
Emerging Roles of Vitamin B12 in Aging and Inflammation

1
Research Center for Translational Medicine, Sirius University of Science and Technology, 354340 Sochi, Russia
2
Division of Immunobiology and Biomedicine, Center for Genetics and Life Sciences, Sirius University of Science and Technology, 354340 Sochi, Russia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2024, 25(9), 5044; https://doi.org/10.3390/ijms25095044
Submission received: 9 April 2024
/
Revised: 28 April 2024
/
Accepted: 29 April 2024
/
Published: 6 May 2024
(This article belongs to the Special Issue Functional Role of Cytokines in Cancer and Chronic Inflammation)
Abstract
:Vitamin B12 (cobalamin) is an essential nutrient for humans and animals. Metabolically active forms of B12-methylcobalamin and 5-deoxyadenosylcobalamin are cofactors for the enzymes methionine synthase and mitochondrial methylmalonyl-CoA mutase. Malfunction of these enzymes due to a scarcity of vitamin B12 leads to disturbance of one-carbon metabolism and impaired mitochondrial function. A significant fraction of the population (up to 20%) is deficient in vitamin B12, with a higher rate of deficiency among elderly people. B12 deficiency is associated with numerous hallmarks of aging at the cellular and organismal levels. Cellular senescence is characterized by high levels of DNA damage by metabolic abnormalities, increased mitochondrial dysfunction, and disturbance of epigenetic regulation. B12 deficiency could be responsible for or play a crucial part in these disorders. In this review, we focus on a comprehensive analysis of molecular mechanisms through which vitamin B12 influences aging. We review new data about how deficiency in vitamin B12 may accelerate cellular aging. Despite indications that vitamin B12 has an important role in health and healthy aging, knowledge of the influence of vitamin B12 on aging is still limited and requires further research.
1. Introduction
Vitamin B12 is the generic name for a group of cobalamins that play an important role in physiological processes. Recent studies elucidate that vitamin B12 may be involved in several pathological processes: change in methylation of DNA [1], aberrant protein post-translational modifications [2], change in microbial composition in the gut [3], change in tumorigenesis probability [4], and modulation of inflammation [5]. We discuss how these B12-associated processes may contribute to acceleration of aging and senescence and to inflammation. Apart from the increased risk of age-related diseases, such as cognitive decline, cardiovascular diseases, osteoporosis, and oxidative stress, which are alleviated by supplementation of vitamin B12 [6,7], we propose that vitamin B12 may play a role in the stimulation of cellular senescence and the pro-inflammatory senescence-associated secretory phenotype through modulation of microbiota, mitochondrial damage, cryptic transcription, and inflammaging (chronic low-grade inflammation in elderly individuals).
Cobalamins consist of an almost planar corrinoid core with a cobalt atom in the center and two ligands orthogonal to the corrinoid plane. The (“lower”) alpha-ligand is 5′,6′-dimethylbenzimidazole, while the “upper” beta-ligands are different in the different forms of cobalamin (Figure 1). The chemical synthesis of vitamin B12 is cumbersome because it includes over 60 steps and provides a 1% yield [8]. Biosynthesis of natural cobalamins is restricted to only few prokaryotes, including human gut residents such as Pseudomonas, Propionibacterium, Bacteroides, Akkermansia, and others [9,10,11]. Humans cannot synthesize vitamin B12 de novo and need to ingest it from animal-based food [9]. Cyanocobalamin is a metabolically inactive vitamin B12 form produced by industrial bacterial strains and is cyanodified during production to improve air stability (Figure 1) [12]. Metabolically active forms of vitamin B12 in mammals are methylcobalamin (MeCbl) and 5-deoxyadenosylcobalamin (coenzyme B12, AdoCbl) (Figure 1). The former is a cofactor for the enzyme methionine synthase, which is active in the cytosol, while the latter is a cofactor for the enzyme methylmalonyl-CoA mutase in mitochondria.
2. Vitamin B12 Transport
On the way to its destination, vitamin B12 forms complexes with several proteins. Orally ingested B12 binds to a protein, haptocorrin, in the upper part of the digestive system, where cobalamin is released from digested proteins by the acidic environment and proteases (Figure 1). The Cbl–haptocorrin complex protects Cbl from this aggressive environment. In the duodenum, haptocorrin is cleaved by pancreatic proteolytic enzymes, liberating cobalamin from the complex. Vitamin B12 then binds to the intrinsic factor, which discriminates it from other corrinoids [13]. In the distal ileum, enterocytes uptake vitamin B12 only in complex with the intrinsic factor. Exocytosed from enterocytes to blood, free vitamin B12 mostly binds to haptocorrin, and a lesser fraction of vitamin B12 binds to transcobalamin [14]. Cells absorb vitamin B12 in complex with transcobalamin, so-called holotranscobalamin, by a transcobalamin receptor, CD320 (Figure 1) [15]. All RNAs for the transcobalamin receptor subunits and transcobalamin are translated by specific ribosomes (denoted as RPS25/eS25) [16]. Renal re-absorption is accomplished by uptake of holotranscobalamin with another B12 receptor, megalin [17]. After endocytosis, transcobalamin releases vitamin B12 in lysosomal acidic pH, and free vitamin B12 is transported to the cytosol via ATP-dependent transport by lipocalin-1 interacting membrane receptor domain containing protein 1 (LMBD1) and adenosine triphosphate (ATP)-binding cassette subfamily D member 4 (ABCD4) [18,19]. Within the cytosol, vitamin B12 binds methylmalonic aciduria and homocystinuria type C protein (MMACHC), which decyanates or dealkylates vitamin B12 depending on its form [20,21]. MMACHC, with the assistance of methylmalonic aciduria and homocystinuria type D protein (MMADHC), carries MMACHC-refined cobalamin to methionine synthase or to mitochondria, with the decision of fate governed by MMADHC [22]. In mitochondria, methylmalonic aciduria type B protein (MMAB) converts cobalamin to AdoCbl in an ATP-dependent manner and delivers it to methylmalonyl-CoA mutase (MMUT) [23], while methylmalonic aciduria type A protein (MMAA) controls the correctness of cobalamin forms inserted into MMUT [24].
Free vitamin B12 and vitamin B12 that has not been absorbed in the small intestine is utilized by resident bacteria further along the digestive path. A significant part of the human gut microbial community (estimated at ~45–55%) relies on external vitamin B12, about 20–40% of human gut microbes are capable of its synthesis (although corrinoids may be needed as precursors), and some do not use it in homeostasis [25,26]. Bacterial B12-dependent enzymes catalyze isomerization, dehalogenation, deamination, dehydration, and methyl transfer reactions [10]. Cobalamin synthesized by bacteria is not a significant source for humans because most of the bacteria reside downstream of the digestive path with respect to host B12 receptors and synthesized quantities are small with respect to daily intake needs [3,26]. Vitamin B12 rather shapes the microbiota in the gut by attenuating the metabolism of affected bacteria [3,11].
Vitamin B12 has complex absorption machinery in the body, and during aging, some parts may deteriorate, thus decreasing B12 bioavailability (Figure 1). Atrophic gastritis, a form of chronic age-associated stomach inflammation, reduces gastric acid secretion necessary for vitamin B12 release from proteins [27]. Moreover, old patients have lower absorption in the small intestine and decreased renal re-absorption of vitamin B12 due to a change in expression of the receptor for the vitamin B12–IF complex in enterocytes and the kidneys [28]. The brains of old subjects have lower levels of MeCbl and AdoCbl than young subjects, despite normal levels of vitamin B12 in blood, thus indicating deterioration of vitamin B12 import into cells and processing [29].
3. B12-Dependent Proteins
Bacteria have a dozen B12-dependent proteins, while in mammals, only two proteins utilize vitamin B12 [3]. Methionine synthase (MS) produces a universal methyl donor, S-adenosyl-methionine (SAM), which is used in most methylation reactions. Mitochondrial methylmalonyl-CoA mutase (MMUT) converts methylmalonyl-CoA to succinyl-CoA in the Krebs cycle for the utilization of branched carbon chains derived from lipids and amino acids.
3.1. Methylmalonyl-CoA Mutase
Methylmalonyl-CoA mutase is a mitochondrial enzyme that uses vitamin B12 in the form of AdoCbl as a cofactor [30,31]. MMUT converts L-methylmalonyl-CoA to succinyl-CoA [32], which then enters the tricarboxylic acid (TCA) cycle (Figure 2). Methylmalonyl-CoA is derived from the catabolism of branched-chain amino acids, lipids, and cholesterol side chains; thus, MMUT connects this metabolism to the TCA cycle (Figure 2). Human MMUT is a homodimer, and every subunit has N- and C-domains [33]. Each subunit of MMUT accepts one molecule of AdoCbl [30]. The N-domain binds the substrate, and the C-domain binds AdoCbl [33]. They are connected through a linker, and the active site is located at the N-/C-domain interface. MMAA ensures selective binding of AdoCbl and no other cobalamins to MMUT [33]. MMUT follows a radical generation mechanism: the Co-C bond in AdoCbl is cleaved along with the generation of a 5′-deoxy-5′-adenosyl radical for isomerization of methylmalonyl-CoA [34]. Reactivation of MMUT occurs due to MMAA, which exchanges an oxidized cofactor [24]. MMAA induces the release of the inactive cofactor and restores MMUT activity through their complex formation.
Disturbed functioning of MMUT due to scarcity of vitamin B12 leads to impaired mitochondrial function caused by methylmalonyl-CoA accumulation and subsequent aberrant acylations of proteins. It has been shown in model mice with liver-specific MMUT knockout and patients with methylmalonic aciduria, a disease caused by mutations in genes encoding vitamin B12-interacting proteins [2]. In this context, the activity of the mitochondrial enzyme carbamoyl phosphate synthetase-1 (CPS-1), which participates in the urea cycle, is severely compromised. The number of mitochondrial DNA copies and transcription of genes coding for the electron transport chain are reduced due to reduced activity of the associated transcription factor and the polymerase. However, the levels of these proteins (mitochondrial transcription factor A, TFAM, and DNA-directed RNA polymerase) do not change. The glycine cleavage system is compromised due to prevention of lipidation of glycine cleavage H protein (GCSH). All of these abnormalities are caused by an increase in the acylations of the corresponding enzymes caused by MMUT malfunction.
