Real-space renormalization group study of the Hubbard model on a non-bipartite lattice

1
Department of Chemistry, Purdue University, West Lafayette, IN 47907, USA
2
The Fritz Haber Research Center for Molecular Dynamics, the Hebrew University, Jerusalem 91904, Israel
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2002, 3(1), 4-16; https://doi.org/10.3390/i3010004
Submission received: 9 October 2001
/
Accepted: 2 December 2001
/
Published: 31 January 2002
(This article belongs to the Special Issue From Nanoclusters to Proteins)
Abstract
:We present the real-space block renormalization group equations for fermion systems described by a Hubbard Hamiltonian on a triangular lattice with hexagonal blocks. The conditions that keep the equations from proliferation of the couplings are derived. Computational results are presented including the occurrence of a first-order metal-insulator transition at the critical value of U/t ≈ 12.5.
1 Introduction
The ability to prepare arrays with made to measure electronic properties has generated much recent activity[1]. This was made possible by the development of synthetic methods for the preparation of, so called, quantum dots. For our purpose these dots act like designer atoms. The properties of individual dots can be designed by the choice of composition and size. Size is a particular variable we wish to emphasize. Quantum dots are aggregates of tens or hundreds of atoms (or molecules) and so are nanoscale building blocks. The size of the dots is however small enough so that, since the highest lying electron(s) are confined to the volume of the dot, the electronic energy levels are still discrete. For a chemist, it is convenient to think of the higher-most electrons of each dot as the valence electron of an atom[2].
These dots can self assemble into a planar array. For, e.g., Ag nanodots, the packing is hexagonal. The reason why adjacent dots in such an array may have an unequal charge distribution is related to their nano-scale size. Unlike the smaller ordinary atoms, a quantum dot does not require a high energy cost to induce an electron transfer from a neighboring dot. This is technically stated by saying that the charging energy of a dot is atypically low. Electrostatic considerations suggest that the charging energy scales inversely with size and this has been experimentally born out. Metallic quantum dots have charging energy of well below an eV while atoms have charging energies of at least several eV’s.
The current attention on architecture with quantum dots building blocks is partly motivated by potential applications of such nanoscale quantum devices. In this paper we explore how corresponding computations can be carried out. The technical problem is that the Coulombic repulsion between two electrons (of opposite spins) that occupy the same dot cannot be described in a one electron approximation. It requires allowing for correlation of electrons. The simplest model Hamiltonian that can describe the Coulombic (or charging energy) effects is due to Hubbard[3]. This paper is a study of exploring a computationally tractable method for solving for the states of the Hubbard Hamiltonian for a lattice. The development makes essential use of the nature of the intended applications. At low temperatures, only those states of the array that are quite close to the ground state are important[4].
The Hubbard model is the simplest model one can use to study the Mott metal-insulator transition, i.e. the transition from a metallic to an insulating phase driven by correlations between electrons[5]. Despite the extensive efforts in studying the Hubbard model [3, 6, 7] there are still difficulties in extending the one-dimensional results to higher-dimensions. The difficulties mainly come from the electron correlations inherent in this model, which becomes more complicated in two and three-dimensions. Several methods have been developed to treat these problems such as exact diagonalization methods[8], quantum Monte-Carlo simulations[9] and a variety of approximation techniques like mean-field theory[10], Green’s function decoupling schemes[11], functional integral formulations [12], variational approach[13] and perturbation expansion [14]. It is already known that the numerical methods suffer from the finite-size effect and perturbation calculations are not useful when dealing with the intermediate-to-strong correlations. In this context, using the real-space renormalization group method seem to be a promising direction because of its nonperturbative nature. In fact, this method has been applied to quantum lattice systems[15]. For example, the density matrix renormalization group[16] has provided a revolutionary way to obtain reliable results for the one-dimensional Hubbard model and the numerical renormalization group for interacting finite Fermi systems[17] and for excitations in atoms[18]. To apply the renormalization group method to higher dimensions, the simplest way is to use the block renormalization group method(BRG)[19]. The BRG method has already been applied to interacting electrons in two-dimensional systems for both bipartite lattices[20] and more recently for nonpartite lattices such as the triangular one[21].
