SELDI-TOF-MS Analysis of Transcriptional Activation Protein Binding to Response Elements Regulating Carcinogenesis Enzymes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Methods
AP-1consensus forward: 5’-[NH2-(CH2)12]-CGCTTGATGAGTCAGCCGGAA-3’ AP-1 consensus reverse: 3’-GCGAACTACTCAGTCGGCCTT-5’ AP-1scrambled forward: 5’-[NH2-(CH2)12]-CAGGCGTGTCTGACTACAGAG-3’ AP-1 scrambled reverse: 3’-GTCCGCACAGACTGATGTCTC-5’Complementary oligonucleotides were annealed at a hybridized concentration of 25 µM in 10 mM K2HPO4/KH2PO4, 10 mM MgCl2 buffer (pH 7.4) at 90o C, 5 min, cooled to ambient temperature for 30 min and stored at 4º C. Annealing buffer was adjusted to pH 9.0 for coupling of the hybridized DNA to the PS20 protein chip array epoxide surface (Ciphergen Biosystems, Fremont, CA) via reaction of the 5'- amine on the leading oligonucleotide to the activated epoxy groups on the chip surface. To each spot 250 pmol oligonucleotide was applied in 10 µL and incubated at 37o C for 1 h in a humid chamber. To block residual active sites on the chip surface, 4 µL 1 M ethanolamine, pH 8.0 in annealing buffer was added for 30 min at 37º C , then washed with buffer (100mM Tris, 250 mM NaCl, 1mM EDTA, pH 7.4).