3.2. Methionine Synthase
Methionine synthase (5-methyltetrahydrofolate-L-homocysteine S-methyltransferase, EC 2.1.1.13; MS) is a cytosolic enzyme that uses MeCbl as a cofactor [35]. The gene has ubiquitous expression, with the highest numbers of transcripts in epithelial tissues and the lowest in the brain [36]. Free vitamin B12 regulates MS expression through a feedback loop with a B12-dependent internal ribosome entry site (IRES) before a coding sequence for MS [37]. MS from E. coli is the most-studied MS; it has 53% homology to the human enzyme [38]. The protein consists of five domains: Cob—a cobalamin-binding domain, Hcy—an L-homocysteine-binding domain, Fol—a folate-binding domain, Act—an S-adenosyl-L-methionine-binding domain, and Cap—a cobalamin-binding domain. The Hcy, Fol, and Cap domains sequentially interact with Cob during the MS catalytic cycle [39,40]. The MS catalytic cycle consists of three steps [41,42,43,44]. In the first step, a cobalamin molecule, the MS cofactor, accepts a methyl group from 5-methyltetrahydrofolate and then transfers it to the sulfur atom in L-homocysteine to produce L-methionine, thus acting as a methyl donor. In the second step, the cobalt ion in the cobalamin is methylated with 5-methyltetrahydrofolate and covalently binds to the N5 atom of tetrahydrofolate. In the third step, tetrahydrofolate dissociation occurs by homolytic cleavage of the Co-N bond and the return of the cobalamin to its initial state. Oxygen and physiological oxidants may cleave the bond between the cobalt ion and the methyl group. In this form, cobalamin cannot enter the catalytic cycle, and the catalytic cycle is terminated after about 2000 repeats. A restoration of enzyme function requires reduction of MS by NADPH reductase (EC 1.16.1.8) and reactivation of cobalamin [45]. Produced L-methionine is a substrate for methionine adenosyltransferase, which converts it to S-adenosyl-L-methionine (SAM)—a universal methyl donor (Figure 3). Demethylation of 5-methyltetrahydrofolate in the folate cycle provides a methyl radical for cobalamin and increases the level of metabolically active B9 (folate) (Figure 3) [46,47]. The MS cycle is dependent on glycine, serine, and threonine catabolism as it supplies 5-methyltetrahydrofolate.
Genes of SAM-dependent enzymes are highly present in eukaryotic genomes; these enzymes occupy about 1% of total eukaryotic proteins, including DNA- and RNA-methyltransferases and histone methyltransferases [48,49]. Methylation of phospholipids is the main way in which SAM is consumed [50]. This process is used for the regulation of cellular methylation potential and creates methyl “sinks” [50]. Methylation reactions are competitively inhibited by their coenzyme product, S-adenosyl-L-homocysteine [51]. Phospholipid methylation also occurs after activation of dopamine receptor D4.4R in an SAM-dependent reaction involving a methionine in the receptors [52]. Thus, the activity of D4.4R also regulates other methylation processes, including DNA methylation [53].
When there is an excess of physiological oxidants (oxidative stress), methionine is misincorporated instead of other amino acids in protein synthesis to provide new functions for proteins or scavenge oxidants in the protein-bound form [54,55]. Free methionine may be oxidized and reduced with the methionine sulfoxide reductase A, which is capable of reducing methionine-(S)-sulfoxide in the free form [56].
A participant in the methionine cycle, homocysteine, is a precursor for glutathione in the transsulfuration pathway. Glutathione is a tripeptide crucial for cellular reduction–oxidation homeostasis [57]. Glutaredoxin enzymes use glutathione for the reduction of disulfide bonds in cells. In healthy cells, a high ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) (close to 98%) helps to maintain intracellular thiols in the reduced state [58].
4. Nucleic Acids and Histone Methylation
Vitamin B12 induces the production of S-adenosylmethyonine (SAM), which is a methyl donor in DNA and histone methylation (Figure 3). DNA methylation is the process of transferring a methyl group from the donor molecule (SAM) to nucleotide bases, which is catalyzed by SAM-dependent DNA-methyltransferases. In mammalian somatic cells, the methyl group is usually covalently bound to C5 of cytosine in CpG dinucleotides, where about 70–80% of CpG dinucleotides in somatic cells are methylated [59]. 5-Methylcytosine can populate ~1% of cellular DNA [60], and some anticancer drugs, such as cisplatin, can specifically interact with it [61]. Cytosine methylation in CpA and CpT dinucleotides was found in stem cells [62]. 6-Methyladenosine is another type of DNA methylation in mammalian cells [63]. It is detected in excess amounts in tumor cells of the esophagus and liver [64,65]. Thus, various nucleotides in DNA can be methylated.
DNA methylation plays a regulatory role in gene expression. The main mechanisms of gene repression due to methylation are inhibition of the binding of transcription factors to DNA in hypermethylated regions and the binding of such regions to repressor methyl-binding proteins [66]. The strongest repression of gene expression occurs when its promoter or the first exon are methylated [67]. Methylation of a gene body positively correlates with its expression level in dividing cells [68]. DNA demethylation has opposite effects on gene expression. Global DNA demethylation mainly occurs during early embryogenesis, stem cell differentiation, and reprogramming [69]; however, aging is accompanied by DNA demethylation [70]. DNA is demethylated with Fe2+/α-ketoglutarate-dependent dioxygenases [71]. α-Hydroxyglutarate is an inhibitor of these enzymes. Mutations in the IDH1 or IDH2 genes lead to the reduction of α-ketoglutarate to α-hydroxyglutarate and subsequent DNA hypermethylation, including tumor suppressor genes, and oncological transformation of cells [72]. ALKBH1 demethylates 6-methyladenosine in DNA and RNA [63,73].
Defects in DNA methylation in cells lead to impaired gene repression and aberrant expression, which disrupt their homeostasis, increase tumor occurrence, and promote cellular aging. For example, methylation of the transposable repetitive sequences Alu and LINE-1 is reduced with age, similarly to cancer cells [70,74,75]. In tumor cells, unlike in normal cells, the methylation regulation by long non-coding RNA is impaired [76]. Methylation of microRNA regulatory regions strictly controls their expression in normal cells, while at the same time, tumor cells [77,78] and aging cells [79] are characterized by abnormal expression of microRNAs associated with relevant processes. Moreover, aging is associated with a drastic change in DNA methylation patterns [80]. Thus, the balance between DNA methylation and demethylation plays a critical role in the normal homeostasis of the cell.
Dynamic post-translational modifications of histones provide regulation of chromatin structure and gene expression [81]. Histones are highly conserved proteins that organize eukaryotic DNA into nucleosomes. Currently, more than 25 different types of post-translational modifications of histones, which can affect their various amino acid residues, are known.
Methyl groups on histones are specific marks for enzymes that interact with DNA and change the structure of the chromatin. In humans, histone methylation is catalyzed by dozens of SAM-dependent methylases [82]. The regulatory function of methylation depends on the position at which it occurs (Table 1). Some modifications may play an ambiguous role, for example, H3K79me3 [83,84], and there is an interplay and correlation between histone post-translational modifications [85]. SAM deficiency may cause impaired histone methylation and loss of heterochromatin, including age-associated loss [86]. Positions H3K4, H3K36, and H3K79 are most frequently methylated, and inhibition of these processes leads to a 2–5-fold increase in intracellular SAM levels [50], which indicates a significant need of SAM for histone methyltransferases. The modification H3K36me3 is a hallmark for exons [87], where it prevents transcription initiation outside promoters [88]. Methylation of DNA regions is specifically inhibited in the presence of H3K4 methylation [89]. For the process of gene imprinting in mammals, the efficiency of the methylation processes of both DNA and histones is important. A knockout of the DNMT3L (DNA (cytosine-5)-methyltransferase 3-like) gene leads to a loss of the allele specificity of the histone repressive modifications H3K9me3, H4K20me3, and H2A/H4R3me2 in imprinting control regions [90].
Histone acetylation is associated with a transformation of heterochromatin regions into euchromatin and an increase in their transcriptional activity. Deacetylation of histones bound to a DNA region is mechanistically associated with hypermethylation of the CG-rich stretch of this region: the methyl-CG-binding protein meCP2 binds to hypermethylated DNA and recruits to nearby nucleosomes histone deacetylases and methyltransferases that methylate histones at the H3K9 position [95]. Histone deacetylation in humans is catalyzed by 18 different deacetylases, including 7 NAD+-dependent deacylases—sirtuins [96]. The strongest deacetylase activity is observed for sirtuins 1–3 [97]. In addition to removal of the acetyl group, sirtuins catalyze the removal of several other non-acetyl acyl modifications from histones and other proteins. SIRT5 has desuccinylase, demalonylase, demethylmalonylase, and other activities [2,98,99]. Sirtuins are involved in the processes of suppressing cellular aging by delaying replicative shortening of telomeres, maintaining genome integrity, and promoting DNA damage repair [100].
Manipulation of histone methylation and acetylation through deletion, inhibition, or overexpression of histone-modifying enzymes prolongs the lifespan of worms and flies and shows phenotypes of healthy aging in human cellular models (reviewed in [101]). DNA methylation and histone modifications, which are partially dependent on vitamin B12 availability, are involved in aging [102].
5. Consequences of Vitamin B12 Deficiency
Vitamin B12 deficiency causes several consequences on organismal, cellular, and molecular levels which resemble some of the phenotypic consequences of aging and senescence (Figure 4).
Currently, there is no universal method for determining B12 deficiency for widespread application which could be considered the “gold standard” [103]. In most cases, total vitamin B12 levels are measured in serum. In clinical practice, measurement of total serum B12 is made using a chemiluminescence (ECL) assay by measuring the competitive binding of purified haptocorrin to vitamin B12 after its release from endogenous proteins [104]. This technique is available for widespread application due to its precision (detection is possible in the range of 50–2000 pg/µL), speed, and efficacy and the availability of high-tech automated systems [105]. A deficiency in biologically available B12 can be indirectly determined by the concentration of the protein that transports it, holotranscobalamin (holoTC) [103]. Concentrations of several small molecules increase due to a deficiency in active forms of B12: MMA (2-methylmalonic acid), due to insufficiency of the MMUT function, and Hcy (homocysteine), due to insufficiency of the MS function (Table 2), which may be quickly and accurately measured with automated systems [103]. However, the serum vitamin B12 concentration, whether detected directly or indirectly, may not correspond to the intracellular vitamin B12 concentration. Even with “adequate levels” of vitamin B12 in blood, concentrations of B12 forms inside cells can be significantly decreased [29]. In laboratory settings, HPLC-UV with internal standards [106] or HPLC strengthened with mass spectrometry or tandem mass spectrometry (HPLC-MS/MS), which do not require internal standards, are routinely used for measurement of vitamin B12. The advantages of HPLC are its high speed, accuracy, sensitivity, and specificity for various cobalamins, allowing quantitative measurements of cyanocobalamin, MeCbl, hydroxocobalamin, other cobalamins, cobinamides, and corrinoids. HPLC methods usually do not require difficult sample preparation [106]. Typically, HPLC and HPLC-MS have a two- to three-order lower limit of detection, thus excluding single-cell analysis for research purposes.