In this paper, we present the real-space block renormalization group equations for fermion systems on a triangular lattice with hexagonal blocks. By using operator transformations, we obtain the conditions that make the renormalized Hamiltonian in the bare Hubbard model have the same structure as the old one, i.e. the renormalization group equations in the parameters space with constant dimension will remain the same, without any proliferation of couplings. Finally, using numerical calculations we show that the metal-insulator transition occurs at the critical value of U/t ≈ 12.5.
2 Real-space renormalization group
In this section, we present a general computational procedure for applying the BRG method for interacting fermions on a lattice using the Hubbard Hamiltonian. As an example we will take the hexagonal block on a triangular lattice. The Hubbard Hamiltonian can be written as follows,
where t is the nearest-neighbor hopping term, U is the local repulsive interaction and µ is the chemical potential. creates(annihilates) an electron with spin σ in a Wannier orbital located at site i; the corresponding number operator is and < > denotes the nearestneighbor pairs. H.c. denotes the Hermitian conjugate.
The essence of the BRG method is to map the above many-particle Hamiltonian on a lattice to a new one with fewer degrees of freedom and with the same low-lying energy levels [22]. Then the mapping is repeated leading to a final Hamiltonian for which an exact solution can be obtained. The procedure can be divided into three steps: First divide the N−site lattice into appropriate ns−site blocks labeled by p(p = 1,2,...,N/ns) and separate the Hamiltonian H into intrablock part HB and interblock HIB
where
and
in which i(p) denotes the ith site on the pth block and i(p,b) denotes the border site of the block p.
The second step is to solve Hp exactly to get the eigenvalues Epi and eigenfunctions Φpi(i = 1,2,...,4ns). Then we can build the eigenfunctions of HB by direct multiplication of Φpi, which can be written as |ΨB(i1,i2,...,iN/ns) >= |Φ1i1 > |Φ2i2 > ...|ΦN/nsiN/ns > (i1,i2,... ∈ {1,2,...,4ns}).
The last step is to treat each block as one site on a new lattice and the correlations between blocks as hopping interactions. The original Hilbert space has four states per site. By following the above procedure one obtains an equivalent Hamiltonian with (4ns)N/ns = 4N, degrees of freedom, which is the same as the original Hamiltonian. But in the realistic case, if we only care about the properties related to some special energy levels of the system it is not necessary to keep all the states for a block to obtain the new Hamiltonian. For example, when studying the metalinsulator-transition[23], we may only need to consider the ground state and the first excited state energies.
The above scheme is a general procedure for applying the BRG method. In order to make the new Hamiltonian more tractable, it is desirable to make it have the same structure as the original one, i.e. the reduction in size should not be accompanied by a proliferation of new couplings. Then we can use the iteration procedures to solve the model. To achieve this goal it is necessary to keep only 4 states in step 2, which can be understood from the following renormalized intrasite Hamiltonian. The four selected states are taken to be
where is the creation(annihilation)operator of the block state and their corre-sponding energies are Ei (i = 1, 2, 3, 4).
Our next task is to rewrite the old Hamiltonian H = HB + HIB in the space spanned by the truncated basis
where the truncated basis is given by
In order to avoid proliferation of additional couplings in H′, the four states kept from the block cannot be arbitrarily chosen. Some definite conditions must be satisfied in order to make H′ have the same structure as H. Substituting H into H′ and using the product of different operators ( see Table I) we can get the expression for Hp
Note that by keeping only four states from the block states in the beginning gives no other extra couplings in the new Hamiltonian.

{kind=link}
{kind=link}
| < 0|′ | <↑ |′ | <↓ |′ | <↑↓ |′ | |
| |0 >′ | ||||
| | ↑>′ | ||||
| | ↓>′ | ||||
| | ↑↓>′ |
∗In this table the product reads , etc.
Comparing the above intrasite Hamiltonian with Eq.(1), we get the next conditions in order to copy the intrasite structure of the old Hamiltonian, i.e. E3 = E4. Because of the additional vacuum energy E1 in the new Hamiltonian we rewrite the intrasite part of Eq.(1) as
where we introduce another parameter K to the original system and Ii is a unit operator. The new intrasite Hamiltonian is given by
Then the renormalized parameters U, µ and K can be obtained from the following relations
in which E1, E2 and E3 are functions of the old parameters t, U, µ, K.