Results and Discussion
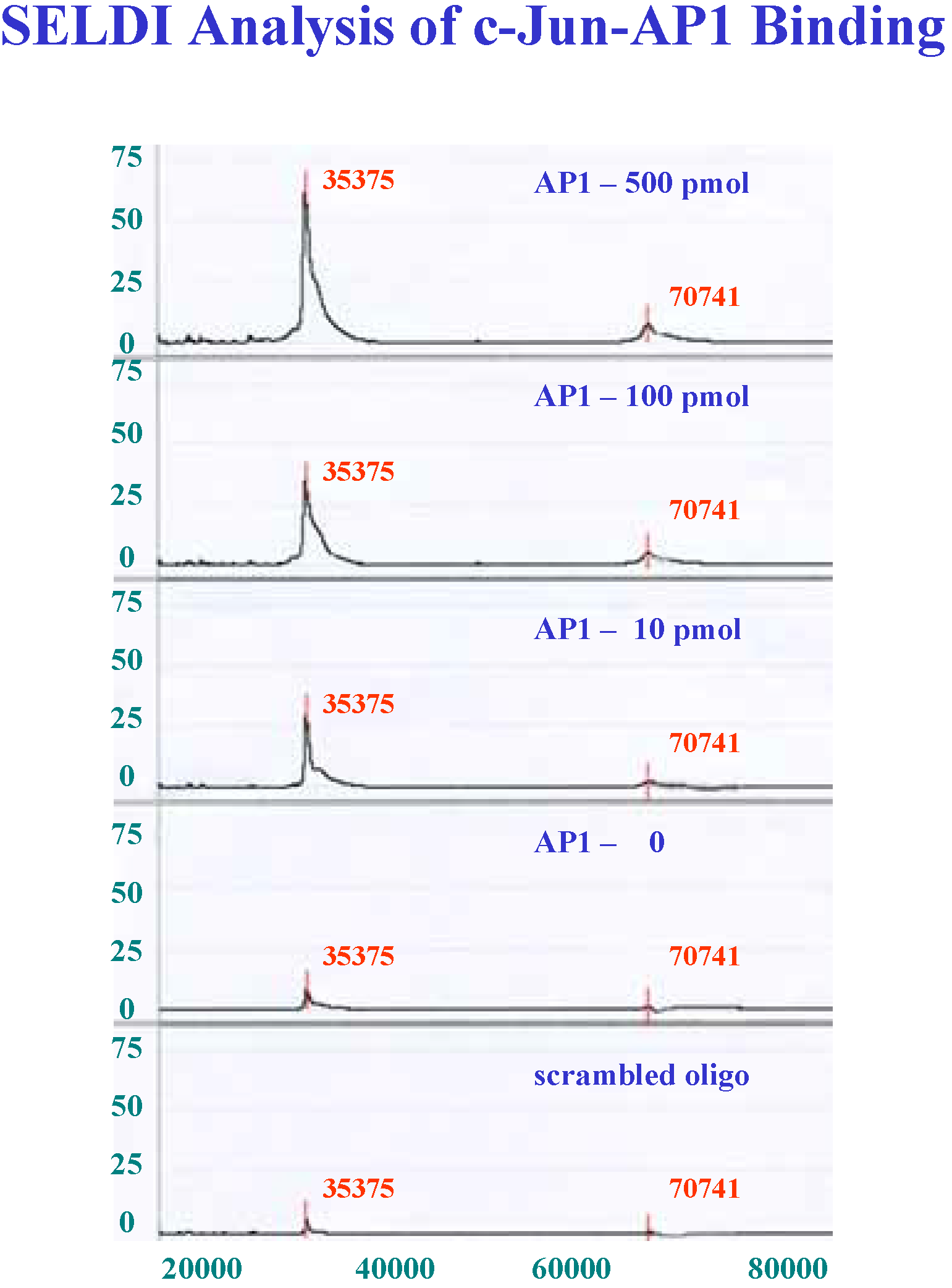
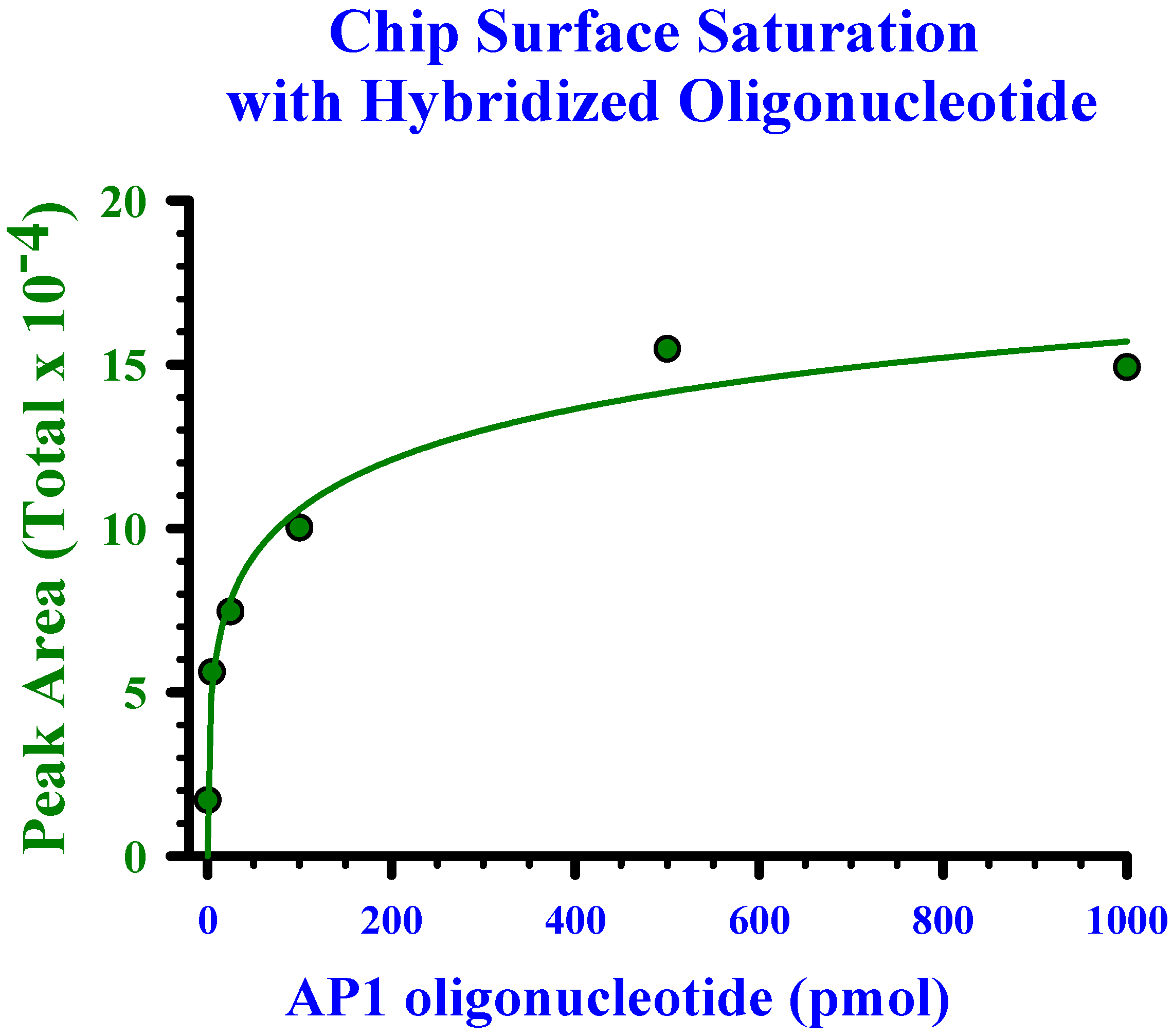
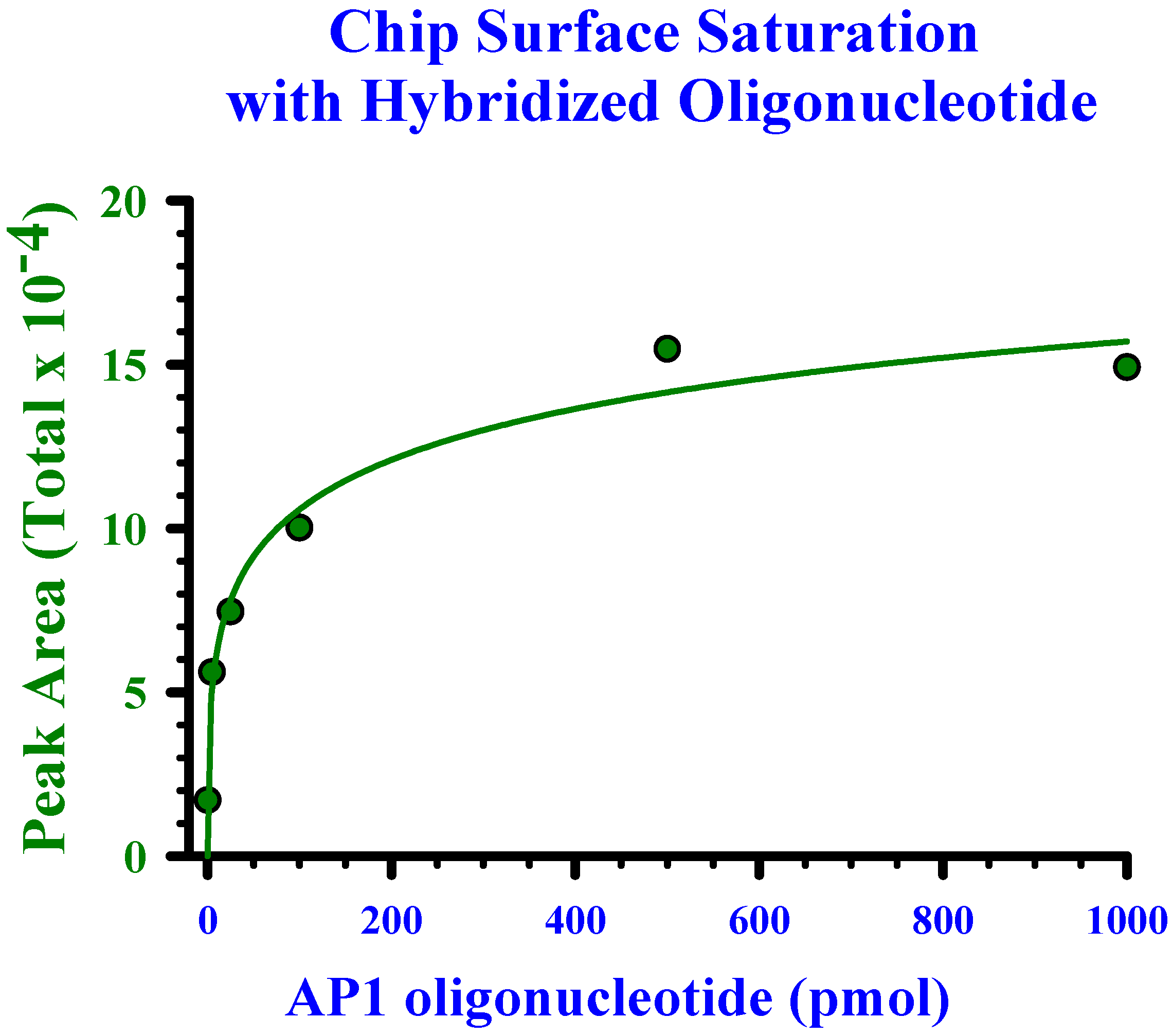
Determination of linearity and saturation of chip surface with oligonucleotide

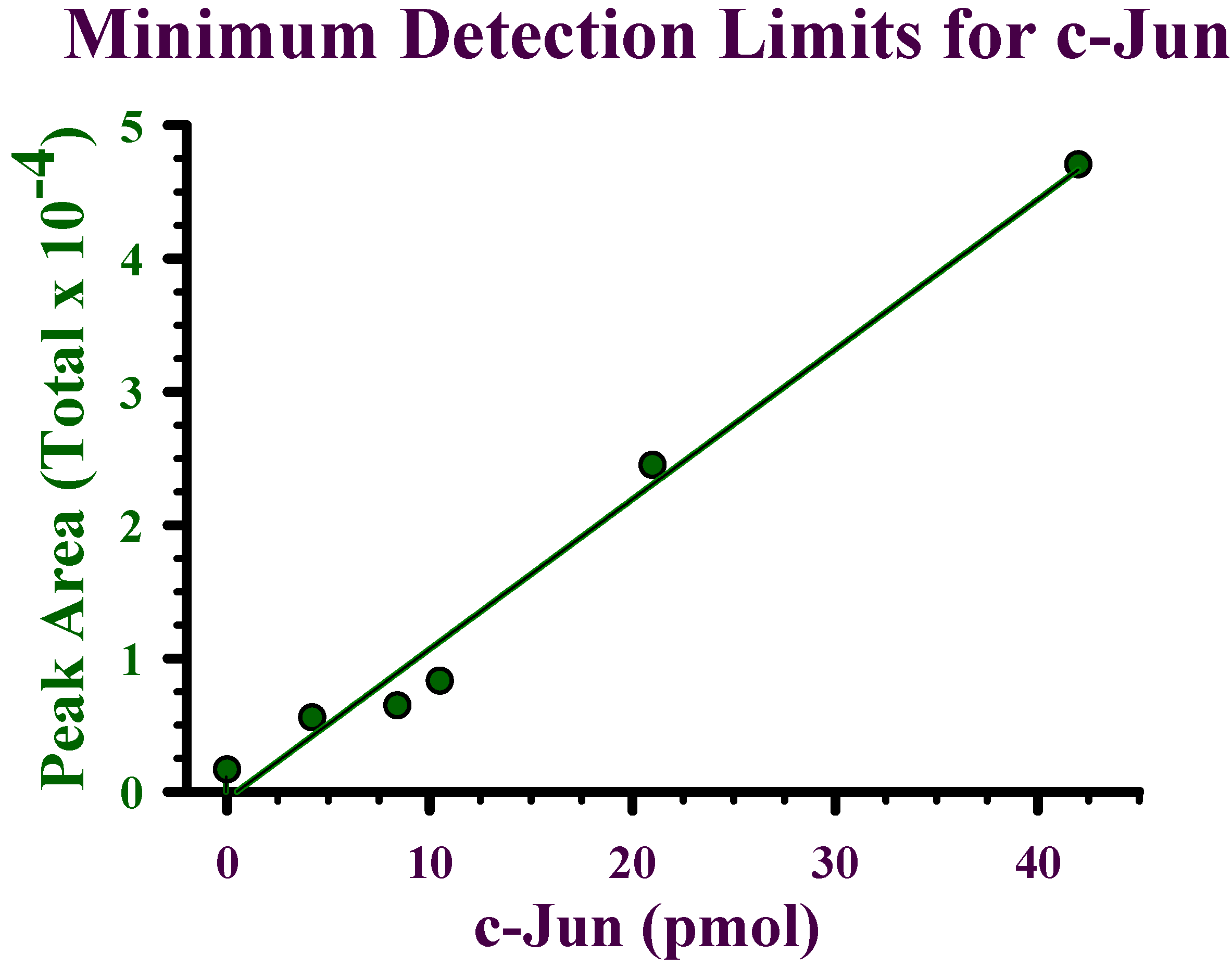
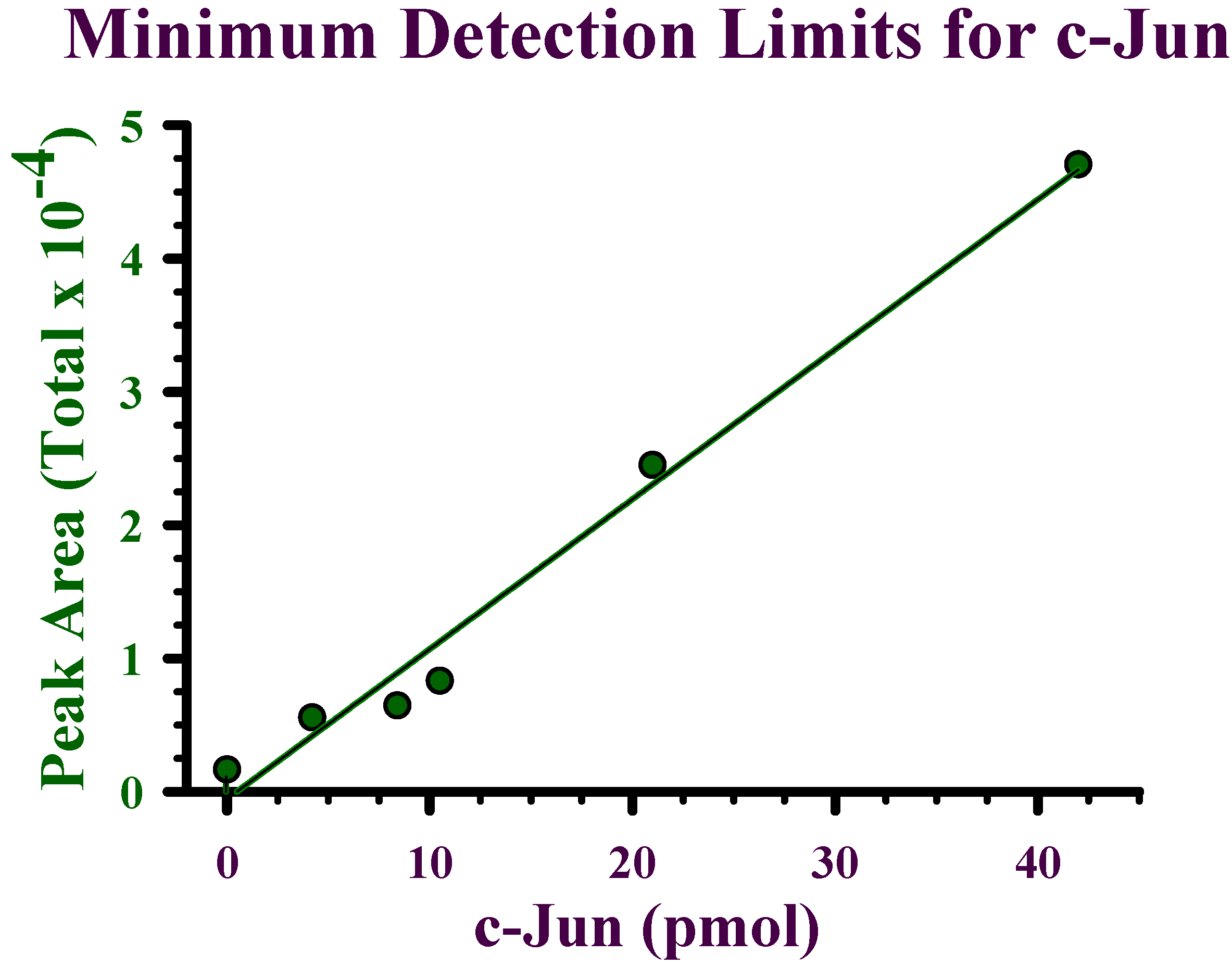
Detection limits for recombinant c-Jun
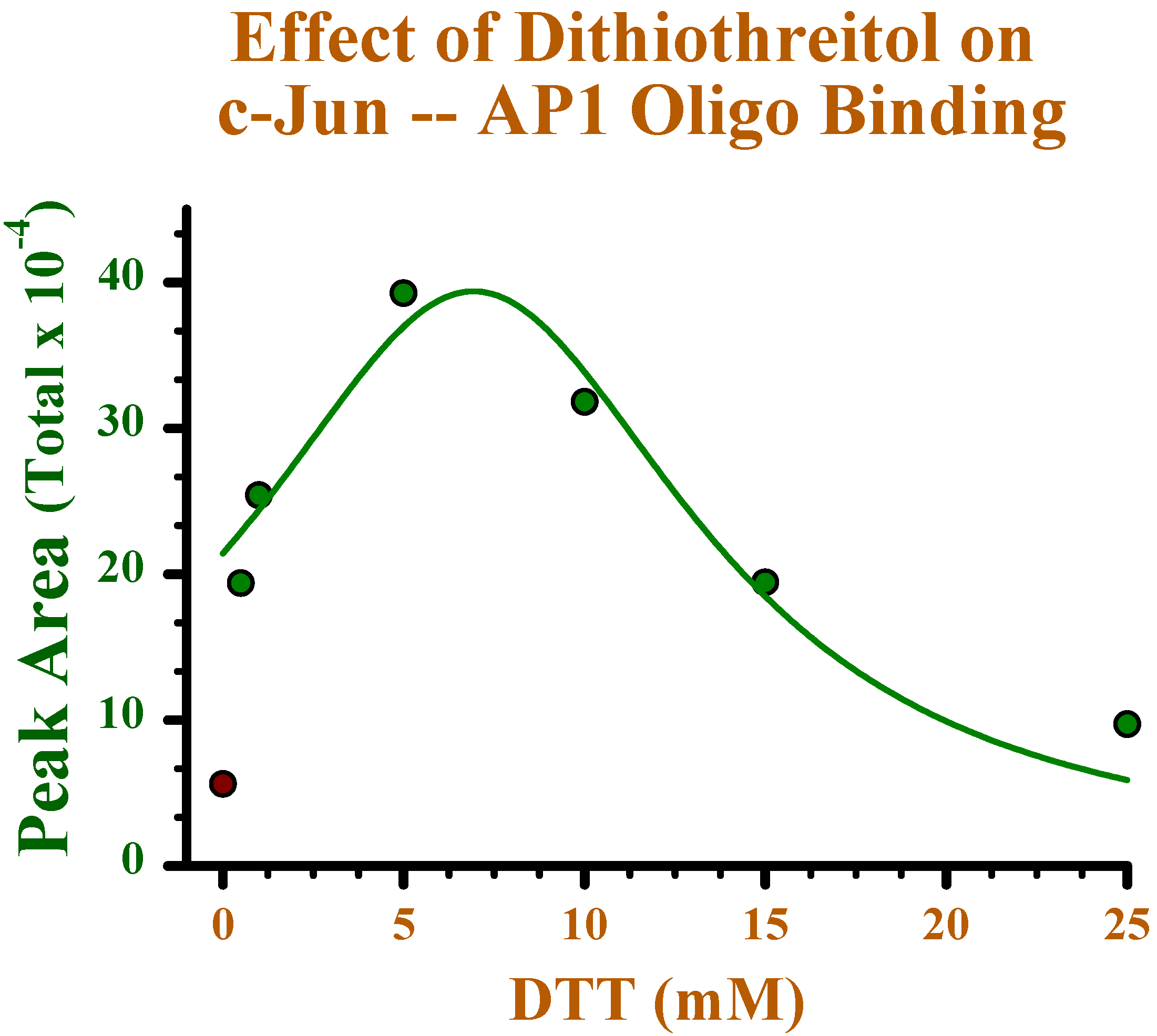
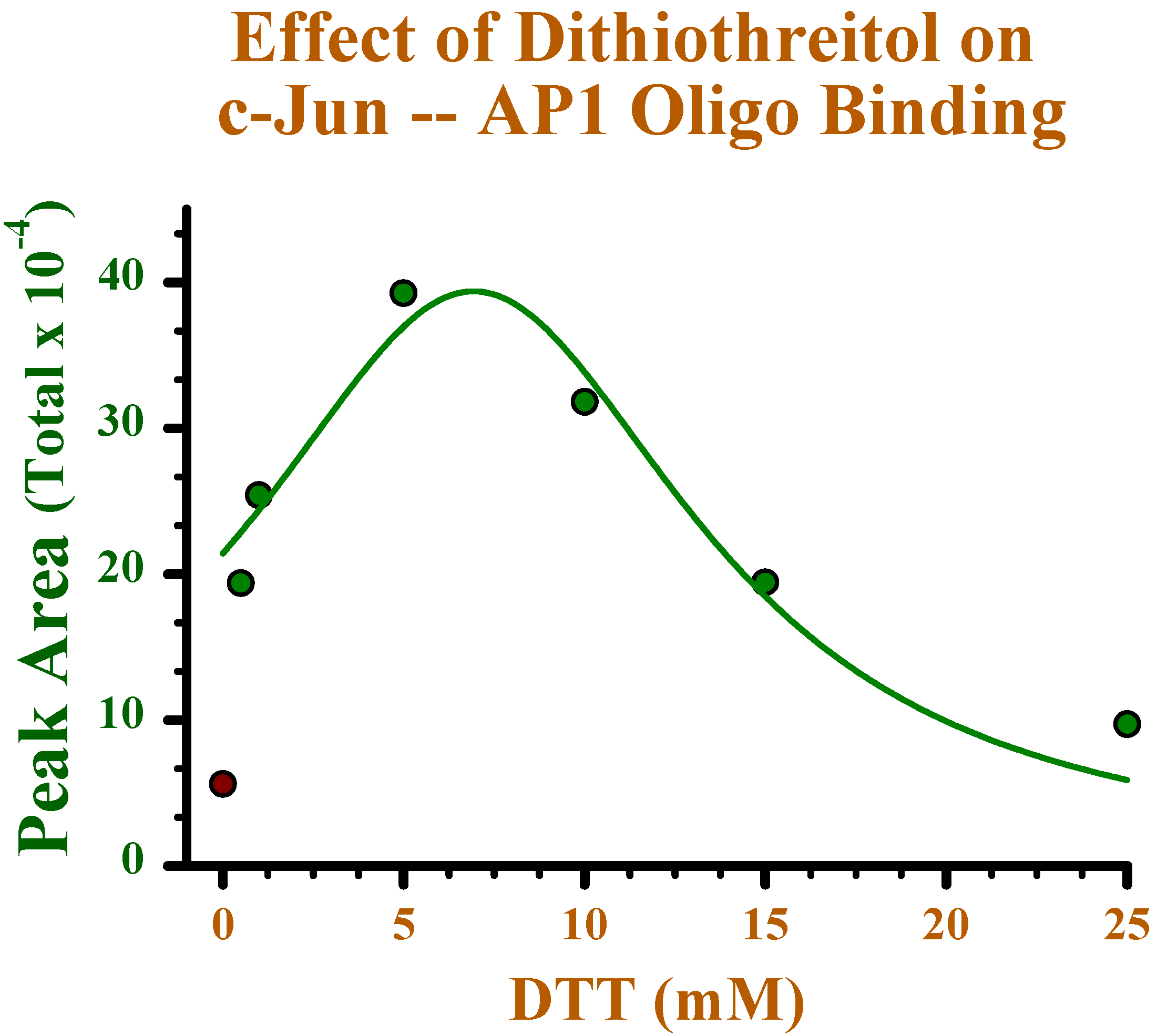
Effect of reducing conditions on binding of c-Jun to oligonucleotide
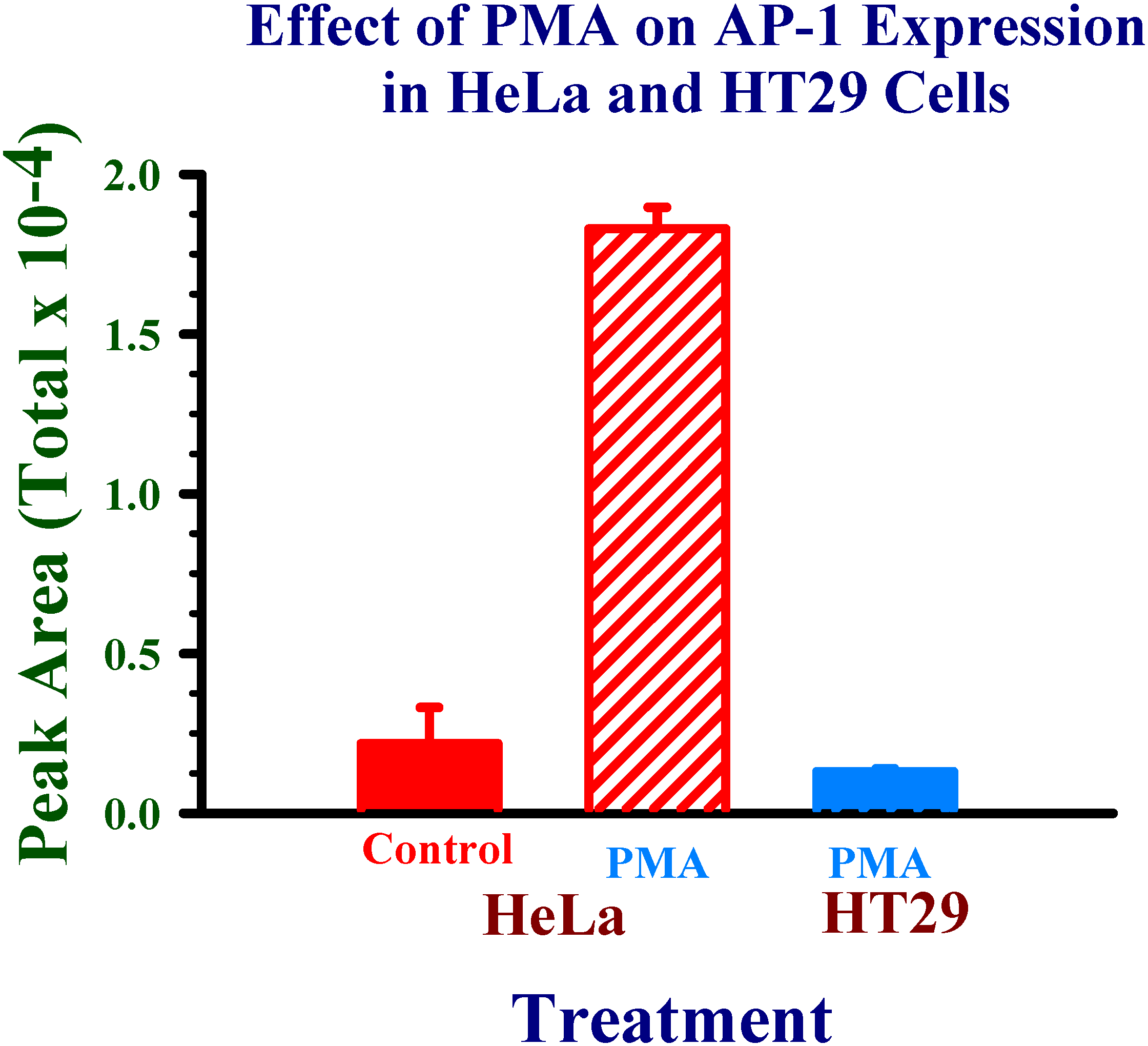
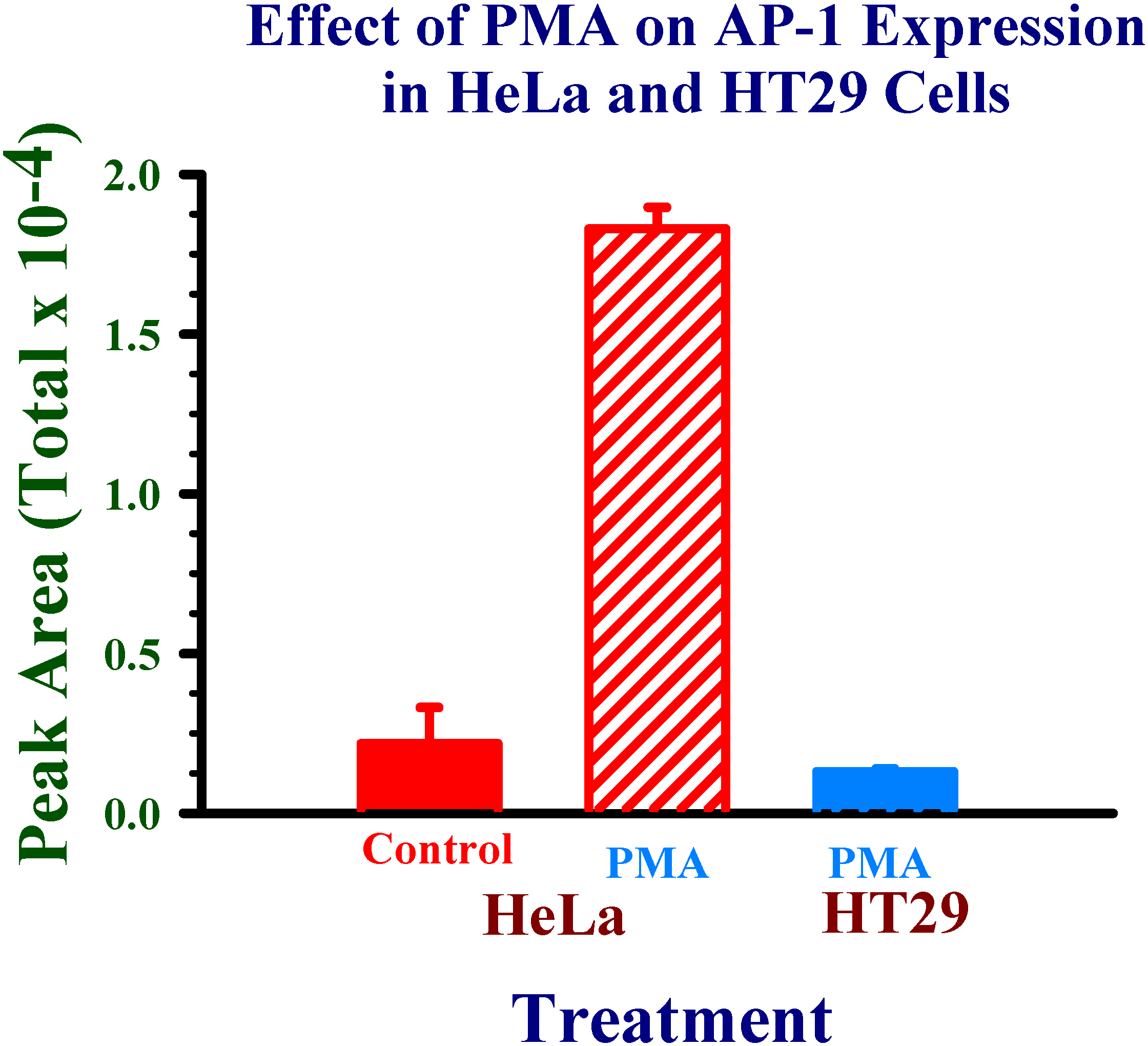
Application of experimental conditions to human cell nuclear protein extracts
Acknowledgments
References
- Parkinson, A. Biotransformation of xenobiotics. In Casarett & Doull’s Toxicology The Basic Science of Poisons; Klaassen, C.D., Ed.; McGraw-Hill Inc.: New York, 2001; pp. 133–237. [Google Scholar]