While in North America the fraction of the population with low serum B12 levels is not greater than 10%, in other regions such fractions can exceed 40% [107]. Marginal vitamin B12 deficiency is reported in 30–80% of the population (Table 2) [107]. In elderly populations around the world, rates of severe and marginal vitamin B12 deficiency are higher [107].
Vitamin B12 deficiency during gestation leads to a number of metabolic disorders in the offspring, which may reduce their lifespan. In Wistar rats, folic acid supplementation along with vitamin B12 deficiency during gestation reduces global DNA methylation and plasma and placental levels of ω-3 docosahexaenoic acid [108]. The offspring of rats fed a vitamin B12-deficient diet have a higher risk of developing obesity, insulin resistance, and elevated levels of triacylglycerides and glucose, which increases the probability of progression of metabolic and cardiovascular diseases with age [109]. They also have reduced resistance to oxidative stress and a lower birth weight, elevated levels of TNFα are found in the blood and adipose tissue, and there are increased levels of cortisol, leptin, and pro-inflammatory IL-6 in blood, while levels of adiponectin and IL-1β, on the contrary, are decreased. In offspring, changes in glucose metabolism and antioxidant status can be reversed with a return of serum B12 to physiological levels after fertilization. In the postnatal period, these changes can be partially, but not absolutely, reversed [109]. These phenomena may be based on a decrease in CpG methylation in non-coding regions before the onset of genes associated with the transport and metabolism of fatty acids, including mitochondrial genes, in the offspring of dams [110]. These consequences can be phenotypically and molecularly reversed when B12 levels in gestational females return to normal [109]. Similar effects have been observed in sheep. Offspring of sheep fed a vitamin B12-deficient diet and folate before fertilization are more likely to be overweight, antigen-hypersensitive, insulin-resistant, and to have high blood pressure than offspring of sheep fed a normal diet [111]. At the stage of fetal development, an altered methylation status of 4% of 1400 CpG islands is observed in sheep liver tissues, which may indicate an epigenetic mechanism for the offspring to acquire metabolic disorders due to B12 deficiency in the mother. Thus, vitamin B12 concentrations are very important in the early stages of ontogenesis.
Deficiency in MeCbl and AdoCbl in cells due to insufficiency of vitamin B12 supply to cells causes promiscuous non-enzymatic post-translational modifications. Scarcity of MeCbl and consequent deterioration in methionine synthase function increase amounts of homocysteine. Homocysteine may be converted to homocysteine thiolactone, which non-enzymatically modifies ε-amino groups of lysines [112], including those on histones H3K27, H3K36, and H3K79 responsible for chromatin structure [113]. In the brains of mice with decreased B12 in their food, expression of histone-modifying enzymes (e.g., H4K20-methylating and Zn2+-dependent histone deacetylase HDAC4) increases [114], and the amount of oxidative stress markers (malondialdehyde and protein carbonyls) rises. Decreased activities of superoxide dismutase and the H2O2-degrading enzyme catalase in such brains lead to these oxidative post-translational modifications [114]. Moreover, vitamin B12 is capable of scavenging superoxide radicals with a rate similar to that of superoxide dismutase [115], and supplementation with B12 helps to reduce the consequences of oxidative stress [116]. A deficiency in AdoCbl, which is obligatory for the activity of MMUT, provokes accumulation of methylmalonyl-CoA. Deficiency in the MMUT, MMAA, and MMAB functions leads to a range of metabolic diseases named methylmalonic acidurias, which have an increased concentration of methylmalonic acid (MMA) in blood as a consequence [117]. MMA and its derivative organic acids cause damage to the central nervous system [118]. On a molecular level, in model mice with a liver-specific knockout of MMUT, abnormal methylmalonylation and malonylation are present on a few proteins [2]. Despite these proteins being mostly localized to mitochondria, some of them are present in other compartments, such as the plasma membrane, the nucleus, lysosomes, and others, indicating that such modifications are not restricted to the mitochondria. Deacylases capable of demethylmalonylaton and demalonylation, such as SIRT1 and SIRT5 [2,98], which have strong activity against methylamolnylation and propionylation, are acyl-modified due to the loss of MMUT activity, thus decreasing their function [2]. In liver samples of patients with MMA abundancy, the activity of several sirtuins, including SIRT1 and SIRT5, is substantially reduced [2]. SIRT5 has a preference for extended acyl groups, such as malonyl, succinyl, propyonyl, and methylmalonyl groups [2,98]; thus, it is not solely methylmalonyl modifications that may accumulate.
At the organismal level, the effects of severe chronic B12 deficiency primarily affect the nervous system, resulting in neurological disorders. Chronic vitamin B12 deficiency leads to behavioral abnormalities in female mice: with a moderate to severe vitamin B12 shortage in the diet, they exhibit more anxious behavior and less maternal care for their offspring than controls with a sufficient B12 level [114]. Abnormal maternal behavior in model mice and a reduction in postnatal period duration are observed when there is a decrease in the expression of the MEST gene due to disturbances in its imprinting, a process dependent on DNA methylation [119]. Impaired histone methylation due to serum B12 deficiency, which was assessed by measuring serum homocysteine and holotranscobalamin levels, led to a disruption of gene imprinting, in particular, to a decrease in the expression of the MEST gene in the placenta of mice [120]. In infectious diseases of the nervous system, such as meningitis, a therapy including vitamin B12 promotes neuronal survival and reduces the inflammatory markers CCR2, CCL3, and IL-1β: bacterial meningitis model rats maintained on a vitamin B12-supplemented diet had fewer apoptotic cells in the hippocampal dentate gyrus than those who did not receive it [121]. In treatment of viral infections in humans, additional amounts of B12 reduce the intensity of pain and subsequently reduce the risk of memory loss and concentration problems [122]. Neuronal damage caused by B12 deficiency may involve increased levels of homocysteine due to lower activity of MS, as in autistic subjects [29]. Accumulation of Hcy in cells increases the probability of their apoptosis and necrosis, which can be attenuated with NMDA and mGluR1 antagonists [123]. The risk of development of mental disorders, such as autism and schizophrenia, is increased if patients have mutations in the genes associated with vitamin B12 transport and one-carbon metabolism: methionine synthase (MTR), methionine synthase reductase (MTRR), transcobalamin (TCN2), and 5,10-methylenetetrahydrofolate reductase (MTHFR), reducing their activity [29]. There is an association between low concentrations of active forms of B12 in the brain and mental disorders such as autism and schizophrenia [29]. Indeed, even normal serum vitamin B12 levels do not guarantee the same cellular B12 concentrations. In older people, the total concentration of B12 isoforms in the brain decreases with age [29]. Moreover, the amount of MeCbl and AdoCbl decreases significantly, and the amount of OHCbl increases, regardless of the normal serum concentration of B12, which may indicate accumulating defects in vitamin B12 processing inside cells. In the brains of old subjects, levels of homocysteine were higher compared to young subjects, and methionine and SAM levels were lower [29]. There is a connection between decreased vitamin B12 levels and decreased cognitive function, as well as worsening symptoms of neurological and psychiatric diseases [124]. In the nervous system, DNA methylation is associated with memory. The age-related decline in the ability to remember information and learn correlates with a decrease in global neuronal DNA methylation [125], and overexpression of Dnmt3a DNA methylase restores cognitive function in aged mice [126]. Since methylation may not be efficient enough when the concentration of SAM in neurons decreases [127] and a decrease in SAM levels is observed with premature aging and numerous age-associated diseases [128], these phenomena may be related to each other and to a deficiency in B12, a cofactor of methionine synthase (Figure 3). Vitamin B12 deficiency worsens the prognosis for patients with age-associated neurodegenerative diseases. Patients with Alzheimer’s disease were found to have higher serum concentrations of Hcy and lower levels of folate and B12 than the healthy control group [129]. Reduced serum levels of vitamin B12 are also associated with the rapid progression of Parkinson’s disease, worsening cognitive ability to move, and cognitive impairment [130]. On the other hand, older patients with these conditions often have impaired B12 absorption, so whether B12 deficiency is a cause or consequence of age-related frailty is not yet established [124].
An increase in the level of B12 in blood plasma may indicate pathological processes in the organism. Solid tumors, myeloproliferative diseases, tumor metastases in the liver, and liver and kidney diseases are possible consequences of elevated serum B12 levels [131]. In particular, long-term elevated plasma B12 concentrations are significantly associated with the occurrence of solid tumors [4]. On the other hand, higher levels of Cbl in plasma are associated with a higher antioxidant capacity, which may be due to direct radical scavenging capacity and also due to participation in the transsulfuration pathway for glutathionine synthesis [132], which is reduced in elderly people [29].
Vitamin B12 is essential for cellular homeostasis, and its deficiency causes many abnormalities, such as oxidative stress, impaired DNA and histone modifications, neurological disorders, and disturbance of signaling pathways such as sirtuin pathways.
6. Role of B12 in Aging, Senescence, and Inflammation
Aging leads to a gradual decline in the organism’s performance, which is reflected on cellular and molecular levels. Prominent hallmarks of aging include pronounced epigenetic changes, impaired mitochondrial function, cellular senescence, chronic inflammation, and others [133]. It is known that deficiency in or low levels of vitamin B12 correlate with worsening symptoms of age-related conditions, such as sarcopenia and dynapenia, cognitive decline, frailty syndrome, and cardiovascular diseases (reviewed in [6,124]). Global expression of the vitamin B12 transporting and processing proteins MMAA, MMUT, MMADHC, and haptocorrin decreases during aging [134], which potentially impairs the absorption of vitamin B12 from the intestine, reduces its processing on the way to MS and MMUT, and additionally decreases the activity of MMUT. Below, we discuss aging- and senescence-associated mechanisms caused by (sub-)deficiency in vitamin B12. However, elevated levels of plasma vitamin B12 increase the overall risk of mortality in elderly individuals [135,136,137], indicating that there is an optimal range of vitamin B12 concentrations.
6.1. Vitamin B12 and Its Role in the Formation of the Senescence Phenotype
Cellular aging is a biological process that leads to a gradual increase in the probability of cell death due to a progressive loss of normal cell function. It consists of two stages: replicative aging, which causes a cell to lose its ability to divide, and senescence, a program induced by cell cycle arrest characterized by a pro-inflammatory phenotype and prevention of proliferation [138]. Telomere shortening and end-to-end chromosome joining in aged cells are the hallmarks of replicative aging [139]. Senescence is characterized by changes in DNA methylation patterns and histone post-translational modifications, aberrant gene expression due to large-scale chromatin rearrangement, disturbances in signaling cascades, mitochondrial dysfunction and related oxidative stress, mitochondrial damage, and decreased NAD+ levels [140]. Senescence is an adaptive mechanism rather than a cause of aging [141].