For the half-filled case, µ = U/2, HB can be expressed as
with the initial value of . By using the particle-hole symmetry, E1 = E2, the renormal-ization group equations for U and K take the form
To illustrate this procedure, let us consider the triangular lattice with hexagonal blocks as shown in figure 1. For this non-bipartite lattice the interaction between blocks can be written as
Figure 1.
Schematic diagram of the triangular lattice with hexagonal blocks. Only two neighboring blocks p and p′ are drawn here. The dotted lines represent the interblock interactions and solid line intrablock ones.
Figure 1.
Schematic diagram of the triangular lattice with hexagonal blocks. Only two neighboring blocks p and p′ are drawn here. The dotted lines represent the interblock interactions and solid line intrablock ones.
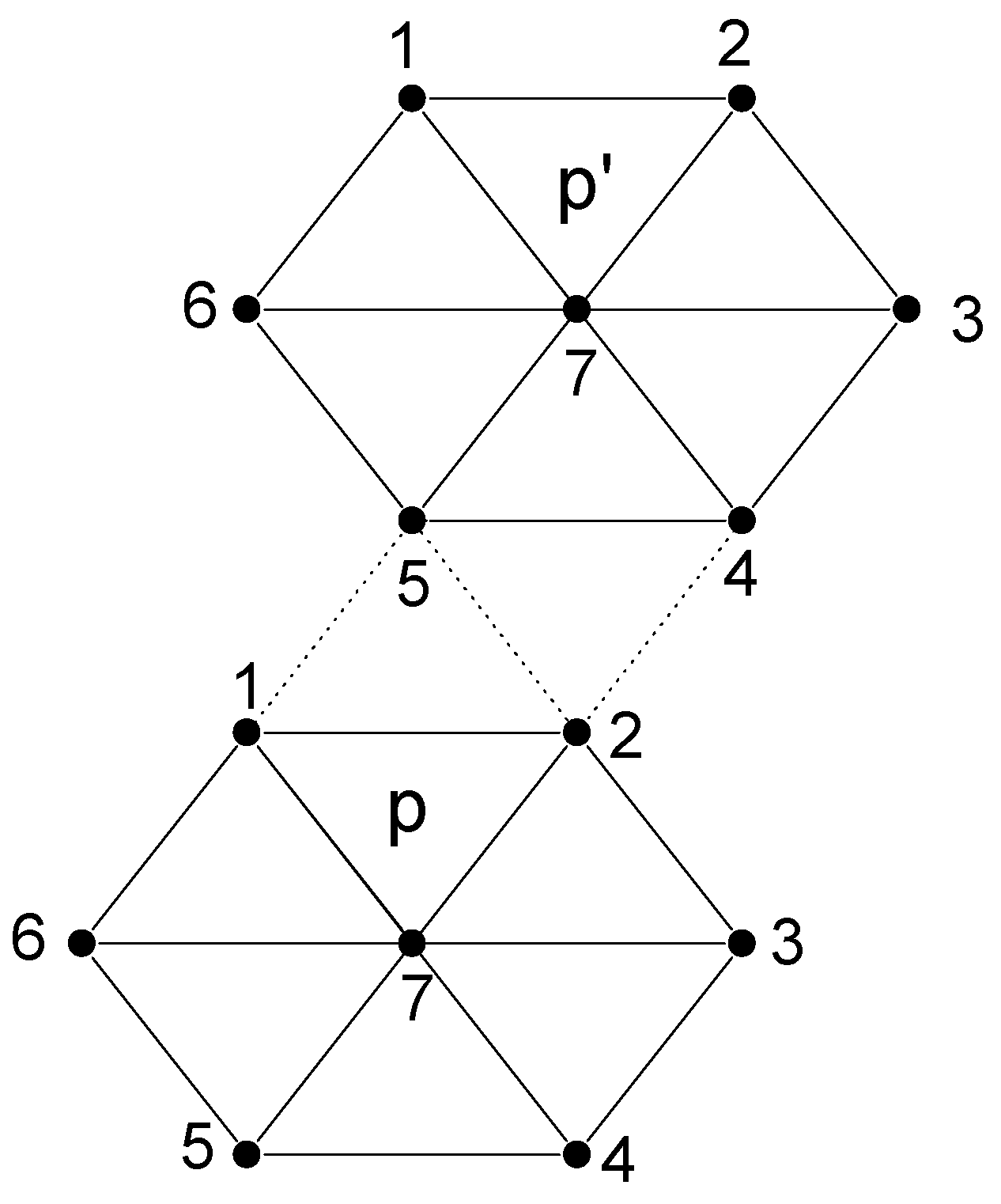
Since we would like to keep Vpp′ of the form
we use the product transformation in Table I to simplify Eq. (20),
It can be easily seen now that in order to make all the extra couplings vanish, it is necessary to make further restrictions upon the selected states,
Using similar calculations to the other neighboring interactions of the block, we can finally obtain the following conditions,
for all the border sites on the block. Then the new hopping term becomes,
where ν represents the number of couplings between neighboring blocks. In figure 1 ν = 3. The last renormalization group equation is readily obtained
Up to now, we have given a general discussion of the conditions under which no proliferation of couplings results from the application of the BRG method to non-partite lattice. Because on the border of non-partite lattice block, there is only one type of sites, the above procedures can be extended to other lattices with different dimensions or blocks without much difficulty.
