Computational details
We performed first principles molecular dynamics simulations [
15] within the Becke-Lee-Yang-Parr gradient-corrected density functional approach [
16]. The valence-core interaction was accounted for via norm-conserving Troullier-Martins pseudopotentials [
17]. In the case of Mg and Ti non-linear core corrections were included [
18], while for a correct description of both the structural and the electronic properties of V the use of semi-core states turned out to be necessary. This implies that the plane wave expansion of the valence wave functions required a cut-off of 40 Ry for Ti-systems [
19], but 80 Ry were needed to describe V-based systems with comparable accuracy. The support consists of a slab of 24 MgCl
2 formula units in a supercell of 12.729 x 11.782 x 28.000 Å
3 with an angle
ab = 72
o; this amounts to a thickness of 6 layers, exposing the (110) lateral cut, which has been shown to provide a strong binding for mononuclear catalytic species [
11]. On one side the slab is kept fixed to the bulk crystal structure, while Ti and V adducts, as well as the olefins and the growing polymer, are placed on the opposite fully relaxed side. The large
z dimension ensures an empty space sufficient to accommodate the active center and the olefin chain and keeps the system far enough from its repeated images, since periodic boundary conditions are imposed. The reaction path is sampled within the Blue Moon ensemble theory [
20] assuming as a reaction coordinate ξ the distance between one of the carbon atoms of the incoming olefin, C
1, and the first carbon atom of the growing polymer C
α directly bound to the metal catalyst M (M = Ti, V)
This holonomic constraint is included in the Car-Parrinello equations of motion by linearly adding to the Lagrangean
LCP the analytical constraint via a Lagrange multiplier λ
ξ(t),
LCP+λ
ξ(t)(ξ-ξ
0). We can write down the standard expression for the free energy
where
KB is the Boltzmann constant and
T the simulation temperature and, from this expression, it is easy to evaluate the first order variation of
F. If we observe that the variation of Car-Parrinello Hamiltonian <
HCP> (constant of motion) is zero and that <ξ> = ξ
0 , our expression reads
Hence, the dynamical average value <λ
ξ> represents the derivative of the free energy with respect to the chosen reaction coordinate (i.e. the constraint force). Integrating this expression between the initial distance
a (reactants) and the final value
b (products) gives us the free energy Δ
F of the process as described in refs. [
9,
20]:
The temperature of the system was controlled via a Nosé-Hoover thermostat chain [
21] and set to 323 K according to experiments [
22]. An electronic fictitious mass of 800 a.u. and an integration step of 5.0 a.u. (0.1208 fs) ensured good control of the conserved quantities.
Results and Discussion
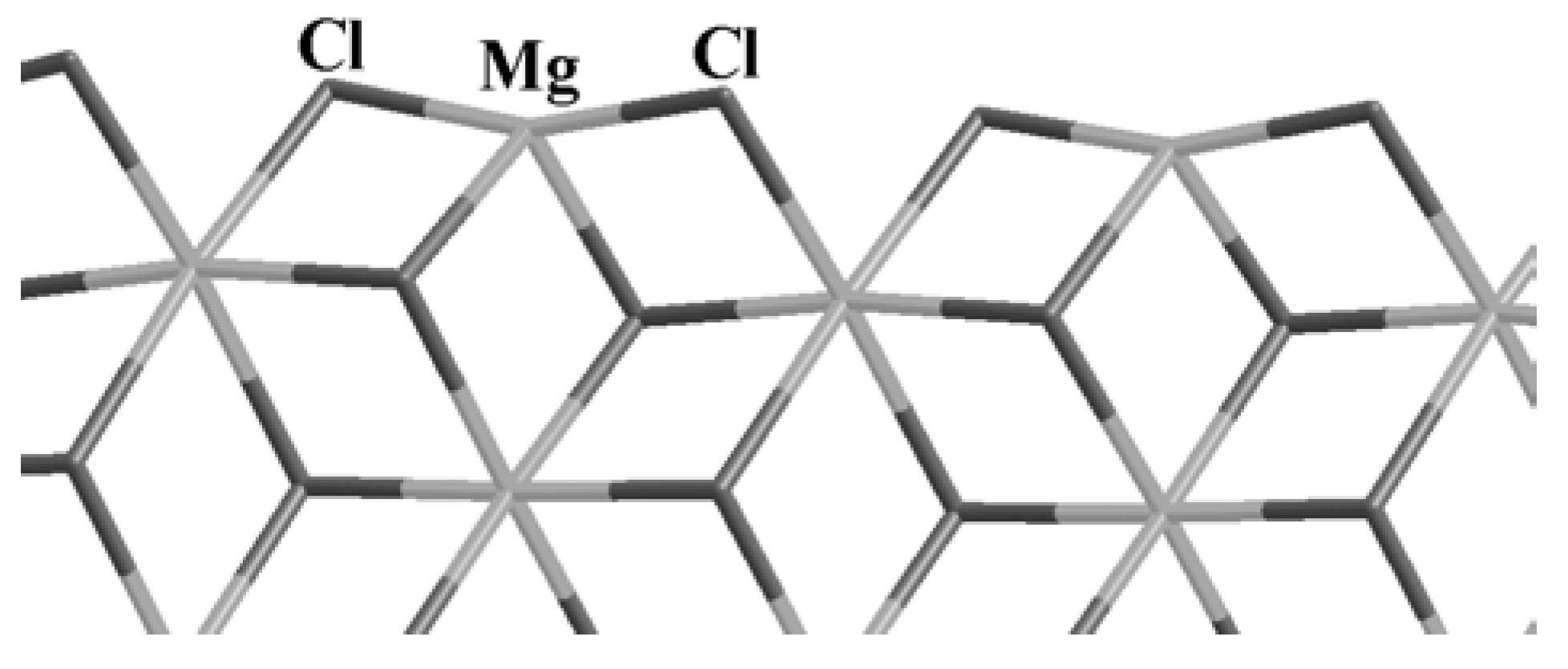
We have already described elsewhere the crystal structure of MgCl
2 [
8]. For the ongoing discussion, we recall that it has a layered structure packed as
ABCABC... and belongs to the
R3m symmetry group. Each layer presents Mg atoms 6-fold coordinated in an octahedral arrangement and the Mg-Cl bond is ionic. The details of a single layer exposing the relaxed (110) surface are shown in
Figure 1. On this surface, the Mg atoms are 4-fold coordinated as a consequence of the cleavage from the bulk. This is the surface that has been shown to provide a very active support for TiCl
4 species.
Figure 1.
The relaxed (110) MgCl2 surface. The Mg atoms are located at the cross points of the light gray sticks, while Cl atoms are represented by the darker segments as indicated by the labels.