Senescent cells have features that are not typical of “young” ones. Senescent cells are characterized by irreversible arrest of the mitotic cycle and dysregulation of signaling through multiple pathways (sirtuin, IGF-1, mTOR, AMPK, FOXO, and NF-kB pathways). Cells that have entered the state of senescence are characterized by specific and increased activity of lysosomal β-galactosidase (senescence-associated β-galactosidase, SA-β-gal) at pH = 6, and this is used for their quantitative and qualitative determination in laboratory experiments [142,143]. Protein p16 is a product of the tumor suppressor gene CDKN2A, an inhibitor of cyclin-dependent kinases 2A and 4, blocking the transition of cells from the G1 to S phases [144]. The expression of the tumor suppressor gene significantly increases with age in most mammalian tissues, and the level of expression in T lymphocytes is a biomarker of aging [145]. Hypermethylation of the promoter of the gene encoding p16 protein increases cell resistance to oxidative stress and prevents premature cell aging induced by oxidative stress, as shown in the example of keratinocytes [146]. The p21 protein is an inhibitor of cyclin-dependent kinases 1 and 2, and its expression results in the arrest of the cell cycle at the G1 or G2/M phases [147]. The senescence state can be observed even without p16 expression if the p21 expression level is high, which has been shown in myocytes [148] and fibroblasts [149]. The protein p53 arrests the cell cycle in G1 and induces p21 expression as a transcription factor [150]. Senescent cells reduce p53 activity [151]. Senescent cells with low p16 expression levels proliferate after p53 inactivation, whereas senescent cells with high levels of p16 expression do not, even when p53 is inactivated [152].
One of the most important characteristics of senescent cells is the change in the secretory phenotype to the senescence-associated secretory phenotype (SASP), which includes increased secretion levels of interleukins (ILs), chemokines, growth and regulatory factors, and metalloproteinases [101,153,154,155]. SASP affects surrounding cells in a variety of ways. For example, some factors may act by inducing cellular senescence, thus limiting tumor progression. SASP promotes the elimination of senescent cells by stimulating the immune system and promoting tissue repair. In that respect, SASP is considered to be a beneficial response of senescent cells in organisms [156,157]. On the other hand, SASP has negative consequences for the organism under normal and pathological conditions. Some SASP factors, depending on the cellular context and environment, have deleterious effects on organisms, such as tissue inflammation and tumor progression [158,159]. In tumors, SASP has been shown to influence various processes: the initiation of epithelial–mesenchymal transition [160,161], induction of stemness [162,163], local tissue invasion [160,161,164], angiogenesis [165], fibroblast activation [166], immunosuppression [159,167], enhanced metastasis [168], and resistance to therapy [169,170,171,172].
Appropriate regulation of cellular senescence and SASP will be beneficial for human health because the elimination of senescent cells reduces age-related disorders and extends the lifespan of organisms [173]. To prevent senescent cell accumulation in tissues, the removal of senescent cells from these tissues may be achieved with the use of senolytics—molecules which selectively eliminate senescent cells [174]. However, the complete removal of senescent cells in tissues causes impaired regeneration of lung and liver tissues, although it improves bone regeneration [175].
Another approach for reducing the number of senescent cells in tissues is to reverse their transition to a senescent state by inducing their pluripotency using Yamanaka factors (OSKM: Oct3/4, Sox2, c-Myc, and Klf4) and subsequent redifferentiation [176]. Induced pluripotent stem cells (iPSCs) are morphologically indistinguishable from human embryonic stem cells (hESCs) and can be reprogrammed from senescent cells. Such cells have comparable telomere lengths and can be re-differentiated into rejuvenated tissue-specific cells [177]. A high level of p16 expression in senescent cells is a factor limiting the efficiency of reprogramming, and the removal of such cells from the tissue significantly increases reprogramming efficiency [178]. Additional supplementation with vitamin B12 improves reprogramming in model mice with tetracycline-inducible expression of OSKM [179].
Maintaining the state of senescence requires adaptation, which is achieved by deep epigenetic rearrangement. These changes are necessary, on the one hand, to maintain permanent cell cycle arrest and avoid apoptosis and, on the other hand, to implement the SASP. In this context, senescent cells are characterized by the formation of specific regions of heterochromatin called senescent-associated heterochromatin foci (SAHF) [180]. However, SAHF are not typical for all senescent cells [102]. SAHF are defined as DAPI-dense nuclear regions characterized by the presence of a central core of condensed chromatin enriched in H3K9me3 and macroH2A. The core is bordered by a peripheral ring containing H3K27me3 [181]. SAHF formation requires the presence of p16 and represents a deep and targeted reorganization of heterochromatin [182]. SAHF formation promotes both gene activation and gene repression. There are other epigenetic features, such as senescence-associated satellite stretching, reactivation of mobile elements and endogenous retroviruses, and lamin modifications, which are reviewed elsewhere [74].
During senescence, the epigenome undergoes significant and consistent changes that are necessary for cellular adaptation. DNA and histone methylation are important changes that together provide dynamic epigenetic control over gene expression [183]. To implement these changes, cells utilize the machinery to add methylation marks to DNA and histones. To highlight the link between B12 and global epigenetic rearrangements in senescent cells, we will discuss the specifics of this program in senescent cells.
Impaired histone modifications appear to play a special role in senescence and aging. Accumulation of H3K9me3/H3K27me3/macroH2A during SAHF block formation is typical for senescent cells undergoing oncogene-induced senescence [184]. Meanwhile, a global decrease in H3K9me2/3 and H4K20me levels but an increase in H3K9me1 levels in gene bodies characterizes replicative senescence in human fibroblasts [185]. In rat hepatocytes, trimethylation of histone H4 at position K20 increases with age [186]. The prevalence of the epigenetic modification H4K20me3 near the promoters of genes is associated with the SASP in cancer cells and reversibly terminates the mitotic cycle [187]. The rarity of H4K20me3 in the same positions in senescent cells may indicate an epigenetic mechanism for the transition of cells to this state when the H4K20 methylation process is disrupted, which may be a consequence of disturbed one-carbon metabolism (Figure 1) [187]. A decrease in H3K9 methylation is observed in Hutchinson–Gilford progeria (premature aging), and the depth of this decrease is proportional to an increase in the level of a defective nuclear protein, lamin A [188,189,190].
The onset of replicative senescence is linked to telomere shortening. Telomere shortening is often a by-product of either oxidative stress or inflammation. Micronutrients with antioxidant properties can protect telomeres from such damage. Vitamin B12 is essential for maintaining DNA integrity through nucleotide biosynthesis and methylation. Plasma B12 levels are associated with telomere length [191,192]. Vitamin B12 supplementation in women was associated with an increase in telomere length [193]. In addition, both low B12 levels associated with Hcy accumulation and high B12 levels associated with inflammation may influence telomere shortening [191]. These experimental data also reveal an unrecognized role of B12 in prevention of telomere shortening and regulation of senescence-associated processes through a positive feedback loop: since B12 is required for DNA methylation and telomere length is epigenetically regulated by DNA methylation, B12 levels appear to be directly reflected in telomere length [194].
A deficiency in vitamin B12 leads to the appearance of cellular senescence. Vitamin B12-deficient astrocytes have increased levels of SA-β-gal. Furthermore, overexpression of p16 and p21 in this model was conditioned by B12 deficiency [195]. Disruption of DNA synthesis due to cobalamin deficiency can probably induce p21 expression [196]. hESCs grown in a medium deficient in vitamins B6, B9, and B12 exhibit a number of phenotypic changes: cell disintegration, expression of senescence-associated β-galactosidase, reduced metabolic activity, and smaller size and number. These changes can be partially reversed by increasing the level of vitamins B6, B9, and B12 from 10–30% of their physiological concentrations to 70%; however, only hESCs that are moderately damaged by vitamin deficiencies are subject to such restoration [197]. Telomere length and mitochondrial DNA copy number decrease as cells age in inverse proportion to their B12 levels [198].
When MMUT function is deteriorated, e.g., in methylmalonic aciduria or in vitamin B12 deficiency, non-enzymatic post-translational modification of proteins such as malonylation and methylmalonylation may change the functions of a few proteins [2]. Such proteins are involved in the innate immunity response, maintenance of nuclear and mitochondrial DNA, RNA stability, and cell division control [2]—the features that change during aging [101]. Some of them directly interact with senescence-associated proteins, for example, those involved in the p53 signaling pathway, yet others, such as MMP13, are part of the SASP. Sirtuin SIRT5 removes acyl post-translational modifications, including those on histones [98]. However, in cases of deterioration of MMUT function, its activity is reduced due to MMUT modification [98]. Levels of global lysine malonylation are higher in the brains of older mice than in those of young mice [199], including proteins with molecular masses close to those of histones. Since histone malonation, especially in H2BK5, is regulated by SIRT5 in the mouse brain and liver, SIRT5 may have a significant role in aging [199]. Vitamin B12 deficiency also causes ER stress due to impaired SIRT1 deacetylation of heat shock factor 1 (HSF1) [200], which can be reversed by the drug metformin [201]. An increase in lifespan, which occurs in model yeast with calorie restriction, does not occur in cells with a mutated SIRT2 homolog gene [202]. During aging, MMA accumulation in blood (which is a characteristic of impaired MMUT function) promotes transformation of cells into aggressive tumor-like states [203]. Thus, decline in MMUT function due to scarcity of AdoCbl leads to impaired mitochondrial function, which is a feature of senescence.
It is important to note that SASP expression is also directly regulated by epigenetic rearrangements during SAHF formation. At the same time, as we have pointed out, it is SASP that is the major negative component of the presence of senescent cells in tumors. It is possible that the effects on vitamin B12 and the understanding of its fundamental effects on SAHF formation will help to regulate epigenetic rearrangements during senescence in the future. This will help to avoid the negative effects of senescent cells in cancer treatment. It is currently known that any type of cancer therapy (immunotherapy, chemotherapy, or radiotherapy) leads to an increase in the pool of senescent cells.
6.2. Vitamin B12 and Epigenetic Age
DNA methylation is used for the determination of the “biological” age of mammalian cells because methylation patterns change with age [204]. Thus, it can be assessed by “epigenetic clocks” by assessing the levels of hypo- and hypermethylation of certain CG regions of the genome [80,204].