3 State Selections
After deriving the conditions for the renormalization group equations, the next task is to select states that satisfy these conditions. At this stage the symmetry properties of the lattice play an important role. From Eqs.(23) and (24), it can be easily seen that if we assume the particle number in the state |0 >′ to be Ne − 1, then in | ↑>′, | ↓>′ and | ↑↓>′, there should be Ne, Ne, Ne + 1 particles respectively. Moreover if the spin in |0 >′ is Sz, the spins for |↑>′,| ↓>′ and | ↑↓>′, should be Sz + 1/2, Sz − 1/2 and Sz. The total electron number Ne and the spin Sz for each block are good quantum numbers since their corresponding operators commute with the Hubbard Hamiltonian. So when we diagonalize the Hubbard Hamiltonian of the selected block, we keep Ne and Sz fixed to be (Ne-1, Sz), (Ne, Sz+1/2), (Ne, Sz-1/2) and (Ne+1, Sz) respectively. Thus we obtain four groups of eigenenergies and eigenstates corresponding to the above quantum numbers. From each group, we select the lowest-energy state to form the final required four states. It should be mentioned that the lowest-energy state has to be selected according to definite special symmetry considerations, which shall be discussed in the next paragraph. In order to obtain the insulating to conducting gap, which is defined to be the energy difference between extracting one electron from the system and adding one electron to it, Ne is selected to be equal to Ns. For Sz, we choose it to be zero so as to make the block have the same spin property as the one-site. So now the renormalized lattice will be composed of N/ns renormalized ”sites” with N/ns ”particles”.
Instead of forcing the above conserved quantities upon the selected states in analogy to the one site properties in a consistent way, here we get them directly from the no-coupling-proliferation conditions. λ does not depend on σ in Eq. (25), this can be guaranteed by the particle-hole symmetry, which means that only in half-filled lattice can the renormalized Hamiltonian have exactly the same form as the original one[24]. Moreover, the irrelevance of λ with respect to the border site i(p,b) can be shown by requiring the selected states to belong to the same irreducible representation of the spatial group of the lattice. For the triangular lattice with hexagonal blocks, the Hamiltonian is invariant under C6v [25]. So if we choose the same one-dimensional irreducible representation of the group C6v for |0>′, | ↑>′, |↓>′ and | ↑↓>′, the conditions can be satisfied.