Figure 1.
The relaxed (110) MgCl2 surface. The Mg atoms are located at the cross points of the light gray sticks, while Cl atoms are represented by the darker segments as indicated by the labels.
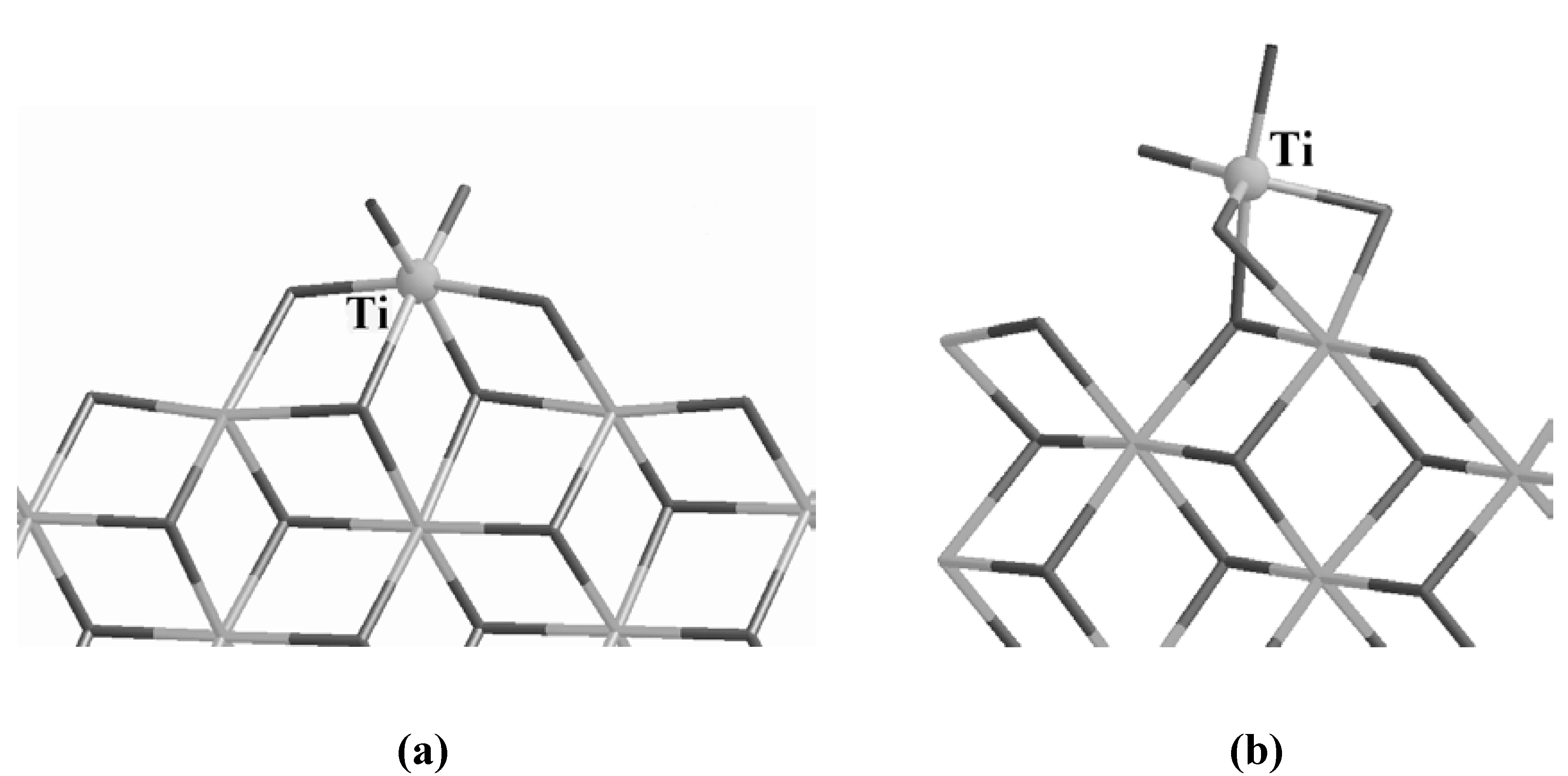
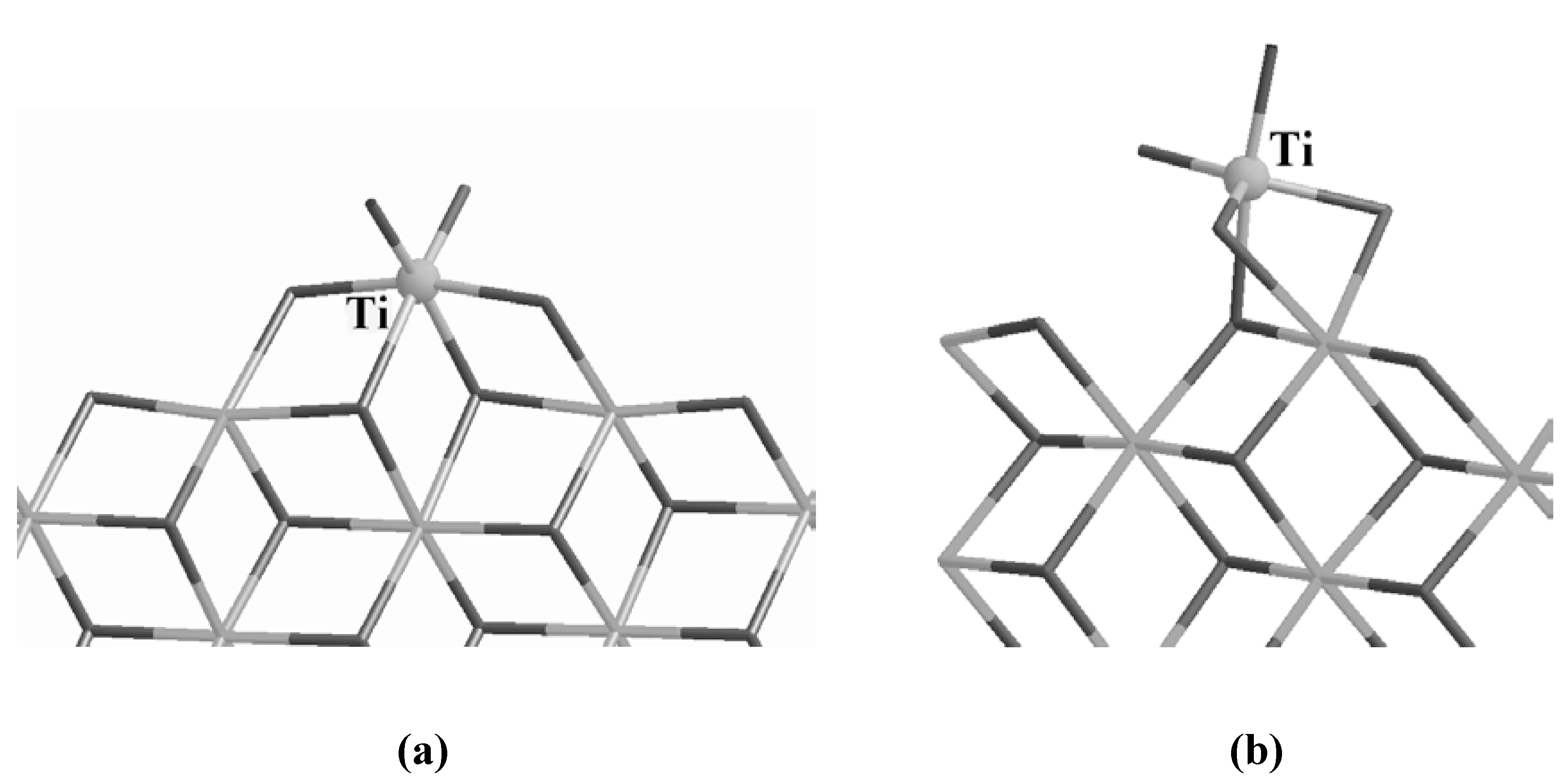
Ti can indeed bind efficiently in octahedral configurations, as reported in
Figures 2 (a) and (b). The configuration (a) was originally proposed by the group of Corradini [
23], while (b) was obtained by simulating the deposition of a TiCl
4 molecule on the (110) surface [
8,
9]. In configuration (a) Ti forms 4 bonds with the support, sits at the center of an octahedron and each vertex is occupied by a Cl atom. In configuration (b) Ti forms only 3 bonds with the substrate, the Ti is still sitting at the center of an octahedron but one Cl is missed at one of the vertices. This is not, however, a drawback. On the
Figure 2.
Corradini’s Ti mononuclear site (a) and the 5-fold Ti site obtained by simulating the deposition of TiCl
4 on the (110) surface. The Ti atom is labeled and evidenced as a ball for clarity. The gray scale code is the same as in
Figure 1.
Figure 2.
Corradini’s Ti mononuclear site (a) and the 5-fold Ti site obtained by simulating the deposition of TiCl
4 on the (110) surface. The Ti atom is labeled and evidenced as a ball for clarity. The gray scale code is the same as in
Figure 1.
contrary, a vacant site is required in order to allow an incoming olefin to approach the catalyst and start the polymerization [
24,
25]. In this respect, the 5-fold coordinated Ti of