In vitro and in vivo, loss of epigenetic information underlies the transition to the senescent state of mammalian cells, which is confirmed by an increase in the levels of its epigenetic marks during aging induced by DNA double-strand breaks (DSBs), such as hypomethylation of H3K27 and its hyperacetylation, and increase in “epigenetic age” [205]. Some of this information is lost due to DNA double-strand break repair mechanisms, even if mutations are not introduced in DNA. Induction of aging by double-strand DNA breaks in mouse cells doubled their epigenetic age with respect to control mice, and after the introduction of OSK factors, the epigenetic age of the mice was reduced by 57%. At the phenotypic level, mice subjected to such induced aging exhibit typical aging hallmarks. In mouse fibroblasts subjected to DSB-induced senescence, histone modification of H3K27me3 decreases, while that of H3K27ac increases. The described changes can be reversed with a time-limited induction of their pluripotency during epigenetic reprogramming with OSK factors [205]. Additional B12 amounts improve the efficiency of cellular reprogramming using OSKM factors in OSKM mice and promote faster regeneration of the intestinal epithelium after ulcerative colitis induced by sodium dextran sulfate [179]. The limiting factor for successful cellular reprogramming is vitamin B12, deficiency in which leads to SAM deficiency, which can impede the global rearrangement of DNA and histone methylation patterns, including H3K36me3 histone modification [51,179]. H3K36me3 prevents cryptic transcription, which is one of the hallmarks of senescent cells; thus, additional vitamin B12 increases translation fidelity during reprogramming [88,179,206].
Whether dietary supplementation with vitamin B12 increases or decreases epigenetic age is controversial. It has been known for more than three decades that global DNA methylation levels decrease with age [207,208,209] as well as in the case of impaired transport of vitamin B12 into cells [210]. A meta-analysis shows that supplementation of B12 with folic acid in adults increases global DNA methylation and changes gene-specific methylation status [211]. In a randomized, double-blinded, controlled clinical trial, after 1 year of treatment, older patients who received supplementation with calcium and vitamins D, B6, B9 and B12 versus calcium and vitamin D tended to have a higher epigenetic age based on the “epigenetic clocks” developed by Weidner and colleagues [212,213]. However, the effect was more pronounced for initially epigenetically “younger” individuals whose epigenetic age was lower than the chronological age at the baseline. This may indicate an influence on DNA methylation and not necessarily on the aging process, since potential confounders, such as 5,10-methenyltetrahydrofolate reductase polymorphism (MTHFR C677T), which causes increased homocysteine and decreased folate concentrations, were not taken into account. In another study with elderly volunteers, supplementation with folic acid and vitamin B12 increased overall DNA methylation, and changes in epigenetic age were dependent on MTHFR genotype and sex [214]. Women with MTHFR C677T exhibited the most pronounced reduction in expected and observed epigenetic age [214]. Despite the study design benefiting from the exclusion of individual-specific confounders due to the collection of blood samples before and after the intervention, the study lacked a placebo group. In another study, supplementation with vitamin B12 and folic acid for 2 years revealed six significantly differentially methylated regions in elderly individuals in comparison to the placebo group [1]. In the study, participants with either MTHFR genotype were equally distributed between the groups. Moreover, the methylation status of 425 regions correlated with the serum vitamin B12 level [1]. Long-term intake of folic acid and vitamin B12 in elderly people leads to changes in DNA methylation of genes involved in normal development and tumorigenesis [1]. Interestingly, in the data provided by Kok et. al., methylation of the 1500 bp upstream region of the gene PRKDC, a DNA damage sensor, is dependent on the serum holotranscobalamin level, and the product of PRKDC is methylmalonyated in the mouse model of MMUT deficiency [2]. In worms, feeding with MeCbl or AdoCbl decreases reproductive age [215]. However, in infants, higher B12 levels and lower levels of Hcy in plasma are correlated with lower epigenetic age [216].
Thus, adequate serum B12 levels fit the antagonistic pleiotropy theory of aging, a beneficial metabolite at a young age becoming detrimental at an older age.
6.3. Role of B12 in Cytokine Regulation and Inflammation
There is growing evidence that B12 levels are associated with markers of inflammation. Patients with high B12 levels and long telomeres have significantly lower levels of inflammatory markers such as IL-6. The authors of a study proposed that patients with high B12 levels and systemic inflammation have an increased risk of death due to accelerated telomere shortening [191]. In patients and in naturally aged mice, a decreased level of serum vitamin B12 is correlated with increased inflammatory markers, such as proinflammatory cytokine IL-6 and reactive C protein [5]. Increased concentrations of IL-6 are associated with all-case mortality in elderly individuals [217]. We hypothesize that this may be related to the senescence and production of SASP. IL-6 is a major component of SASP and a cytokine that plays a negative role in oncogenesis [218,219]. However, data from human cohorts are contradictory. While some studies have found an association, others have not. Lee et al. reported a reverse association between low vitamin B12 status and high IL-6 levels among people with diabetes [220]. In contrast, treatment of 150 women with vitamin supplements containing vitamin B12 for an average of 7 years had no effect on IL-6 and markers of endothelial dysfunction [220]. In the literature, vitamin B12 is discussed as an anti-inflammatory agent because of suppression of nuclear factor-κB (NF-kB), inhibition of nitric oxide synthase, and stimulation of oxidative phosphorylation [221,222]. The major cytokine that triggers the NF-kB pathway is TNF [223]. Increased blood levels of cobalamin are associated with inflammatory diseases and TNF levels [224]. Cobalamin levels also modulate TNF levels in the cerebrospinal fluid of patients [225]. This is complemented by several studies in cobalamin-deficient rats showing increased levels of some neurotoxic molecules, including TNF, in cerebrospinal fluid [226,227]. TNF plays an important role in inflammatory reactions and aging processes and in the activation of the SASP program [228,229].
Vitamin B12 may also alleviate inflammation in elderly individuals in another way. Itaconate is converted from aconitate derived from the Krebs cycle by the enzyme IRG1 (Aconitate decarboxylase 1, ACOD1) mainly expressed in macrophages [230]. Itaconate in its acid form is cell-permeable since the intracellular concentration of itanocyl-CoA, a product of itaconate, increases after incubation of cells with media supplemented with itaconate [231]. Itanocyl-CoA and citramyl-CoA bind to the active site of MMUT instead of methylmalonyl-CoA and irreversibly damage MMUT-bound AdoCbl [231]. This also prevents the damaged enzyme from being repaired [232]. Inability to recover MMUT and AdoCbl will deplete cobalamin inside the cells and disturb their metabolism. Given that itaconate may pass through the cell membrane and travel to adjacent cells, it will cause a local intracellular reduction in AdoCbl if the affected cells express IRG1. There is chronic, low-grade inflammation in aging, which is known as inflammaging [233]. In macrophages of aged mice, expression of Irg1 is higher than in young mice and leads to accumulation of itaconate inside cells [234]. Dietary supplementation of vitamin B12 together with other essential micronutrients decreases inflammaging [235]. Inflammaging may aggravate or even partially cause the consequences of cellular vitamin B12 deficiency.
Taken together, these data suggest that cobalamin modulates the expression of certain cytokines and growth factors, but few studies have been conducted at the cellular level as opposed to in human groups. This is due to the availability and widespread use of vitamin B12 as a nutritional supplement.
6.4. Microbiota, Vitamin B12, and Aging
Since vitamin B12 is ingested from food in the intestine, microbial community members compete for vitamin B12 with the host and each other (Figure 1). Only few species in human microbiota are able to produce cobalamin themselves from preceding molecules, namely, cobanamides [9]. During aging, microbial communities as well as metabolic pathway activities alter [236]. The composition of the microbiota has been used for age prediction and correlates with diet [237]. A more recent model for age prediction based on fecal microbiota data links age with metabolic pathway regulation, such as that of the vitamin B12 synthesis pathway, which is predicted to decrease “age” and be associated with healthy aging [238]. Despite the indications that humans can absorb minute amounts of B12 independently of the intrinsic factor after an intrinsic factor system overload or when it has defects [239,240], increased intestine vitamin B12 levels may not provide significant amounts of B12 to the host due to the inefficiency of such passive diffusion (~1% of total vitamin B12 excess). Cobalamin receptors are located in the small intestine, while most of the vitamin B12-synthesizing bacteria reside downstream in the digestive tract in the colon [241]. Moreover, cobalamin constitutes only a tiny fraction of all corrinoids, at least in feces [242]. MMUT and MS can utilize cobalamin analogues for catalysis in vitro with a lower affinity and reaction rate; however, whether other corrinoids can reach MMUT in vivo for partial restoration of B12 deficiency is not known. Transfer of microbiota from young to old mice or from the wild type to prematurely aged mice prolongs their lifespan [243,244]. Moreover, oral supplementation with the Akkermansia muciniphila MucT bacterium only, which is more represented in feces of young mice, was sufficient for lifespan extension in prematurely aged mice. While A. muciniphila is not a vitamin B12 synthesizer, increased vitamin B12 concentration shifts its metabolism to decrease production of succinate and increase production of propionate and acetate [11]. However, some other species from the Akkermansia genus, including other Akkermansia muciniphila strains, are able to synthesize vitamin B12 de novo [11]. Interestingly, the Akkermansia genus as well as a B12-synthesizing Bilophila are also substantially increased in the microbiota of centenarians [245]. Increased cobalamin synthesis probably reshapes microbiota composition and metabolic pathways rather than being absorbed by the host [3]. Vitamin B12 tunes the gut microbiota, shifting the community towards the production of metabolites able to pass through cell membranes or helping to maintain the mucosal layer [3,243,246]. On the other hand, the microbiota, especially commensal Gram-negative bacteria, may accelerate immunosenescence in the colon. Overstimulation of B cells in the colon by lipopolysaccharide spawns their senescence, resulting in decreased amounts and diversity of microbiota shaping IgA [247]. Continuous exposure of macrophages to LPS may lead to the production of itocanate, which might deplete AdoCbl in nearby cells [231]. Some injuries, like colitis, an inflammation in the colon caused by altered microbiota, require partial dedifferentiation of cells for recovery, a process similar to cellular reprogramming. This process demands increased amounts of vitamin B12 because levels of holotranscobalamin in the serum decrease [179].
Thus, ingested vitamin B12 modulates microbiota in ways that are important for healthy aging.
7. Conclusions
In light of recent studies, vitamin B12 emerges as a modulator of aging. It is clear that deficiency in vitamin B12 disturbs cellular homeostasis by decreasing the capacity to fight oxidative stress, support of one-carbon and energy metabolism, and maintenance of proper epigenetic features (Figure 4). Metformin, initially an antidiabetic drug, is in clinical trials for use as an antiaging drug. It alleviates all hallmarks of aging [248] and regulates global DNA methylation through accumulation of SAM [249]. In a clinical study, metformin usage led to vitamin B12 deficiency [250], which may indicate an increased demand for vitamin B12 in cells. Scarcity of vitamin B12 exacerbates age-related cognitive decline, post-translational protein modifications, inflammaging, and reduced capacity for regeneration and potential for cellular reprogramming. High amounts of vitamin B12 are beneficial during gestation; however, they may have detrimental effects in elderly people due to the increasing probability of cancer and death, which fits the antagonistic pleiotropy theory of aging. During aging, transcription fidelity decreases, which may affect vitamin B12 ingestion and transport to cells [251,252]. Nonetheless, other possible mechanisms may also exist, such as deprivation of vitamin B12 caused by inflammaging, at least in macrophages. Adequate levels of vitamin B12 are needed for healthy aging, and antiaging interventions (i.e., cellular reprogramming) require an additional supply of B12. There is a need for standardization in determining vitamin B12 deficiency. We find it interesting to determine the role of B12 in senescence programs and its fundamental role in the implementation of epigenetic regulation programs, including SASP. Research studies on aging and age-related pathologies will benefit from methods allowing dynamic and specific assessments of cobalamins, including genetically encoded sensors with single-cell resolution, such as SenVitAL and its derivatives [253,254].
