: adsorption energy, equilibrium Cu – NO bond distance and angle, intramolecular NO bond length.
Table 3.
Calculated properties of adsorption complexes between NO molecule and CuZSM-5: adsorption energy, equilibrium Cu – NO bond distance and angle, intramolecular NO bond length.
| Centre in M7 | Eads kcal/mol | RCu-NO Å | Cu-N-O angle (deg) | RNO Å |
|---|
| Cu2+-NO | 40 | 1.80 | 128 | 1.15 |
| Cu+-NO | 24 | 1.84 | 132 | 1.19 |
Table 4 lists parameters of the charge distribution and NO stretching frequencies in addition to simple geometrical factors. It is clearly visible that divalent copper withdraws electrons from the sorbed molecule and, in consequence, intramolecular NO bond becomes stronger (shorter by 0.01 Å). whereas monovalent copper donates electrons to the molecule and weakens the intramolecular bond, which becomes by 0.03 Å longer. More critical inspection of the charge redistribution indicates, however, that the appropriate copper centre itself cannot be treated as the sole element of the active site. The calculations show that Cu
2+ centre acquires only 0.07 e while NO looses 0.25 e, which must be redistributed over the framework serving as an electron reservoir Accordingly, Cu
+ looses 0.08 e on interaction, only partly on behalf of NO and the remainder must be again redistributed over the framework. Thus the MFI framework acts as a generalised soft ligand which serves as the buffer for electron redistribution processes and strongly influences the properties of the active site.