Figure 2 (b) already has one of the prerequisites to become an active center. The difference in the number of bonds between the Ti atom and the support accounts for the different binding energies of the two sites, being 40.3 kcal/mol for the Corradini configuration and 29.4 kcal/mol for the 5-fold geometry.
The activation of Ti species is achieved experimentally by introducing the alkylating donor Al(C
2H
5)
3 on the pristine TiCl
4/MgCl
2 system. This molecule has the effect of removing dangling Cl atoms from the Ti center and replacing them with a hydrocarbon group that will act as a polymer chain initiator. Once this occurs, the system is ready to begin the polymerization reaction. The main phases for the insertion of an ethylene (C
2H
4) monomer in the metal-carbon bond are sketched in
Figure 3 according to the scheme proposed by Cossee and Arlman [
24,
25].
Figure 3.
The reaction path leading to the insertion of an ethylene molecule in an active Ti catalytic center. The reaction proceeds along the direction indicated by the arrows and the open square indicates the vacant site. The Ti-H dashed lines in the lower panels represent schematically the agostic interaction described in the text.
Figure 3.
The reaction path leading to the insertion of an ethylene molecule in an active Ti catalytic center. The reaction proceeds along the direction indicated by the arrows and the open square indicates the vacant site. The Ti-H dashed lines in the lower panels represent schematically the agostic interaction described in the text.
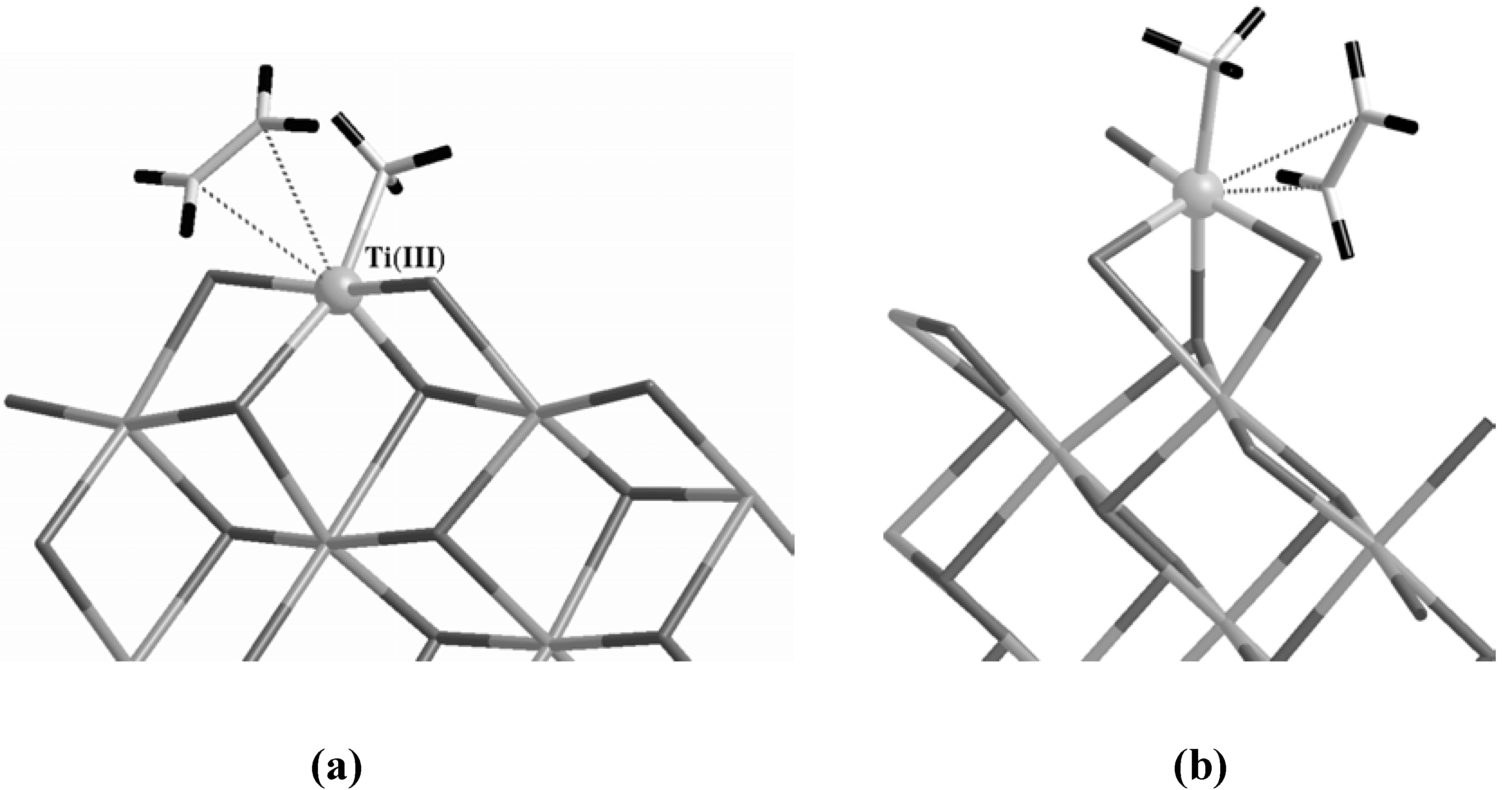
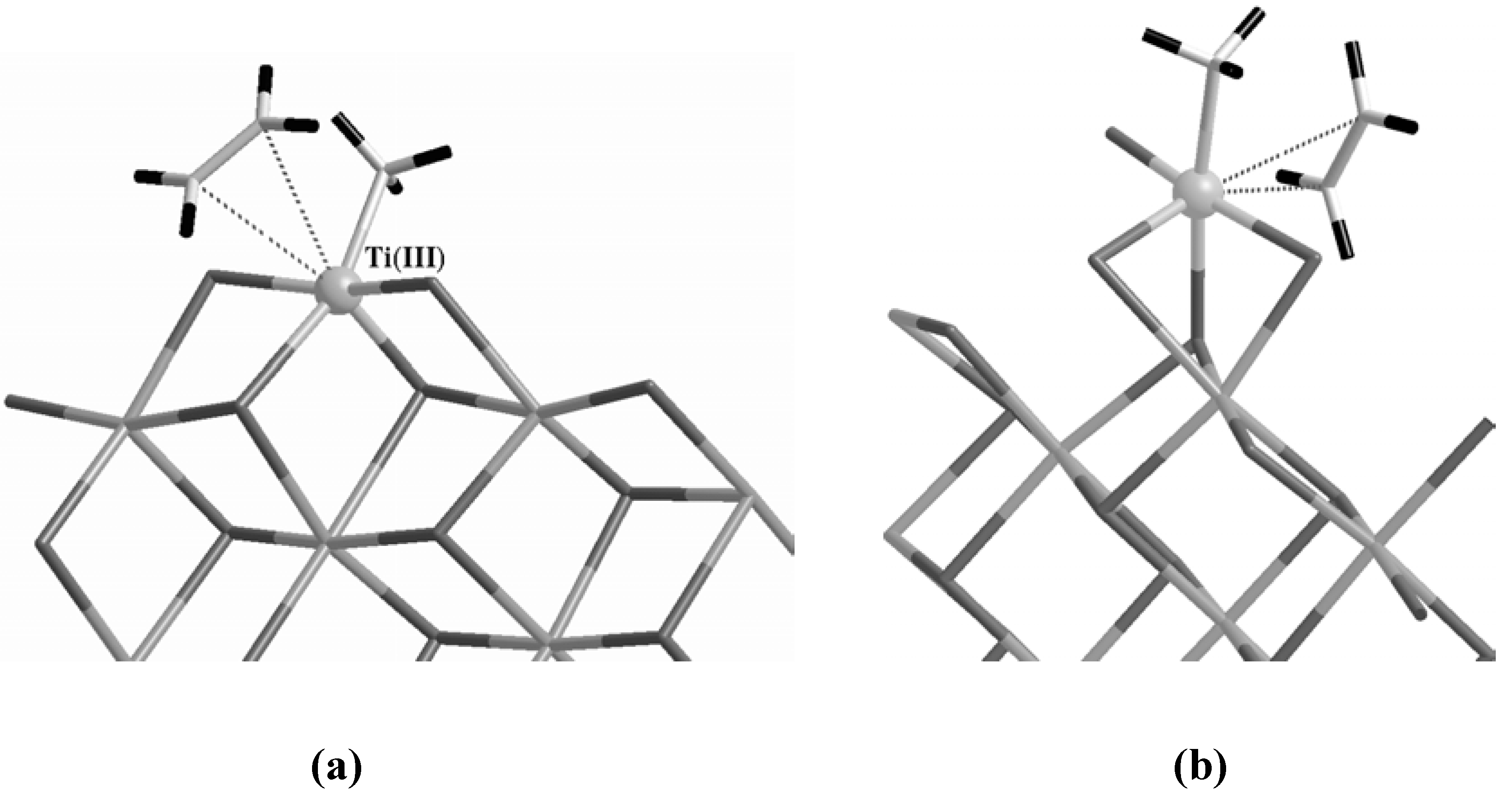
In order to reproduce an active site with the right characteristics, in the case of the Corradini center we replaced one of the two dangling Cl with a methyl group and removed the other dangling Cl to fulfill the requirement of a vacant site (