- Hayes, J.D.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a coordinately regulated defence against oxidative stress. Free Radic. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef] [PubMed]
- Prochaska, H.J.; De Long, M.J.; Talalay, P. On the mechanisms of induction of cancer-protective enzymes: a unifying proposal. Proc. Natl. Acad. Sci. USA 1985, 82, 8232–8236. [Google Scholar] [CrossRef] [PubMed]
- Talalay, P.; De Long, M.J.; Prochaska, H.J. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc. Natl. Acad. Sci. USA 1988, 85, 8261–8265. [Google Scholar] [CrossRef] [PubMed]
- Rushmore, T.H.; Pickett, C.B. Glutathione-S-transferases, structure, regulation, and therapeutic implications. J. Biol. Chem. 1993, 268, 11475–11478. [Google Scholar] [PubMed]
- Baba, T.; Mimura, J.; Gradin, K.; Kuroiwa, A.; Watanabe, T.; Matsuda, Y.; Inazawa, J.; Sogawa, K.; Fujii-Kuriyama, Y. Structure and expression of the Ah receptor repressor gene. J. Biol. Chem. 2001, 276, 33101–33110. [Google Scholar] [CrossRef] [PubMed]
- Barouki, R.; Morel, Y. Repression of cytochrome P450 1A1 gene expression by oxidative stress: mechanisms and biological implications. Biochem. Pharmacol. 2001, 61, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Ryu, J.H.; Kim, S.G. Activation of phosphatidylinositol 3-kinase by oxidative stress leads to the induction of microsomal epoxide hydrolase in H4IIE cells. Toxicol. Lett. 2001, 121, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Carmo-Fonseca, M. The contribution of nuclear compartmentalization to gene regulation. Cell 108, 513–52. [CrossRef]
- Kazlauskas, A.; Poellinger, L.; Pongratz, I. Two distinct regions of the immunophilin-like protein XAP2 regulate dioxin receptor function and interaction with hsp90. J. Biol Chem. 2002, 277, 11795–11801. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, M.; Pongratz, I.; Wilhelmsson, A.; Gustafsson, J.A.; Poellinger, L. Ligand-dependent recruitment of the Arnt coregulator determines DNA recognition by the dioxin receptor. Mol. Cell. Biol. 1993, 13, 2504–2514. [Google Scholar] [PubMed] [Green Version]
- Whitlock, J.P., Jr. Induction of Cytochrome P4501A1. Ann. Rev. Pharmacol. Toxicol. 1999, 39, 103–125. [Google Scholar] [CrossRef]