Table 4.
Effect of NO interaction with CuZSM-5: charge on copper cation and adsorbed NO, changes in NO bond lengths (ΔRNO), calculated and measured NO stretching frequency (νNO).
Table 4.
Effect of NO interaction with CuZSM-5: charge on copper cation and adsorbed NO, changes in NO bond lengths (ΔRNO), calculated and measured NO stretching frequency (νNO).
| | QCu | QNO | ΔRNO Å | νNO (DFT) cm-1 | νNO (IR) cm-1 |
|---|
| Cu2+-NO | +0.43 | +0.25 | -0.01 | 1904 | 1895a) |
| Cu+-NO | +0.36 | -0.03 | +0.03 | 1754 | 1809b) |
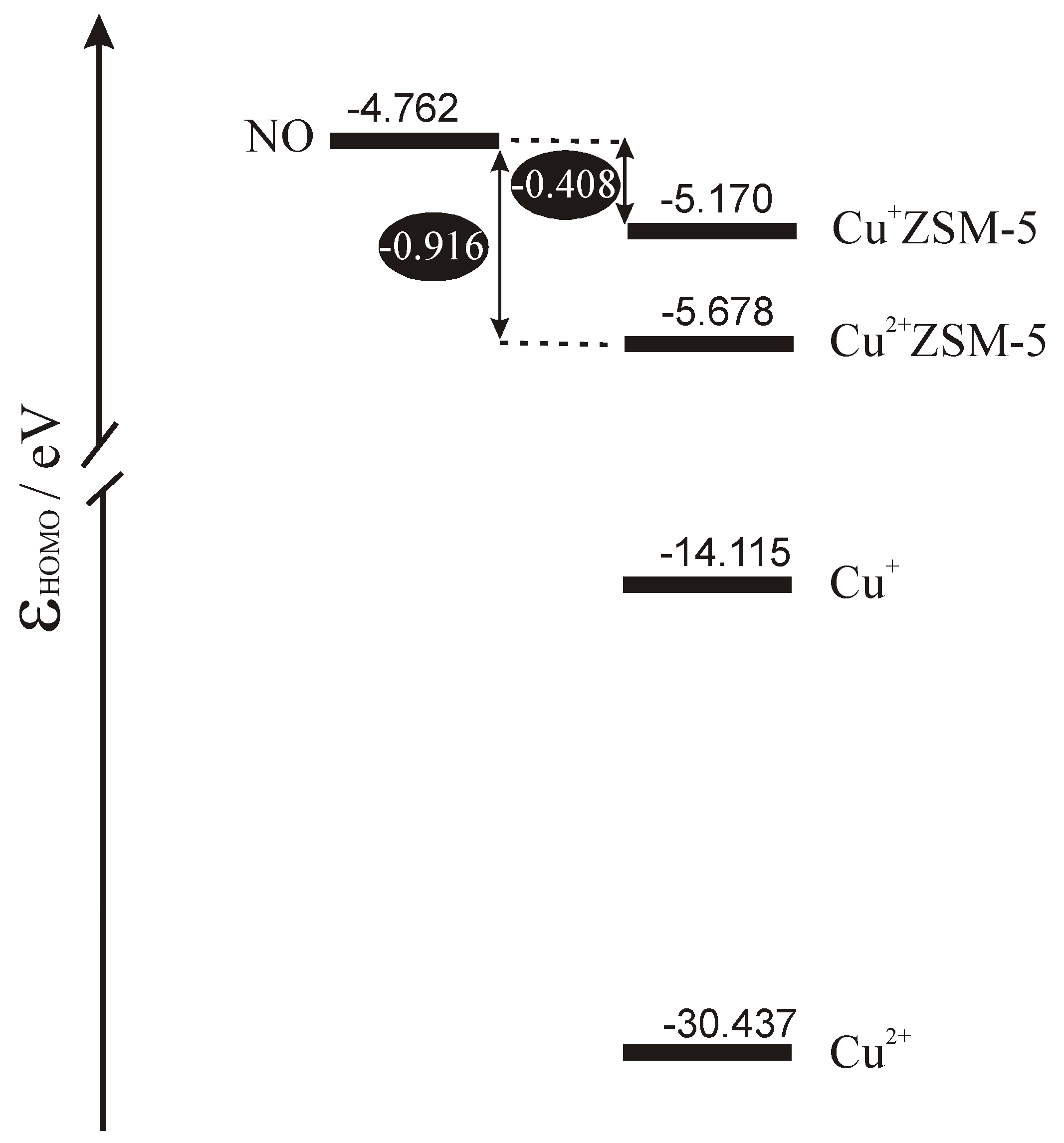
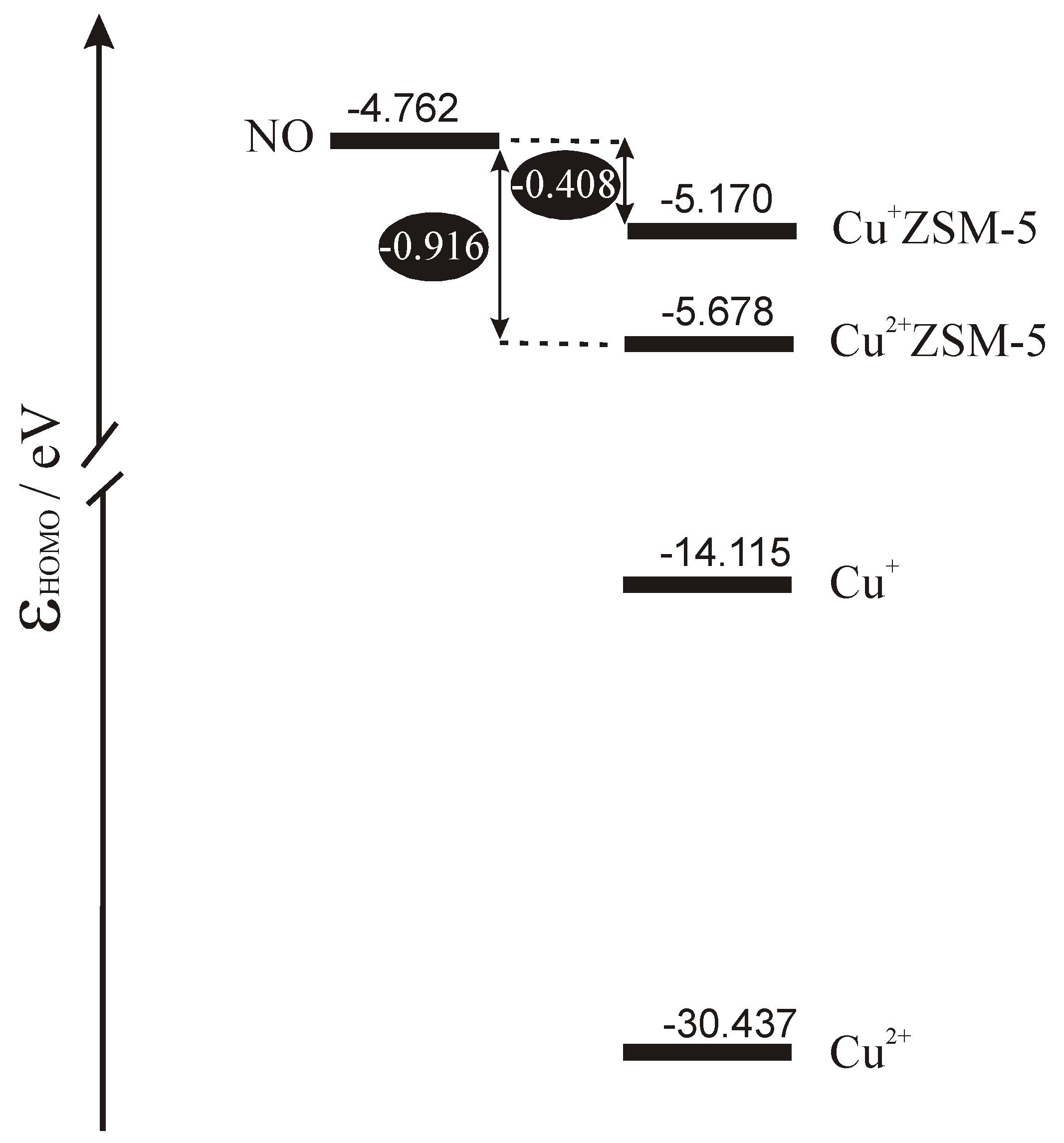
Scheme 1 shows the energetic diagram for the highest occupied orbitals in the isolated subsystems calculated by DFT. To enrich the discussion also bare copper cations are included in the scheme. Inspection of the diagram allows for sketching the interpretation of electron phenomena occurring in the catalytic system composed of copper site in ZSM-5 and the NO molecule. Taking the position of the highest occupied level in the subsystem as the crude approximation of its Fermi level we can conclude that for bare copper cations the only process expected on interaction would be complete transfer of electrons from the molecule to the cation. Copper ligation by bridging oxygens from the MFI framework leads to significant shift of the Fermi level upwards which diminishes copper tendency to accept electrons and triggers out the increase of its donor properties. In the case of Cu
+ZSM-5 Fermi level approaches very closely the energy of semioccupied orbital of NO which enables electron backdonation. As the character of the highest occupied orbital of NO is strongly antibonding donation of electrons must lead to strong destabilisation of NO bond and, in consequence, to the activation of the molecule.
The same effects show in the NO stretching frequency, which is a good experimental measure of the bond strength. The values of ν
NO measured by others [
24,
25] and by us [
16] in IR spectroscopy, and calculated from our basket model [
26,
27] are listed in columns 5 and 6 in
Table 4. Measured and calculated by us NO stretching frequency is equal to 1876 cm
-1 and 1912 cm
-1, respectively [
26,
27]. In IR experiment blue shift of ν
NO is observed for Cu
2+ZSM-5 and substantial red shift is observed for Cu
+ZSM-5. The frequencies calculated for the extended model of copper active sites in ZSM-5 agree well with their experimental counterparts, which gives further validation of the model and supports the interpretation of the activation mechanism of NO over Cu
+ZSM-5 sketched in the preceding paragraph.


{kind=link}
{kind=link}
{kind=link}