Figure 4 (a)). On the other hand, since a vacant site is already present in the case of the 5-fold configuration, we simply substituted one of the exposed Cl atoms with a CH
3 unit as in
Figure 4 (b).
Figure 4.
The active Corradini (a) and 5-fold (b) sites forming a π-complex with an ethylene molecule. The Ti atom is evidenced as a gray ball while H atoms are the terminal black sticks on both the ethylene and the methyl group bound to Ti. The label in parentheses in (a) indicates the formal oxidation state of Ti.
Figure 4.
The active Corradini (a) and 5-fold (b) sites forming a π-complex with an ethylene molecule. The Ti atom is evidenced as a gray ball while H atoms are the terminal black sticks on both the ethylene and the methyl group bound to Ti. The label in parentheses in (a) indicates the formal oxidation state of Ti.
The insertion of an olefin in the metal-carbon bond was studied by gradually shortening the distance between C
α and C
1 as shown in
Figure 5 (a), (b) and (c). The figure refers to the case of the 5-fold site and shows the π-complex, the transition state corresponding to the zero crossing of the constraint force <λ
ξ> and the final product for the first olefin insertion respectively. The reaction path for the Corradini site is very similar and a full discussion is reported in the quoted literature. We call attention only to the main points: the ethylene spontaneously – i.e. in a barrierless way – forms a complex by approaching the Ti on the vacant site. Then a barrier, that can range from 6 to 12 kcal/mol, has to be overcome in order to insert the monomer in the polymer chain. This passes across a ring structure like the one
Figure 5.