4 Numerical Calculations and discussions
The insulating to conducting gap ∆g[7] and the ground state energy per site Eg for the half-filled Hubbard model on a triangular lattice with hexagonal blocks can be written as
and
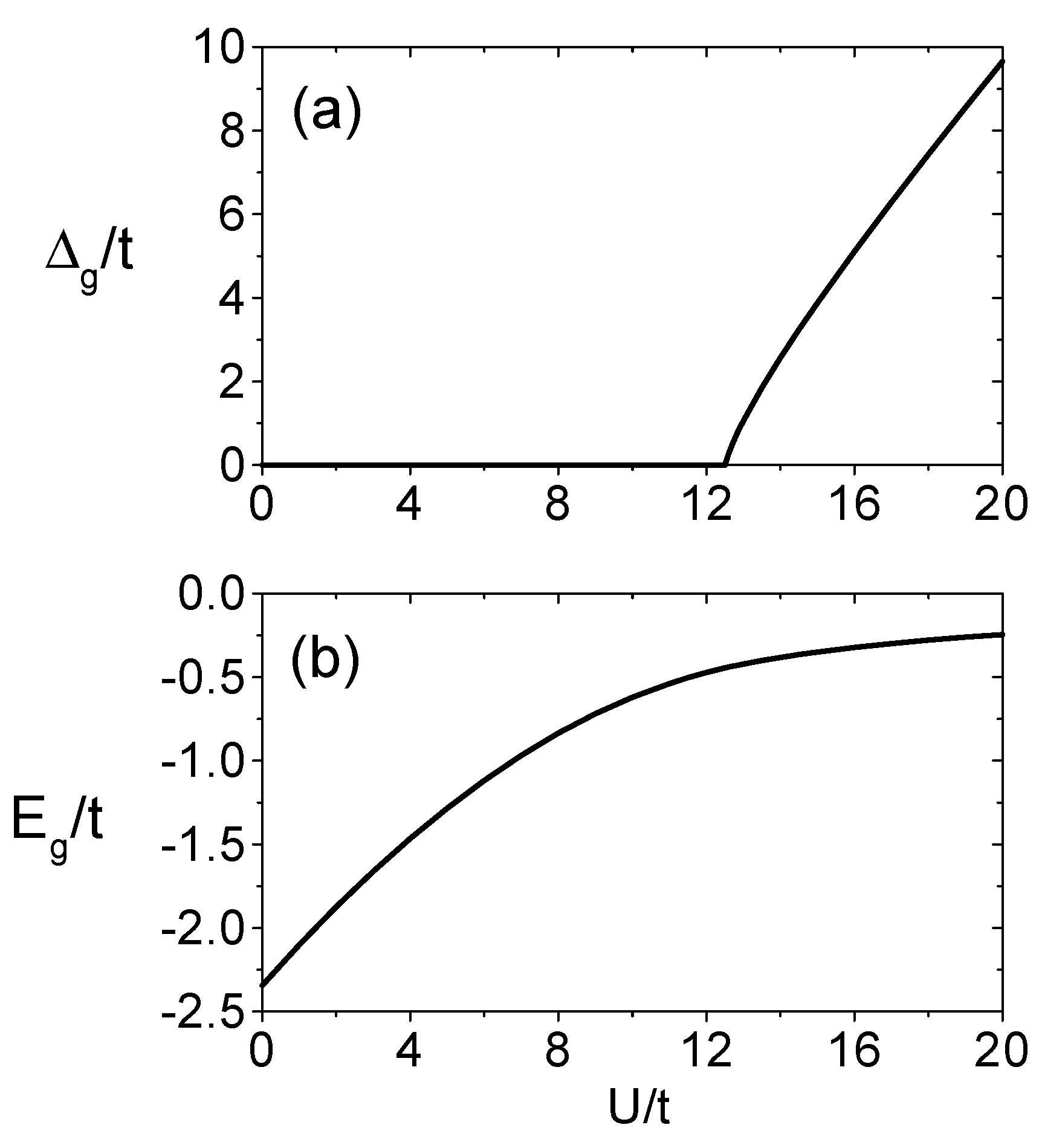
where n denotes the number of iterations in the renormalization equations. As n → ∞, the lattice containing 7n sites will be renormalized to one ’site’, which can be solved exactly to obatin the above equations. U/2 is added in Eq.(29) in order to compensate the chemical potential energy substracted in the original Hamiltonian. The metal-insulator-transition for this lattice can be examined by considering the energy gap ∆ as a function of (U/t). If the insulating gap disappears the system exhibits metallic behavior. Otherwise it is insulating.
Figure 2 shows the numerical results for the scaled gap ∆g/t as a function of (U/t). There is a first-order phase transition at (U/t)c ≈ 12.5. This finding is quite different from the case of half-filled square lattice where an insulating gap exists for arbitrarily small values of U/t. This is because there is a perfect nesting of the Fermi surface on a square lattice, which makes the model unstable toward antiferromagnetism as soon as a nonzero U is turned on, driving the system to an insulating state.