- Pollenz, R.S.; Barbour, E.R. Analysis of the complex relationship between nuclear export and aryl hydrocarbon receptor-mediated gene regulation. Mol. Cell. Biol. 2000, 20, 6095–6104. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.; Poynter, M.E.; Baeuerle, P.A. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radic. Biol. Med. 2000, 28, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.S.; Kennedy, T.P.; Whorton, A.R.; Sturrock, A.B.; Huecksteadt, T.P.; Ghio, A.J.; Hoidal, J.R. Reactive oxygen species from NAD(P)H:quinone oxidoreductase constitutively activate NF-kappaB in malignant melanoma cells. Am. J. Physiol. Cell Physiol. 2001, 280, 659–676. [Google Scholar]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366. [Google Scholar] [CrossRef] [PubMed]
- Moffat, G.J.; McLaren, A.W.; Wolf, C.R. Transcriptional and post-transcriptional mechanisms can regulate cell-specific expression of the human Pi-class glutathione S-transferase gene. Biochem. J. 1997, 15, 91–95. [Google Scholar]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Toone, W.M.; Morgan, B.A.; Jones, N. Redox control of AP-1-like factors in yeast and beyond. Oncogene 2001, 20, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Oberley, L.W. Redox regulation of transcriptional activators. Free Radic. Biol. Med. 1996, 21, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, T.; Watanabe, J.; Kawajiri, K. Characterization of the LxxLL motif in the aryl hydrocarbon receptor: effects on subcellular localization and transcriptional activity. J. Biochem. (Tokyo) 2002, 131, 79–85. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, J.; Hankinson, O. A mutation in the aryl hydrocarbon receptor (AHR) in a cultured mammalian cell line identifies a novel region of AHR that affects DNA binding. J. Biol. Chem. 1997, 272, 31845–31854. [Google Scholar] [CrossRef] [PubMed]
- Forde, C.E.; Gonzales, A.D.; Smessaert, J.M.; Murphy, G.A.; Shields, S.J.; Fitch, J.P.; McCutchen-Maloney, S.L. A rapid method to capture and screen for transcription factors by SELDI mass spectrometry. Biochem. Biophys. Res. Commun. 