The main phases of the ethylene insertion process: the π-complex (a), the transition state (b) and the final product (c). The reaction coordinate is the distance between Cα and C1 while Ha indicates the H atom that gives rise to the agostic interaction with Ti. H atoms of the olefin and the polymer chain are evidenced for clarity as black balls.
Figure 5.
The main phases of the ethylene insertion process: the π-complex (a), the transition state (b) and the final product (c). The reaction coordinate is the distance between Cα and C1 while Ha indicates the H atom that gives rise to the agostic interaction with Ti. H atoms of the olefin and the polymer chain are evidenced for clarity as black balls.
shown in
Figure 5 (b), which represents the transition state. This occurs when our reaction coordinate ξ reaches the value of 2.100 Å. At this point the constraint force passes across zero and changes sign, indicating that we have overcome the energy barrier and the repulsion is now becoming an attraction between C
1 and C
α , which find it energetically more favorable to form a bond. In this ring structure the methyl group of the original metal-carbon bond tilts in such a way that the distance of one of its H atoms from Ti shortens to 1.959 Å (it was 2.627 Å in
Figure 5 (a)) and a weak interaction with Ti, not involving any electron transfer, occurs. This is evidenced in
Figure 5 (b) by the label H
a. Such a phenomenon is known in the chemical literature as
agostic interaction and its role is to reduce the steric hindrance with the incoming monomer and, hence, favor its approach to the catalyst.
At the transition point, the C atom formerly bound to Ti and indicated as C
α in
Figure 5 (a) becomes over-coordinated, having 3 bonds with H, one bond with Ti plus the newly formed C
α-C
1 bond, representing our reaction coordinate [
29]. The longer and weaker metal-carbon bond gradually cleaves and eventually breaks, thus completing the insertion process as in
Figure 5 (c). The complexation energies for both the Corradini and the 5-fold site, as well as the insertion barrier, are reported in
Table 1. Experimental data on the activation energy, which is the rate-limiting step of the polymerization, range from 6 to 12 kcal/mol [
26,
27,
28], according to the procedures adopted both in the preparation of the catalyst and in the polymerization process. These values are in agreement with the present findings and provide support to the outcome of the simulation.
Table 1.
Complexation energies and activation barrier for the insertion of an ethylene molecule in the Ti and V active catalytic sites, as obtained by the simulations. The complexation energy in parentheses in the V column refers to the V site that is destabilized, as explained in the text.