On the triangular lattice, for the lack of perfect nesting, metal-insulator-transition takes place at a finite value of U/t. But because there is no exact solution to the two-dimensional Hubbard model, there are still controversies upon the exact value for this critical point. The situation becomes more complicated and subtle because of the frustrations inherent in the triangular lattice, which may induce a nontrivial competitions among different magnetic phases. In the Hartree-Fock calculations by Krishnamurthy et al.[26, 27] they found that for small U/t the system is a paramagnetic metal which turns to a metal with incommensurate spiral spin-density wave at Uc1/t = 3.97.
Figure 2.
U/t dependence of (a) the insulating gap and (b) the ground-state energy per site.
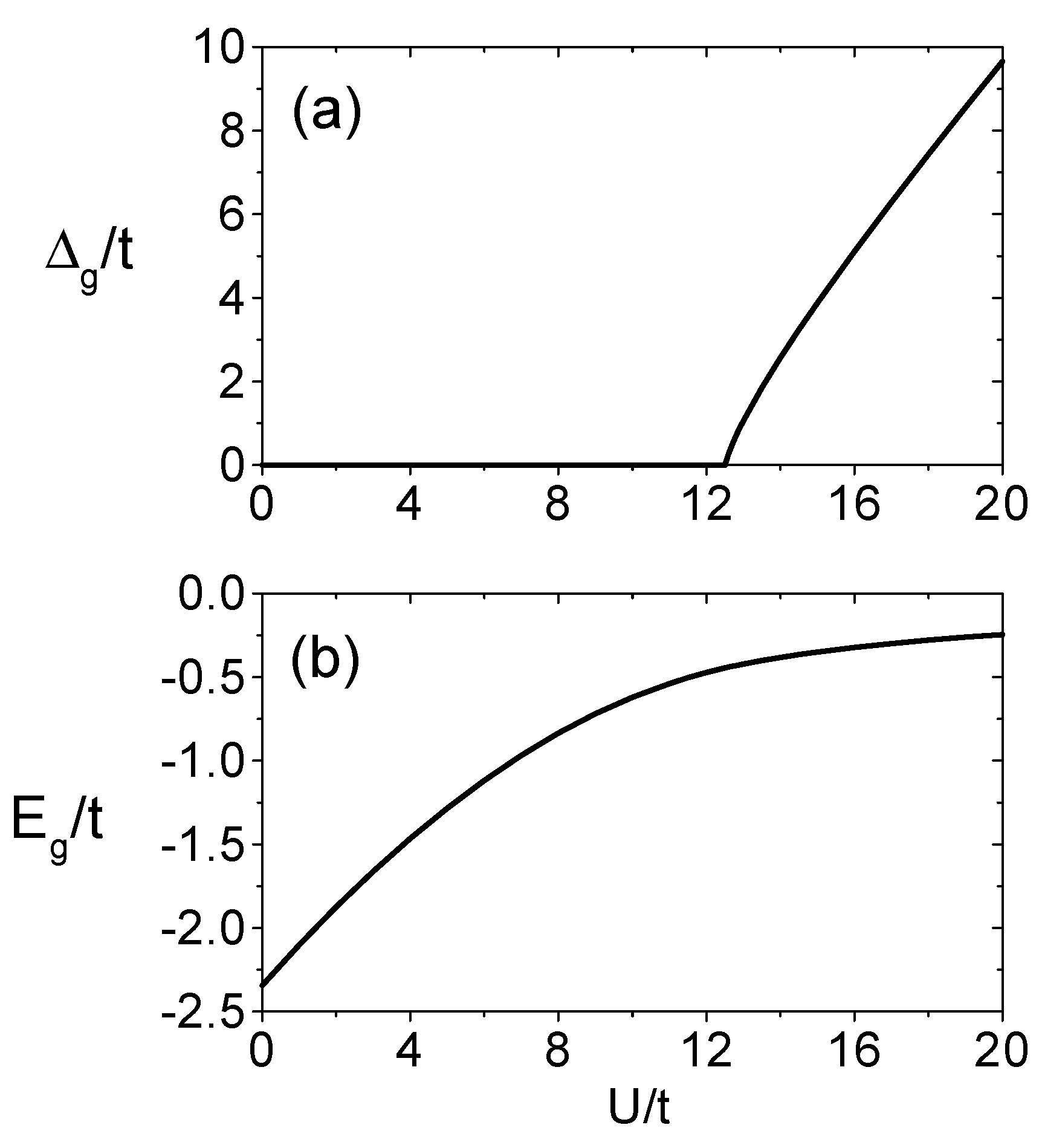
Two successive first-order phase transitions occur at Uc2/t = 4.45, a semi-metallic linear spindensity wave is stabilized and a first-order metal-insulator-transition to an anti-ferromagnetic insulator occurs at Uc3/t = 5.27. Capone et al.[28] obtained qualitatively similar phase transitions by using the Kotliar-Ruckenstein slave-boson technique. That is, the weak-coupling paramagnetic metal continuously evolves into a spiral metal at Uc1/t = 6.68, which crosses the linearly polarized spin-density-wave ground state at Uc2/t = 6.84. The latter phase undergo a further first-order transition toward an anti-ferromagnetic insulator at Uc3/t = 7.68. The exact diagonalization results exhibit a first-order transition between the paramagnetic metal and the anti-ferromagnetic insulator at U/t = 12.07, without intermediate ”exotic” phases[29]. Our results for the critical value of (U/t) ≈ 12.5 for the metal-insulator-transition and the ground state energy as a function of U/t are in agreement with results obtained by the exact diagonalization method[29].