Author Contributions
Conceptualization, N.A.K.; writing—original draft preparation, S.Y.S., D.A.B. and N.A.K.; writing—review and editing, S.Y.S., D.A.B. and N.A.K.; visualization, S.Y.S.; project administration, N.A.K.; funding acquisition, N.A.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, grant number № 23-75-01141, https://rscf.ru/en/project/23-75-01141/, accessed on 1 April 2024.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Kok, D.E.G.; Dhonukshe-Rutten, R.A.M.; Lute, C.; Heil, S.G.; Uitterlinden, A.G.; van der Velde, N.; van Meurs, J.B.J.; van Schoor, N.M.; Hooiveld, G.J.E.J.; de Groot, L.C.P.G.M.; et al. The Effects of Long-Term Daily Folic Acid and Vitamin B12 Supplementation on Genome-Wide DNA Methylation in Elderly Subjects. Clin. Epigenet. 2015, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Head, P.E.; Myung, S.; Chen, Y.; Schneller, J.L.; Wang, C.; Duncan, N.; Hoffman, P.; Chang, D.; Gebremariam, A.; Gucek, M.; et al. Aberrant Methylmalonylation Underlies Methylmalonic Acidemia and Is Attenuated by an Engineered Sirtuin. Sci. Transl. Med. 2022, 14, eabn4772. [Google Scholar] [CrossRef] [PubMed]
- Degnan, P.H.; Taga, M.E.; Goodman, A.L. Vitamin B12 as a Modulator of Gut Microbial Ecology. Cell Metab. 2014, 20, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, V.; Chabrun, F.; Lacout, C.; Ghali, A.; Capitain, O.; Patsouris, A.; Lavigne, C.; Urbanski, G. Persistent Elevation of Plasma Vitamin B12 Is Strongly Associated with Solid Cancer. Sci. Rep. 2021, 11, 13361. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-López, I.; Kovatcheva, M.; Casas, R.; Toledo, E.; Fitó, M.; Ros, E.; Estruch, R.; Serrano, M.; Lamuela-Raventós, R.M. Higher Circulating Vitamin B12 Is Associated with Lower Levels of Inflammatory Markers in Individuals at High Cardiovascular Risk and in Naturally Aged Mice. J. Sci. Food Agric. 2024, 104, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.F.; Ward, M.; Hoey, L.; McNulty, H. Vitamin B12 and Ageing: Current Issues and Interaction with Folate. Ann. Clin. Biochem. 2013, 50, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, K.; Apostolopoulos, V. B Vitamins and Ageing. In Biochemistry and Cell Biology of Ageing: Part I Biomedical Science; Harris, J.R., Korolchuk, V.I., Eds.; Subcellular Biochemistry; Springer: Singapore, 2018; Volume 90, pp. 451–470. ISBN 9789811328343. [Google Scholar]
- Woodward, R.B. The Total Synthesis of Vitamin B12. Pure Appl. Chem. 1973, 33, 145–178. [Google Scholar] [CrossRef]
- Martens, H.; Barg, M.; Warren, D.; Jah, J.-H. Microbial Production of Vitamin B12. Appl. Microbiol. Biotechnol. 2002, 58, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Degnan, P.H.; Barry, N.A.; Mok, K.C.; Taga, M.E.; Goodman, A.L. Human Gut Microbes Use Multiple Transporters to Distinguish Vitamin B12 Analogs and Compete in the Gut. Cell Host Microbe 2014, 15, 47–57. [Google Scholar] [CrossRef]
- Kirmiz, N.; Galindo, K.; Cross, K.L.; Luna, E.; Rhoades, N.; Podar, M.; Flores, G.E. Comparative Genomics Guides Elucidation of Vitamin B12 Biosynthesis in Novel Human-Associated Akkermansia Strains. Appl. Environ. Microbiol. 2020, 86, e02117-19. [Google Scholar] [CrossRef]
- Balabanova, L.; Averianova, L.; Marchenok, M.; Son, O.; Tekutyeva, L. Microbial and Genetic Resources for Cobalamin (Vitamin B12) Biosynthesis: From Ecosystems to Industrial Biotechnology. Int. J. Mol. Sci. 2021, 22, 4522. [Google Scholar] [CrossRef] [PubMed]
- Fedosov, S.N.; Fedosova, N.U.; Kräutler, B.; Nexø, E.; Petersen, T.E. Mechanisms of Discrimination between Cobalamins and Their Natural Analogues during Their Binding to the Specific B12-Transporting Proteins. Biochemistry 2007, 46, 6446–6458. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Rasmussen, M.R.; Andersen, C.B.F.; Nexø, E.; Moestrup, S.K. Vitamin B12 Transport from Food to the Body’s Cells—A Sophisticated, Multistep Pathway. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 345–354. [Google Scholar] [CrossRef]
- Quadros, E.V.; Nakayama, Y.; Sequeira, J.M. The Protein and the Gene Encoding the Receptor for the Cellular Uptake of Transcobalamin-Bound Cobalamin. Blood 2009, 113, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of MRNAs Genome-Wide. Mol. Cell 2017, 67, 71–83.e7. [Google Scholar] [CrossRef] [PubMed]
- Birn, H.; Willnow, T.E.; Nielsen, R.; Norden, A.G.W.; Bönsch, C.; Moestrup, S.K.; Nexø, E.; Christensen, E.I. Megalin Is Essential for Renal Proximal Tubule Reabsorption and Accumulation of Transcobalamin-B12. Am. J. Physiol.-Ren. Physiol. 2002, 282, F408–F416. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Gailus, S.; Miousse, I.R.; Suormala, T.; Sagné, C.; Toliat, M.R.; Nürnberg, G.; Wittkampf, T.; Buers, I.; Sharifi, A.; et al. Identification of a Putative Lysosomal Cobalamin Exporter Altered in the CblF Defect of Vitamin B12 Metabolism. Nat. Genet. 2009, 41, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Coelho, D.; Kim, J.C.; Miousse, I.R.; Fung, S.; du Moulin, M.; Buers, I.; Suormala, T.; Burda, P.; Frapolli, M.; Stucki, M.; et al. Mutations in ABCD4 Cause a New Inborn Error of Vitamin B12 Metabolism. Nat. Genet. 2012, 44, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gherasim, C.; Banerjee, R. Decyanation of Vitamin B12 by a Trafficking Chaperone. Proc. Natl. Acad. Sci. USA 2008, 105, 14551–14554. [Google Scholar] [CrossRef]
- Hannibal, L.; Kim, J.; Brasch, N.E.; Wang, S.; Rosenblatt, D.S.; Banerjee, R.; Jacobsen, D.W. Processing of Alkylcobalamins in Mammalian Cells: A Role for the MMACHC (CblC) Gene Product. Mol. Genet. Metab. 2009, 97, 260–266. [Google Scholar] [CrossRef]
- Suormala, T.; Baumgartner, M.R.; Coelho, D.; Zavadakova, P.; Kožich, V.; Koch, H.G.; Berghaüser, M.; Wraith, J.E.; Burlina, A.; Sewell, A.; et al. The CblD Defect Causes Either Isolated or Combined Deficiency of Methylcobalamin and Adenosylcobalamin Synthesis. J. Biol. Chem. 2004, 279, 42742–42749. [Google Scholar] [CrossRef]
- Padovani, D.; Labunska, T.; Palfey, B.A.; Ballou, D.P.; Banerjee, R. Adenosyltransferase Tailors and Delivers Coenzyme B12. Nat. Chem. Biol. 2008, 4, 194–196. [Google Scholar] [CrossRef]
- Padovani, D.; Banerjee, R. A G-Protein Editor Gates Coenzyme B12 Loading and Is Corrupted in Methylmalonic Aciduria. Proc. Natl. Acad. Sci. USA 2009, 106, 21567–21572. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.A.; Arzamasov, A.A.; Khoroshkin, M.S.; Iablokov, S.N.; Leyn, S.A.; Peterson, S.N.; Novichkov, P.S.; Osterman, A.L. Micronutrient Requirements and Sharing Capabilities of the Human Gut Microbiome. Front. Microbiol. 2019, 10, 1316. [Google Scholar] [CrossRef] [PubMed]
- Magnúsdóttir, S.; Ravcheev, D.; de Crécy-Lagard, V.; Thiele, I. Systematic Genome Assessment of B-Vitamin Biosynthesis Suggests Co-Operation among Gut Microbes. Front. Genet. 2015, 6, 129714. [Google Scholar] [CrossRef] [PubMed]
- van Asselt, D.Z.; de Groot, L.C.; van Staveren, W.A.; Blom, H.J.; Wevers, R.A.; Biemond, I.; Hoefnagels, W.H. Role of Cobalamin Intake and Atrophic Gastritis in Mild Cobalamin Deficiency in Older Dutch Subjects. Am. J. Clin. Nutr. 1998, 68, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Pannérec, A.; Migliavacca, E.; De Castro, A.; Michaud, J.; Karaz, S.; Goulet, L.; Rezzi, S.; Ng, T.P.; Bosco, N.; Larbi, A.; et al. Vitamin B12 Deficiency and Impaired Expression of Amnionless during Aging. J. Cachexia Sarcopenia Muscle 2018, 9, 41–52. [Google Scholar] [CrossRef]
- Zhang, Y.; Hodgson, N.W.; Trivedi, M.S.; Abdolmaleky, H.M.; Fournier, M.; Cuenod, M.; Do, K.Q.; Deth, R.C. Decreased Brain Levels of Vitamin B12 in Aging, Autism and Schizophrenia. PLoS ONE 2016, 11, e0146797. [Google Scholar] [CrossRef]
- Cannata, J.J.B.; Focesi, A.; Mazumder, R.; Warner, R.C.; Ochoa, S. Metabolism of Propionic Acid in Animal Tissues. J. Biol. Chem. 1965, 240, 3249–3257. [Google Scholar] [CrossRef]
- Frenkel, E.P.; Kitchens, R.L. Intracellular Localization of Hepatic Propionyl-CoA Carboxylase and Methylmalonyl-CoA Mutase in Humans and Normal and Vitamin B12 Deficient Rats. Br. J. Haematol. 1975, 31, 501–513. [Google Scholar] [CrossRef]
- Wolfle, K.; Michenfelder, M.; Konig, A.; Hull, W.E.; Retey, J. On the Mechanism of Action of Methylmalonyl-CoA Mutase. Change of the Steric Course on Isotope Substitution. Eur. J. Biochem. 1986, 156, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Kochan, G.; Muniz, J.R.C.; Wu, X.; Gileadi, C.; Ugochukwu, E.; Krysztofinska, E.; Gravel, R.A.; Oppermann, U.; Yue, W.W. Structures of the Human GTPase MMAA and Vitamin B12-Dependent Methylmalonyl-CoA Mutase and Insight into Their Complex Formation. J. Biol. Chem. 2010, 285, 38204–38213. [Google Scholar] [CrossRef]
- Marsh, E.N.G.; Meléndez, G.D.R. Adenosylcobalamin Enzymes: Theory and Experiment Begin to Converge. Biochim. Biophys. Acta BBA Proteins Proteom. 2012, 1824, 1154–1164. [Google Scholar] [CrossRef]