Table 1.
Complexation energies and activation barrier for the insertion of an ethylene molecule in the Ti and V active catalytic sites, as obtained by the simulations. The complexation energy in parentheses in the V column refers to the V site that is destabilized, as explained in the text.
| | Ti Corradini | Ti 5-fold | V |
| Complexation energy (kcal/mol) | 8.4 | 7.8 | 4.7 (4.3) |
| Insertion barrier (kcal/mol) | 14.9 | 6.7 | 28.1 |
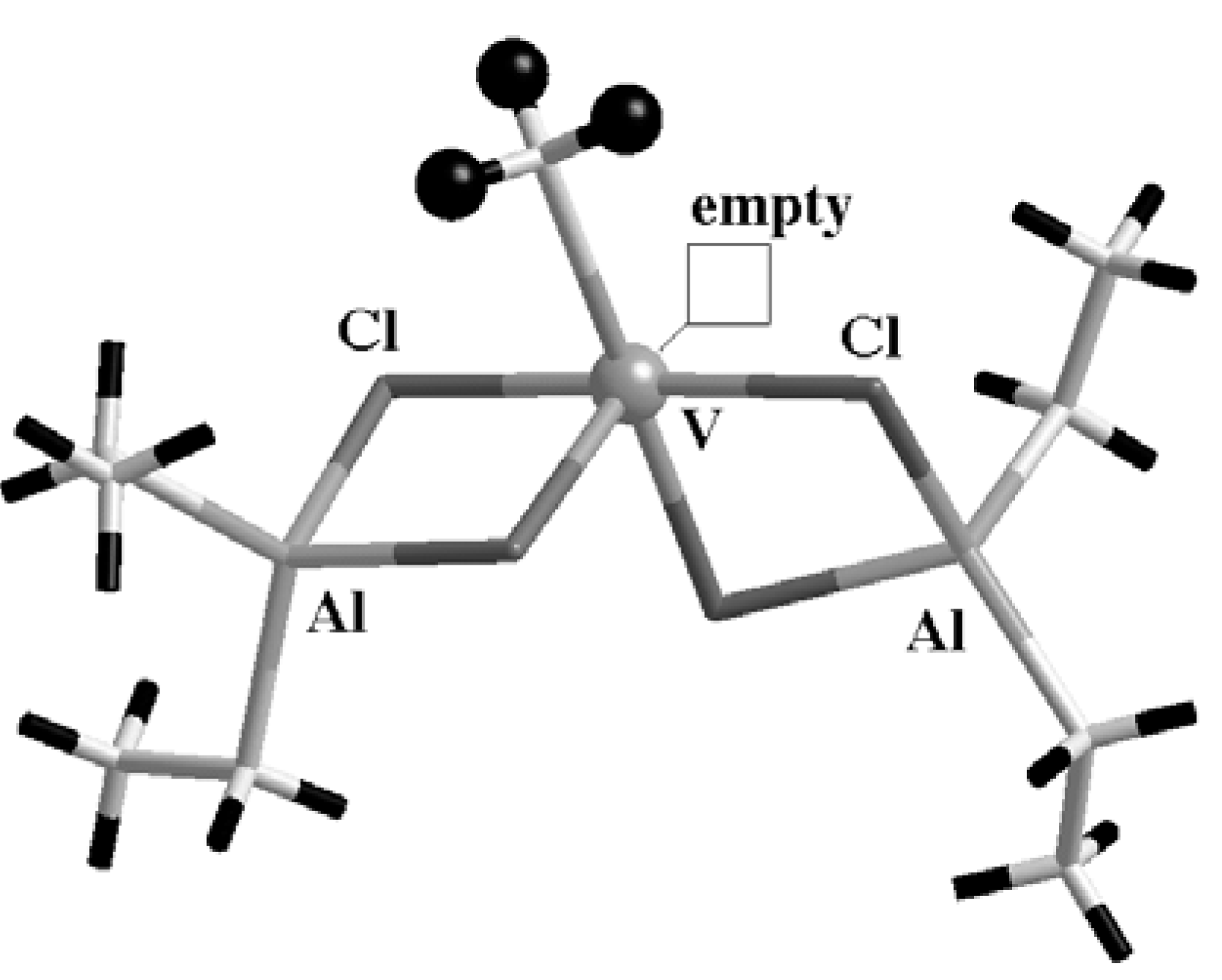
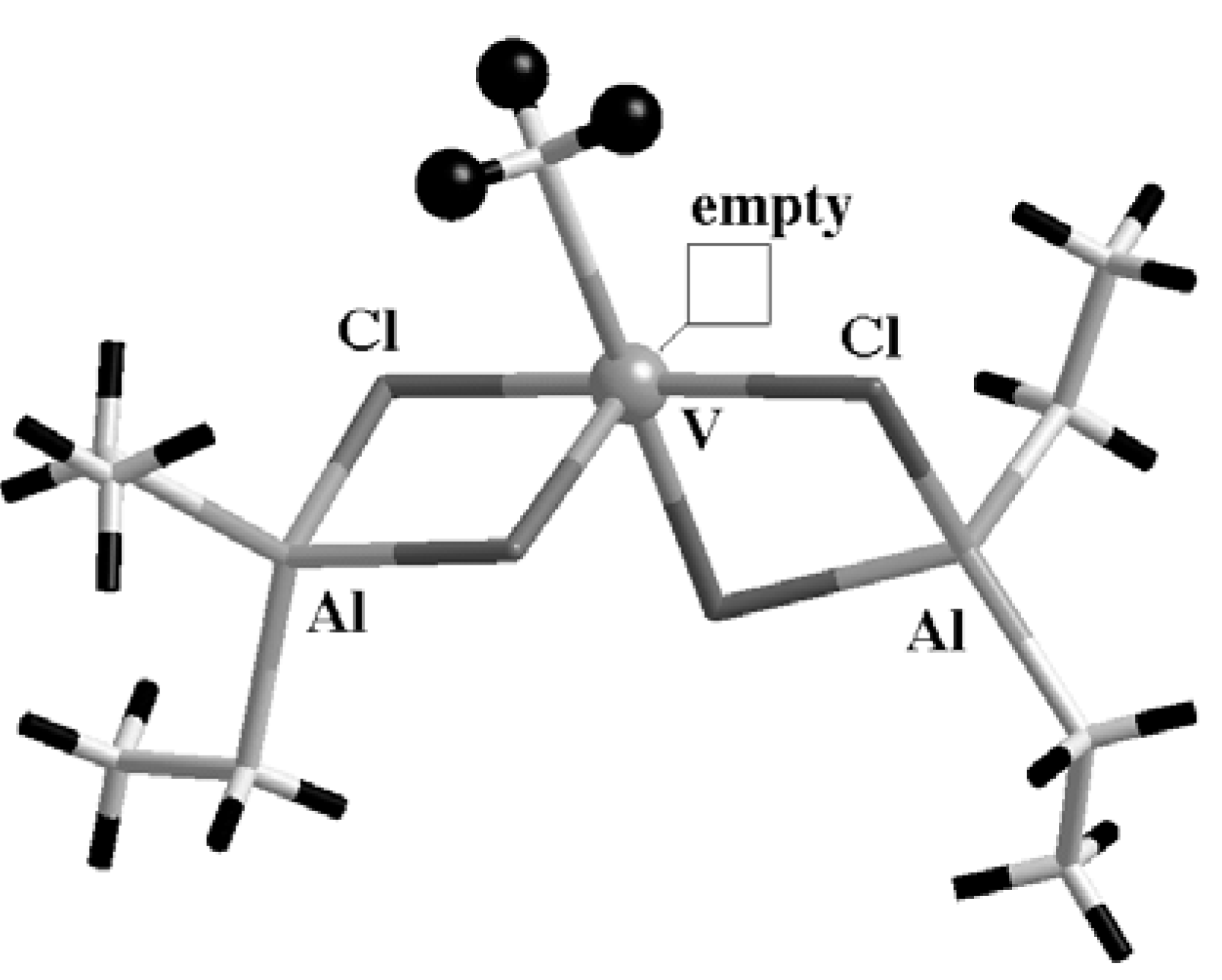
Very recently we made an attempt at understanding the unique role of Ti in heterogeneous systems by inspecting what would happen if one replaces Ti with another transition metal. Looking at the literature on ZN systems and at the periodic table, V immediately follows Ti both chemically and historically. In fact, a few years after the discovery of isotactic polymerization of propene by Ti, a pioneering work of Natta and coworkers [
30] and, later on, further studies from the groups of Zambelli and Corradini [
14] pointed out that a homogeneous system, such as the one shown in
Figure 6, is able to polymerize propylene in a syndiotactic, or sometimes atactic, polymer chain. By looking at the proposed geometry of the V-based catalyst, it is easy to notice a remarkable analogy with the Corradini model for Ti in the heterogeneous case shown in
Figure 4 (a). Also the V atom is hexacoordinated and
Figure 6.
The V-based homogeneous catalyst proposed as an active center. Four Cl atoms bridge V and the Al atoms of the ligand, and the geometry of the site closely resembles the octahedral Corradini Ti site on the (110) surface of MgCl2 (see text for details). Only the H atoms of the methyl group, representing the direction of the growing polymer, have been evidenced as black balls; other H are the black sticks.
Figure 6.
The V-based homogeneous catalyst proposed as an active center. Four Cl atoms bridge V and the Al atoms of the ligand, and the geometry of the site closely resembles the octahedral Corradini Ti site on the (110) surface of MgCl2 (see text for details). Only the H atoms of the methyl group, representing the direction of the growing polymer, have been evidenced as black balls; other H are the black sticks.
surrounded by four Cl atoms and one hydrocarbon ligand (CH
3), while a coordination vacancy ensures the empty site on which the olefin can approach the active center. Since Cl atoms belong to bridge bonds with the support (either Al or Mg atoms), V is in the formal oxidation state III.