2002, 290, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, G.; Vairetti, M.; Stivala, L.; Mirabelli, F.; Richelmi, P.; Orrenius, S. Demon-stration of nuclear compartmentalization of glutathione in hepatocytes. Proc. Natl. Acad. Sci. USA 1992, 89, 4412–4416. [Google Scholar]
- Voehringer, D.W.; McConkey, D.J.; McDonnell, T.J.; Brisbay, S.; Meyn, R.E. Bcl-2 expression causes redistribution of glutathione to the nucleus. Proc. Natl. Acad. Sci. USA 1998, 95, 2956–2960. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Murata, M.; Sachi, Y.; Nakamura, H.; Takeuchi, J.; Mori, K.; Yodoi, J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999, 274, 27891–27897. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nishiyama, Y.; Okada, K.; Hirota, K.; Matsui, M.; Yodoi, J.; Hiai, H.; Toyokuni, S. Induction and nuclear translocation of thioredoxin by oxidative damage in the mouse kidney: independence of tubular necrosis and sulfhydryl depletion. Lab. Invest. 1997, 77, 145–155. [Google Scholar] [PubMed]
- Kadonaga, J.T.; Tjian, R. Affinity purification of sequence-specific DNA binding proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 5889–5893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2002 by MDPI (http://www.mdpi.org).
Share and Cite
Bischoff, S.R.; Kahn, M.B.; Powell, M.D.; Kirlin, W.G. SELDI-TOF-MS Analysis of Transcriptional Activation Protein Binding to Response Elements Regulating Carcinogenesis Enzymes. Int. J. Mol. Sci. 2002, 3, 1027-1038. https://doi.org/10.3390/i3101027
Bischoff SR, Kahn MB, Powell MD, Kirlin WG. SELDI-TOF-MS Analysis of Transcriptional Activation Protein Binding to Response Elements Regulating Carcinogenesis Enzymes. International Journal of Molecular Sciences. 2002; 3(10):1027-1038. https://doi.org/10.3390/i3101027
Chicago/Turabian StyleBischoff, Steve R., Mahfuz B. Kahn, Michael D. Powell, and Ward G. Kirlin. 2002. "SELDI-TOF-MS Analysis of Transcriptional Activation Protein Binding to Response Elements Regulating Carcinogenesis Enzymes" International Journal of Molecular Sciences 3, no. 10: 1027-1038. https://doi.org/10.3390/i3101027