- Bassila, C.; Ghemrawi, R.; Flayac, J.; Froese, D.S.; Baumgartner, M.R.; Guéant, J.-L.; Coelho, D. Methionine Synthase and Methionine Synthase Reductase Interact with MMACHC and with MMADHC. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 103–112. [Google Scholar] [CrossRef]
- The GTEx Consortium; Aguet, F.; Anand, S.; Ardlie, K.G.; Gabriel, S.; Getz, G.A.; Graubert, A.; Hadley, K.; Handsaker, R.E.; Huang, K.H.; et al. The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Banerjee, R. A B12-Responsive Internal Ribosome Entry Site (IRES) Element in Human Methionine Synthase. J. Biol. Chem. 2005, 280, 32662–32668. [Google Scholar] [CrossRef]
- Chen, L.H.; Liu, M.-L.; Hwang, H.-Y.; Chen, L.-S.; Korenberg, J.; Shane, B. Human Methionine Synthase. J. Biol. Chem. 1997, 272, 3628–3634. [Google Scholar] [CrossRef]
- Mendoza, J.; Purchal, M.; Yamada, K.; Koutmos, M. Structure of Full-Length Cobalamin-Dependent Methionine Synthase and Cofactor Loading Captured in Crystallo. Nat. Commun. 2023, 14, 6365. [Google Scholar] [CrossRef] [PubMed]
- Watkins, M.B.; Wang, H.; Burnim, A.; Ando, N. Conformational Switching and Flexibility in Cobalamin-Dependent Methionine Synthase Studied by Small-Angle X-Ray Scattering and Cryoelectron Microscopy. Proc. Natl. Acad. Sci. USA 2023, 120, e2302531120. [Google Scholar] [CrossRef]
- Bandarian, V.; Pattridge, K.A.; Lennon, B.W.; Huddler, D.P.; Matthews, R.G.; Ludwig, M.L. Domain Alternation Switches B(12)-Dependent Methionine Synthase to the Activation Conformation. Nat. Struct. Biol. 2002, 9, 53–56. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Amaratunga, M.; Drennan, C.L.; Scholten, J.D.; Sands, R.H.; Ludwig, M.L.; Matthews, R.G. Mutations in the B12-Binding Region of Methionine Synthase: How the Protein Controls Methylcobalamin Reactivity. Biochemistry 1996, 35, 2464–2475. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kozlowski, P.M. Mechanistic Insights for Formation of an Organometallic Co–C Bond in the Methyl Transfer Reaction Catalyzed by Methionine Synthase. J. Phys. Chem. B 2013, 117, 16044–16057. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Matthews, R.G. Protonation State of Methyltetrahydrofolate in a Binary Complex with Cobalamin-Dependent Methionine Synthase. Biochemistry 2000, 39, 13880–13890. [Google Scholar] [CrossRef] [PubMed]
- Drummond, J.T.; Huang, S.; Blumenthal, R.M.; Matthews, R.G. Assignment of Enzymic Function to Specific Protein Regions of Cobalamin-Dependent Methionine Synthase from Escherichia Coli. Biochemistry 1993, 32, 9290–9295. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Darnell, A.M.; Reilly, M.F.; Kunchok, T.; Joesch-Cohen, L.; Rosenberg, D.; Ali, A.; Rees, M.G.; Roth, J.A.; Lewis, C.A. Methionine Synthase Is Essential for Cancer Cell Proliferation in Physiological Folate Environments. Nat. Metab. 2021, 3, 1500–1511. [Google Scholar] [CrossRef] [PubMed]
- West, A.A.; Caudill, M.A.; Bailey, L.B. Folate. In Present Knowledge in Nutrition; Elsevier: Amsterdam, The Netherlands, 2020; pp. 239–255. [Google Scholar]
- Hashimoto, H.; Vertino, P.M.; Cheng, X. Molecular Coupling of DNA Methylation and Histone Methylation. Epigenomics 2010, 2, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Tu, B.P. Sink into the Epigenome: Histones as Repositories That Influence Cellular Metabolism. Trends Endocrinol. Metab. 2018, 29, 626–637. [Google Scholar] [CrossRef]
- Ye, C.; Sutter, B.M.; Wang, Y.; Kuang, Z.; Tu, B.P. A Metabolic Function for Phospholipid and Histone Methylation. Mol. Cell 2017, 66, 180–193.e8. [Google Scholar] [CrossRef] [PubMed]
- Shyh-Chang, N.; Locasale, J.W.; Lyssiotis, C.A.; Zheng, Y.; Teo, R.Y.; Ratanasirintrawoot, S.; Zhang, J.; Onder, T.; Unternaehrer, J.J.; Zhu, H.; et al. Influence of Threonine Metabolism on S -Adenosylmethionine and Histone Methylation. Science 2013, 339, 222–226. [Google Scholar] [CrossRef]
- Sharma, A.; Kramer, M.L.; Wick, P.F.; Liu, D.; Chari, S.; Shim, S.; Tan, W.; Ouellette, D.; Nagata, M.; DuRand, C.J.; et al. D4 Dopamine Receptor-Mediated Phospholipid Methylation and Its Implications for Mental Illnesses Such as Schizophrenia. Mol. Psychiatry 1999, 4, 235–246. [Google Scholar] [CrossRef]
- Hodgson, N.W.; Waly, M.I.; Trivedi, M.S.; Power-Charnitsky, V.-A.; Deth, R.C. Methylation-Related Metabolic Effects of D4 Dopamine Receptor Expression and Activation. Transl. Psychiatry 2019, 9, 295. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pan, T. Methionine Mistranslation Bypasses the Restraint of the Genetic Code to Generate Mutant Proteins with Distinct Activities. PLoS Genet. 2015, 11, e1005745. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Weiss, S.J.; Levine, R.L. Methionine Oxidation and Reduction in Proteins. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Kuschel, L.; Hansel, A.; Schönherr, R.; Weissbach, H.; Brot, N.; Hoshi, T.; Heinemann, S.H. Molecular Cloning and Functional Expression of a Human Peptide Methionine Sulfoxide Reductase (HMsrA). FEBS Lett. 1999, 456, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.B.; Butterfield, D.A. Measurement of Oxidized/Reduced Glutathione Ratio. In Protein Misfolding and Cellular Stress in Disease and Aging; Bross, P., Gregersen, N., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 648, pp. 269–277. ISBN 978-1-60761-755-6. [Google Scholar]
- Li, E.; Zhang, Y. DNA Methylation in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Wang, R.Y.-H. 5-Methylcytosine in Eukaryotic DNA. Science 1981, 212, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Dubini, R.C.A.; Mayer, P.; Rovó, P.; Daumann, L.J. Formation of Cisplatin Adducts with the Epigenetically Relevant Nucleobase 5-Methylcytosine. Eur. J. Inorg. Chem. 2021, 2021, 30–36. [Google Scholar] [CrossRef]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG Methylation Is Prevalent in Embryonic Stem Cells and May Be Mediated by DNA Methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA Methylation on N6-Adenine in Mammalian Embryonic Stem Cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, M.; Guo, M. DNA N6-Methyladenine Epigenetic Modification Elevated in Human Esophageal Squamous Cell Carcinoma: A Potential Prognostic Marker. Discov. Med. 2020, 29, 85–90. [Google Scholar] [PubMed]
- Lin, Q.; Chen, J.; Yin, H.; Li, M.; Zhou, C.; Hao, T.; Pan, T.; Wu, C.; Li, Z.; Zhu, D.; et al. DNA N6-Methyladenine Involvement and Regulation of Hepatocellular Carcinoma Development. Genomics 2022, 114, 110265. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA Methylation of the First Exon Is Tightly Linked to Transcriptional Silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Rappolee, D.A.; Ruden, D.M. Epigenetic Reprogramming in Mice and Humans: From Fertilization to Primordial Germ Cell Development. Cells 2023, 12, 1874. [Google Scholar] [CrossRef]
- Bollati, V.; Schwartz, J.; Wright, R.; Litonjua, A.; Tarantini, L.; Suh, H.; Sparrow, D.; Vokonas, P.; Baccarelli, A. Decline in Genomic DNA Methylation through Aging in a Cohort of Elderly Subjects. Mech. Ageing Dev. 2009, 130, 234–239. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Kanazhevskaya, L.Y.; Fedorova, O.S. DNA Demethylation in the Processes of Repair and Epigenetic Regulation Performed by 2-Ketoglutarate-Dependent DNA Dioxygenases. Int. J. Mol. Sci. 2021, 22, 10540. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Liu, F.; Clark, W.; Luo, G.; Wang, X.; Fu, Y.; Wei, J.; Wang, X.; Hao, Z.; Dai, Q.; Zheng, G.; et al. ALKBH1-Mediated TRNA Demethylation Regulates Translation. Cell 2016, 167, 816–828.e16. [Google Scholar] [CrossRef]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.A.; Neretti, N.; Sedivy, J.M. Genomes of Replicatively Senescent Cells Undergo Global Epigenetic Changes Leading to Gene Silencing and Activation of Transposable Elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Jansz, N. DNA Methylation Dynamics at Transposable Elements in Mammals. Essays Biochem. 2019, 63, 677–689. [Google Scholar] [PubMed]
- Yang, Z.; Xu, F.; Wang, H.; Teschendorff, A.E.; Xie, F.; He, Y. Pan-Cancer Characterization of Long Non-Coding RNA and DNA Methylation Mediated Transcriptional Dysregulation. EBioMedicine 2021, 68, 103399. [Google Scholar] [CrossRef]
- Lujambio, A.; Esteller, M. How Epigenetics Can Explain Human Metastasis: A New Role for MicroRNAs. Cell Cycle 2009, 8, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Maruyama, R.; Yamamoto, E.; Kai, M. DNA Methylation and MicroRNA Dysregulation in Cancer. Mol. Oncol. 2012, 6, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Munk, R.; Panda, A.C.; Grammatikakis, I.; Gorospe, M.; Abdelmohsen, K. Senescence-Associated MicroRNAs. Int. Rev. Cell Mol. Biol. 2017, 334, 177–205. [Google Scholar] [PubMed]