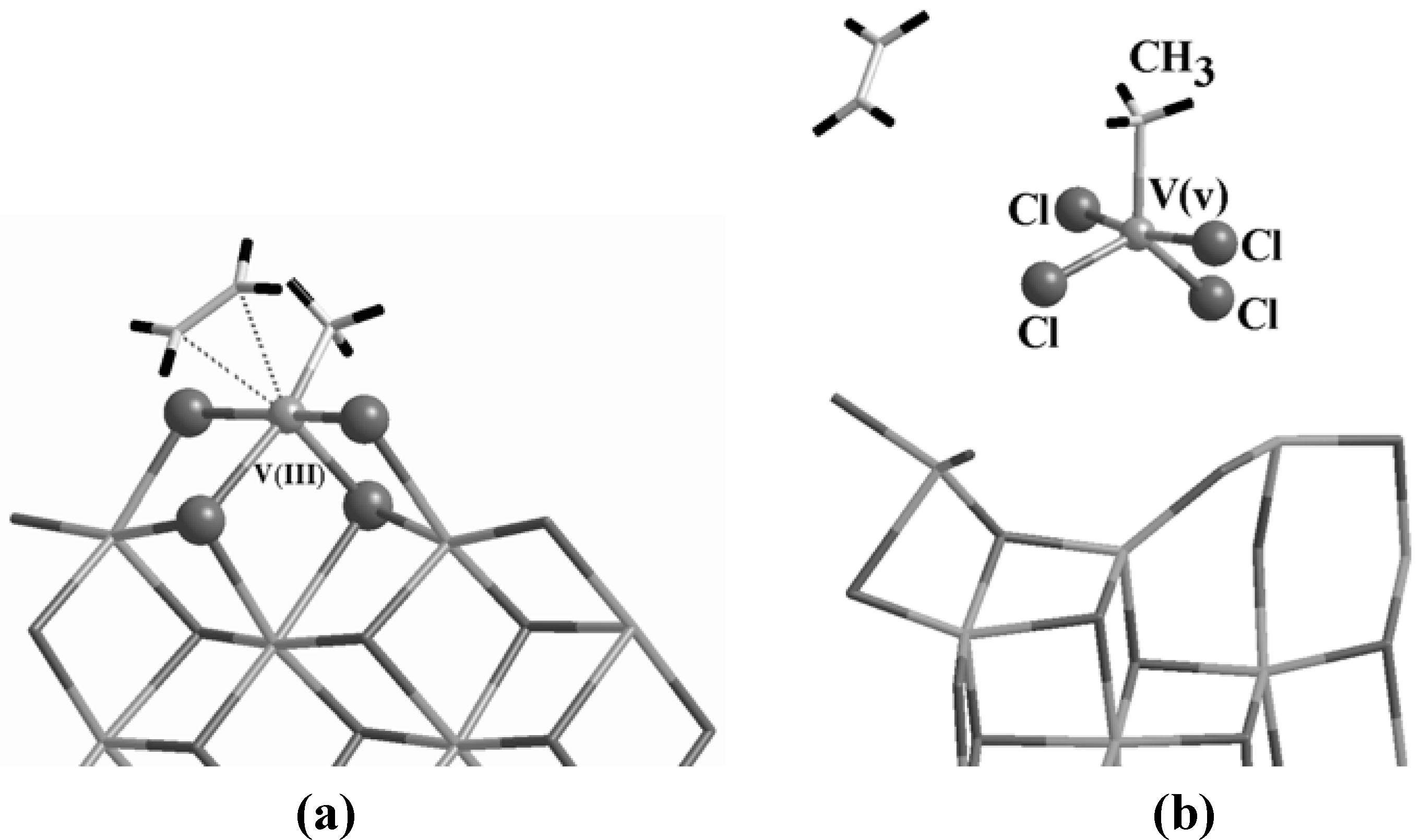
On this basis, we constructed a model of active site, reported in
Figure 7 (a), by analogy with both the V homogeneous system and the classical Ti heterogeneous case, by keeping in mind that the electronic structure and hence the chemistry of V are nevertheless different from Ti: V has an ionic radius of 0.59 Å, which represents a non-negligible difference with respect to the Ti and Mg ionic radii reported in the introduction. Furthermore V is 5-fold, having two 4
s and three 3
d electrons in the valence shell. Differences in both the binding and the behavior of the site during the catalytic activity have then to be expected.
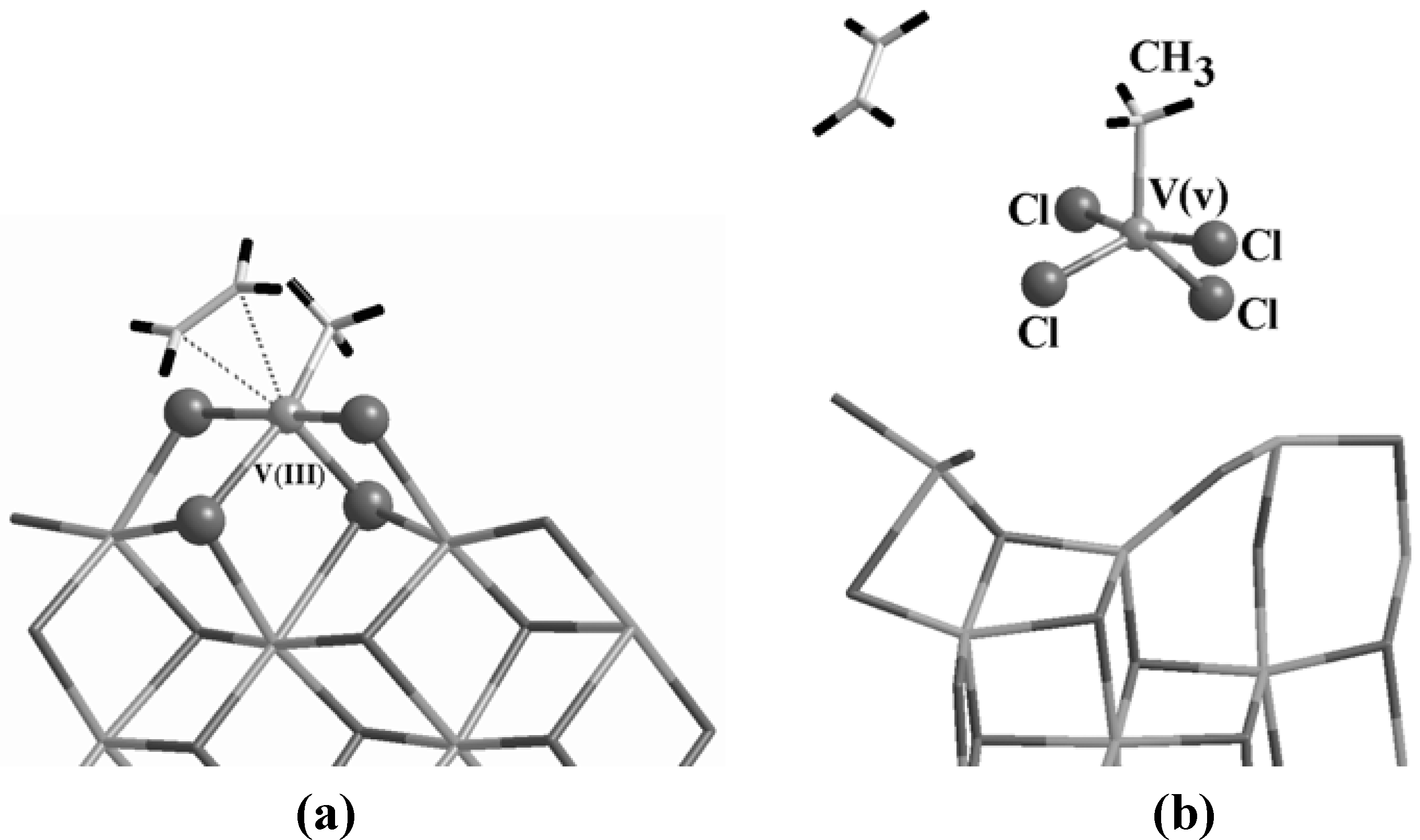
Figure 7.