In summary, we find that there is a first-order phase metal-insulator-transition, at (U/t)c = 12.5 in full agreement with the exact diagonalization result U/t = 12.07.
5 Concluding Remarks
A practical renormalization method for an array described by a Hubbard Hamiltonian has been presented. Specifically, we could find conditions under which the renormalized Hubbard Hamiltonian on a nonpartite lattice has no proliferation of coupling. The method is especially suited for getting the lowest excited states. An application demonstrating the identification of a first order metal-insulator transition has been discussed in detail. In future work we intend to build upon the present results in two directions. First, to apply them to the computation of the transport properties of lattices of quantum dots. We also hope to be able to incorporate into the method one essential aspect of quantum dots that, so far, is not included. This is the (small but finite) variability of the chemical potential. This results from the size fluctuations of dots as they are prepared in the laboratory. In real-space BRG method, how to truncate the states for one block is of central importance to the accuracy of the final results. In this paper, four states for each block are selected from the requirement of non-proliferations of parameters. This greatly simply the theoretical handling of this problem while keeping the main related physics. But for two-dimensional system, how to judge the convergence when more states are kept is still an open problem in this direction. More work is under way.
Acknowledgements
We are delighted to dedicate this paper to Professor R.S. Berry on the occasion of his 70th birthday. We acknowledge the financial support of the National Science Foundation and the Office of Naval Research.
References
- Markovich, G; Collier, C. P.; Henrichs, S. E.; Remacle, F.; Levine, R. D.; Heath, J. R. Accounts of Chemical Research 1999, 32, 415–423.
- Remacle, F.; Levine, R. D. Proc. Natl. Acad. Sci. USA 2000, 97, 553.Remacle, F.; Levine, R. D. ChemPhysChem. 2001, 2, 20.
- Hubbard, J. Proc. R. Soc. Longdon A 1963, 276, 238, A 1964, 277, 237;A 1964, 281, 401.
- Remacle, F.; Beverly, K.C.; Heath, J. R.; Levine, R. D. J. Phys. Chem. in press.
- Mott, N. F. Metal-Insulator Transitions; Taylor and Francis: London, 1990. [Google Scholar]
- Lieb, E. H. arXiv: cond-mat/9311033, 1993.Tasaki, H. arXiv: cond-mat/9512169, 1997.arXiv: cond-mat/9712219, 1997; and references therein.
- Lieb, E.; Wu, F. Phys. Rev. Lett. 1968, 20, 1445.
- Senechal, D.; Perez, D.; Pioro-Ladriere, M. Phys. Rev. Lett. 2000, 84, 522. [PubMed]
- White, S. R.; Scalapino, D. J.; Sugar, R. L.; Loh, E. Y., Jr.; Gubernatis, J. E.; Scalettar, R. T. Phys. Rev. B 1989, 40, 50.
- Penn, D. Phys. Rev. 1966, 142, 350.
- Tahir-Kheli, R.; Jarrett, H. Phys. Rev. 1968, 180, 544.Roth, L. Phys. Lett. 1981, 20, 1431.Kishore, R.; Joshi, S. Phys. Rev. 1969, 186, 484.Arai, T.; Parinellok, M. Phys. Rev. Lett. 1971, 27, 1226.
- Cyrot, M. J. Phys. (Paris) 1972, 33, 125.Kimball, J.; Schriieffer, J. R. Int. Conf. on Magnetism. Chicago(1971).Cyrot, M. Phys. Rev. Lett. 1970, 25, 871.Phil. Mag. 1971, 1031.Cyrot, M.; Lacour-Gayet, P. J. Phys. C 1974, 7, 400.
- Gutzwiller, M. C. Phys.Rev. 1965, 137, A1726.Phys. Rev. Lett. 1963, 10, 159.Langer, W.; Plischke, M.; Mattis, D. Phys. Rev.B 1969, 23, 149.Kaplan, T.; Bari, R. J. Appl. Phys. 1970, 41, 875.Brinkman, W.; Rice, T. M. Phys. Rev.B 1970, 2, 4302.Chao, A. Phys. Rev.B 1971, 4, 4034.
- Harris, A. B.; Lange, R. V. Phys. Rev. 1967, 157, 295.Hubbard, J. Proc. Roy, Soc(London) A 1966, 296, 100.Kanamori, J. Progr. Theor. Phys. (Kyoto) 1963, 30, 275.Esterling, D.; Lange, R. V. Rev. Mod. Phys. 1968, 40, 796.
- Hirsch, J. E. Phys. Rev. B 1979, 20, 3907.Phys. Rev. B 1980, 22, 5259.
- White, S. R. Phys. Rev. Lett. 1992, 69, 2863.Phys. Rev. B. 1993, 48, 10345.
- Tokuyasu, T.; Kamal, M.; Murthy, G. Phys. Rev. Lett. 1993, 71, 4202. [PubMed]
- Murthy, G; Kais, S. Chem. Phys. Lett. 1998, 290, 199.
- Dasgupta, C.; Pfeuty, P. J. Phys. C 1981, 14, 717.Jullien, R.; Pfeuty, R.; Fields, J. N.; Doniach, S. Phys. Rev. B 1978, 18, 3568.
- Perez-conde, J.; Pfeuty, P. Phys. Rev. B 1993, 47, 856.Vanderzande, C. J. Phys. A: Math. Gen. 1985, 18, 889.
- Bhattacharyya, B.; Sil, S. J. Phys.:Condens. Matter 1999, 11, 3513.
- Topics in current physics. In Real-space renormalization; Burkhardt, T. W.; van Leeuwen, J. M. J. (Eds.) Springer-Verlag: Berlin Heidelberg New York, 1982.
- Imada, M.; Fujimori, A.; Tokura, Y. Rev. Mod. Phys. 1998, 70, 1039.
- Bhattacharyya, B.; Sil, S. Phys. Lett. A 1993, 180, 299.
- Boardman, A. D.; O’Connor, D. E.; Young, P. A. Symmetry and its application in science; John Wiley and Sons: New York, 1973. [Google Scholar]
- Krishnamurthy, H. R.; Jayaprakash, C.; Sarker, S.; Senzel, W. Phys. Rev. Lett. 1990, 64, 950. [PubMed]
- Jayaprakash, C.; Krishnamurthy, H. R.; Sarker, S.; Wenzel, W. Europhys. Lett. 1991, 15, 625.
- Capone, M.; Capriotti, L.; Becca, F.; Caprara, S. Phys. Rev. 2001, B63, 0854104.
- Brinkman, W. F.; Rice, T. M. Phys. Rev. B 1970, 2, 4302.
©2002 by MDPI. Reproduction for noncommercial purposes permitted.
Share and Cite
MDPI and ACS Style
Wang, J.X.; Kais, S.; Levine, R.D. Real-space renormalization group study of the Hubbard model on a non-bipartite lattice. Int. J. Mol. Sci. 2002, 3, 4-16. https://doi.org/10.3390/i3010004
AMA Style
Wang JX, Kais S, Levine RD. Real-space renormalization group study of the Hubbard model on a non-bipartite lattice. International Journal of Molecular Sciences. 2002; 3(1):4-16. https://doi.org/10.3390/i3010004
Chicago/Turabian StyleWang, J. X., Sabre Kais, and R. D. Levine. 2002. "Real-space renormalization group study of the Hubbard model on a non-bipartite lattice" International Journal of Molecular Sciences 3, no. 1: 4-16. https://doi.org/10.3390/i3010004