The model for an active V center supported on MgCl
2 (a) as deduced by analogies with the homogeneous system of
Figure 6. During the dynamics, the complex is destabilized as in (b) and the V reverts to an unbound 5-fold molecule leaving the support.
Figure 7.
The model for an active V center supported on MgCl
2 (a) as deduced by analogies with the homogeneous system of
Figure 6. During the dynamics, the complex is destabilized as in (b) and the V reverts to an unbound 5-fold molecule leaving the support.
Indeed this is the case. The proposed configuration is stable prior to reduction and alkylation and, once activated, a complex with ethylene, whose geometry is reported in
Figure 7 (a), could be formed in a barrierless way. However, the complexation energy is only 4.3 kcal/mol, more than a factor of two weaker than for Ti (see
Table 1). Furthermore, after ~0.6 ps of free dynamics, this complex is destabilized (
Figure 7 (b)), the ethylene is released and the V(III) takes from the support the Cl atoms that in
Figure 7 (a) have been evidenced as balls. The V adduct eventually reverts to a V(v) molecule leaving the support that, in turn, reconstructs.
However, this has not to be regarded as a negative result. On the contrary, it seems to be in agreement with experimental evidence. Early works of the group of Zambelli [
14,
30] have shown that the polymerization of propene by a V-based system is not completely regiospecific, but rather consits of a binary copolymer in which propene molecules are chemically bound to each other in head-to-tail and tail-to-head units. This experimental fact does not agree with the symmetry of the assumed hexacoordinated model [
14]. In fact, this observation induced the authors of Ref. [
14] to reexamine their assumptions and eventually a pentacoordinated V(III) model was shown to be more appropriate to interpret the experimental outcome.
In our case, however, the large MgCl2 substrate does not allow us to infer that something similar might hold. For this reason, we chose to follow an unbiased approach by looking at the formation of the active center starting from the precursor molecule used in experiments. Hence, we simulated the deposition of a VOCl3 molecule on the (110) surface of the support. This is the most common V-based molecule and represents the standard precursor used in any preparation of homogeneous systems. When the molecule comes in contact with the active surface, it forms stable bonds with the substrate with a binding energy of ~20.0 kcal/mol.
Another well-known experimental fact is that a V-based catalyst always presents a V in the formal oxidation state III [
14,
31,
32], so that we have to remove and substitute with hydrocarbons the dangling atoms on the pristine VOCl
3 adduct deposited on the surface in order to activate the site. Reduction and alkylation were simulated by eliminating all the dangling Cl and replacing the oxygen with a methyl group. The resulting active center is sketched in
Figure 8 (a), where the π-complex with an ethylene monomer is shown. It is interesting to observe that the geometry of this site is such that three Cl atoms form a bridge to the Mg of the support, one coordination site is the metal-carbon bond and the olefin occupies a fifth empty site. Hence, our V(III) center, that turned out to be stable upon dynamics, has a pentacoordinated structure. This is in complete agreement with the results for homogeneous systems suggested by Zambelli and Allegra [
14].
By performing a constrained dynamics analogous to what we did for Ti, we were able to insert the olefin in the V-CH
3 bond, observing no destabilization phenomena. The main phases are reported in
Figure 8 (b) and (c), referring to the transition state and to the final product respectively. Initially, a π-complex is formed in a barrierless way, very similar to the case of Ti. However, the complexation
Figure 8.
A stable active V site obtained by depositing a VOCl3 molecule on the MgCl2 (110) surface and removing all the dangling atoms. (a) the p-complex, (b) the transition state and (c) the final product show how this site can polymerize without giving rise to destabilization phenomena.
Figure 8.
A stable active V site obtained by depositing a VOCl3 molecule on the MgCl2 (110) surface and removing all the dangling atoms. (a) the p-complex, (b) the transition state and (c) the final product show how this site can polymerize without giving rise to destabilization phenomena.
energy of an ethylene molecule is again lower than for Ti, amounting to ~4.7 kcal/mol. The insertion of the olefin in the metal-carbon bond follows a path analogous to the case of Ti, but with a much higher activation barrier, namely 28.1 kcal/mol.
Larger insertion barriers for V, as well as any other reaction occurring at a V catalytic center, such as chain termination, are not unexpected as reported by the group of Ziegler [
13,
33,
34] and can be partially ascribed to the more filled
d band of V (
d3) with respect to Ti (
d2). This enhances the coulombic repulsion with the π-bond of the incoming monomer. The consequence is that the reaction rate, and hence the production efficiency, is largely reduced with respect to Ti.
As another word of warning, we notice that, contrary to Ti sites like the 5-fold one, the coordination sphere around V is rather empty. Although this has little relevance for the polymerization of ethylene, if we wish to polymerize propene we have to face the problem of the stereoregularity of the polymeric chain, i.e. the insertion of subsequent monomers with the same orientation in the growing ploymer. In the case of the 5-fold site, we have shown that the Cl atoms bridging the Ti and the support are such that they ensure an
indirect tacticity control for steric reasons [
10]. In the case of the proposed V site, this is not the case. Work is still in progress, but we can anticipate that the approaching olefin can reorient rather freely during the complexation phase, thus jeopardizing any stereoselectivity.
This is in agreement with the experimental results on the primary 1,1 insertion of propylene as operated by V-based systems [
35,
36,
37]. The produced polymer, in fact, is found to be non-stereospecific, indicating a rather large freedom in the selection of the olefin enantioface already in the approaching stage. As a consequence, the stereoregularity of the polymer is strongly depressed, if not totally absent.
This seems to suggest that a tacticity control could be recovered only in the presence of stereomodifiers, underscoring the importance of cocatalysts. As a matter of fact, the engineering of the cocatalysts has become one of the major issues in the industrial design of catalytic systems.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}