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Strahl, B.D. Interpreting the Language of Histone and DNA Modifications. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Nimura, K.; Ura, K.; Kaneda, Y. Histone Methyltransferases: Regulation of Transcription and Contribution to Human Disease. J. Mol. Med. 2010, 88, 1213–1220. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.-E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 Recruitment and H3K79 Methylation Are Ubiquitously Coupled with Gene Transcription in Mammalian Cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The Interplay of Histone Modifications—Writers That Read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Kang, W.K.; Florman, J.T.; Araya, A.; Fox, B.W.; Thackeray, A.; Schroeder, F.C.; Walhout, A.J.M.; Alkema, M.J. Vitamin B12 Produced by Gut Bacteria Modulates Cholinergic Signalling. Nat. Cell Biol. 2024, 26, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Kolasinska-Zwierz, P.; Down, T.; Latorre, I.; Liu, T.; Liu, X.S.; Ahringer, J. Differential Chromatin Marking of Introns and Expressed Exons by H3K36me3. Nat. Genet. 2009, 41, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA Methylation Prevents Spurious Transcription Initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.-P.; Allis, C.D. DNMT3L Connects Unmethylated Lysine 4 of Histone H3 to de Novo Methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef]
- Henckel, A.; Nakabayashi, K.; Sanz, L.A.; Feil, R.; Hata, K.; Arnaud, P. Histone Methylation Is Mechanistically Linked to DNA Methylation at Imprinting Control Regions in Mammals. Hum. Mol. Genet. 2009, 18, 3375–3383. [Google Scholar] [CrossRef]
- Benevolenskaya, E.V. Histone H3K4 Demethylases Are Essential in Development and Differentiation. Biochem. Cell Biol. 2007, 85, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.M.; Andrews, R.M.; Flicek, P.; Dillon, S.C.; Karaöz, U.; Clelland, G.K.; Wilcox, S.; Beare, D.M.; Fowler, J.C.; Couttet, P. The Landscape of Histone Modifications across 1% of the Human Genome in Five Human Cell Lines. Genome Res. 2007, 17, 691–707. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac Separates Active from Poised Enhancers and Predicts Developmental State. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Pradeepa, M.M.; Grimes, G.R.; Kumar, Y.; Olley, G.; Taylor, G.C.; Schneider, R.; Bickmore, W.A. Histone H3 Globular Domain Acetylation Identifies a New Class of Enhancers. Nat. Genet. 2016, 48, 681–686. [Google Scholar] [CrossRef]
- Fuks, F.; Hurd, P.J.; Deplus, R.; Kouzarides, T. The DNA Methyltransferases Associate with HP1 and the SUV39H1 Histone Methyltransferase. Nucleic Acids Res. 2003, 31, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.-S. Histone Deacetylase Inhibitors: Potential in Cancer Therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Feldman, J.L.; Baeza, J.; Denu, J.M. Activation of the Protein Deacetylase SIRT6 by Long-Chain Fatty Acids and Widespread Deacylation by Mammalian Sirtuins. J. Biol. Chem. 2013, 288, 31350–31356. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Lu, Z.; Xie, Z.; Cheng, Z.; Chen, Y.; Tan, M.; Luo, H.; Zhang, Y.; He, W.; Yang, K. The First Identification of Lysine Malonylation Substrates and Its Regulatory Enzyme. Mol. Cell. Proteom. 2011, 10, M111.012658. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Lee, J.-H.; Lee, H.-Y.; Min, K.-J. Sirtuin Signaling in Cellular Senescence and Aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Paluvai, H.; Di Giorgio, E.; Brancolini, C. The Histone Code of Senescence. Cells 2020, 9, 466. [Google Scholar] [CrossRef]
- Harrington, D.J. Methods for Assessment of Vitamin B12. In Laboratory Assessment of Vitamin Status; Elsevier: Amsterdam, The Netherlands, 2019; pp. 265–299. ISBN 978-0-12-813050-6. [Google Scholar]
- Sobczyńska-Malefora, A.; Delvin, E.; McCaddon, A.; Ahmadi, K.R.; Harrington, D.J. Vitamin B12 Status in Health and Disease: A Critical Review. Diagnosis of Deficiency and Insufficiency—Clinical and Laboratory Pitfalls. Crit. Rev. Clin. Lab. Sci. 2021, 58, 399–429. [Google Scholar] [CrossRef]
- İspir, E.; Serdar, M.A.; Ozgurtas, T.; Gulbahar, O.; Akın, K.O.; Yesildal, F.; Kurt, İ. Comparison of Four Automated Serum Vitamin B12 Assays. Clin. Chem. Lab. Med. 2015, 53, 1205–1213. [Google Scholar] [CrossRef]
- Hannibal, L.; Axhemi, A.; Glushchenko, A.V.; Moreira, E.S.; Brasch, N.E.; Jacobsen, D.W. Accurate Assessment and Identification of Naturally Occurring Cellular Cobalamins. Clin. Chem. Lab. Med. 2008, 46, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.-L.; Brito, A.; Guéant, J.-L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.-H.; et al. Vitamin B12 Deficiency. Nat. Rev. Dis. Primer 2017, 3, 17040. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Dangat, K.; Kale, A.; Sable, P.; Chavan-Gautam, P.; Joshi, S. Effects of Altered Maternal Folic Acid, Vitamin B12 and Docosahexaenoic Acid on Placental Global DNA Methylation Patterns in Wistar Rats. PLoS ONE 2011, 6, e17706. [Google Scholar] [CrossRef]
- Kumar, K.A.; Lalitha, A.; Reddy, U.; Chandak, G.R.; Sengupta, S.; Raghunath, M. Chronic Maternal Vitamin B12 Restriction Induced Changes in Body Composition & Glucose Metabolism in the Wistar Rat Offspring Are Partly Correctable by Rehabilitation. PLoS ONE 2014, 9, e112991. [Google Scholar]
- Tanwar, V.S.; Ghosh, S.; Sati, S.; Ghose, S.; Kaur, L.; Kumar, K.A.; Shamsudheen, K.V.; Patowary, A.; Singh, M.; Jyothi, V.; et al. Maternal Vitamin B12 Deficiency in Rats Alters DNA Methylation in Metabolically Important Genes in Their Offspring. Mol. Cell. Biochem. 2020, 468, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, K.D.; Allegrucci, C.; Singh, R.; Gardner, D.S.; Sebastian, S.; Bispham, J.; Thurston, A.; Huntley, J.F.; Rees, W.D.; Maloney, C.A.; et al. DNA Methylation, Insulin Resistance, and Blood Pressure in Offspring Determined by Maternal Periconceptional B Vitamin and Methionine Status. Proc. Natl. Acad. Sci. USA 2007, 104, 19351–19356. [Google Scholar] [CrossRef]
- Sharma, G.S.; Kumar, T.; Singh, L.R. N-Homocysteinylation Induces Different Structural and Functional Consequences on Acidic and Basic Proteins. PLoS ONE 2014, 9, e116386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bai, B.; Mei, X.; Wan, C.; Cao, H.; Dan, L.; Wang, S.; Zhang, M.; Wang, Z.; Wu, J.; et al. Elevated H3K79 Homocysteinylation Causes Abnormal Gene Expression during Neural Development and Subsequent Neural Tube Defects. Nat. Commun. 2018, 9, 3436. [Google Scholar] [CrossRef]
- Ghosh, S.; Sinha, J.K.; Khandelwal, N.; Chakravarty, S.; Kumar, A.; Raghunath, M. Increased Stress and Altered Expression of Histone Modifying Enzymes in Brain Are Associated with Aberrant Behaviour in Vitamin B12 Deficient Female Mice. Nutr. Neurosci. 2020, 23, 714–723. [Google Scholar] [CrossRef]
- Suarez-Moreira, E.; Yun, J.; Birch, C.S.; Williams, J.H.H.; McCaddon, A.; Brasch, N.E. Vitamin B12 and Redox Homeostasis: Cob(II)Alamin Reacts with Superoxide at Rates Approaching Superoxide Dismutase (SOD). J. Am. Chem. Soc. 2009, 131, 15078–15079. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Bahnson, E.M.; Wilder, J.; Siletzky, R.; Hagaman, J.; Nickekeit, V.; Hiller, S.; Ayesha, A.; Feng, L.; Levine, J.S.; et al. Oral High Dose Vitamin B12 Decreases Renal Superoxide and Post-Ischemia/Reperfusion Injury in Mice. Redox Biol. 2020, 32, 101504. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.; Leonard, J.V.; Baumgartner, M.R. Causes of and Diagnostic Approach to Methylmalonic Acidurias. J. Inherit. Metab. Dis. 2008, 31, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Cudré-Cung, H.-P.; Zavadakova, P.; do Vale-Pereira, S.; Remacle, N.; Henry, H.; Ivanisevic, J.; Tavel, D.; Braissant, O.; Ballhausen, D. Ammonium Accumulation Is a Primary Effect of 2-Methylcitrate Exposure in an in Vitro Model for Brain Damage in Methylmalonic Aciduria. Mol. Genet. Metab. 2016, 119, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, L.; Viville, S.; Barton, S.C.; Ishino, F.; Keverne, E.B.; Surani, M.A. Abnormal Maternal Behaviour and Growth Retardation Associated with Loss of the Imprinted Gene Mest. Nat. Genet. 1998, 20, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Sapehia, D.; Mahajan, A.; Singh, P.; Kaur, J. High Dietary Folate and Low Vitamin B12 in Parental Diet Disturbs the Epigenetics of Imprinted Genes MEST and PHLDA2 in Mice Placenta. J. Nutr. Biochem. 2023, 118, 109354. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz, K.B.; Cavalcante-Silva, V.; Lopes, F.L.; Rocha, G.A.; D’Almeida, V.; Coimbra, R.S. Vitamin B12 Is Neuroprotective in Experimental Pneumococcal Meningitis through Modulation of Hippocampal DNA Methylation. J. Neuroinflammation 2020, 17, 96. [Google Scholar] [CrossRef] [PubMed]
- Batista, K.S.; Cintra, V.M.; Lucena, P.A.; Manhães-de-Castro, R.; Toscano, A.E.; Costa, L.P.; Queiroz, M.E.; de Andrade, S.M.; Guzman-Quevedo, O.; Aquino, J.d.S. The Role of Vitamin B12 in Viral Infections: A Comprehensive Review of Its Relationship with the Muscle–Gut–Brain Axis and Implications for SARS-CoV-2 Infection. Nutr. Rev. 2022, 80, 561–578. [Google Scholar] [CrossRef] [PubMed]
- Yeganeh, F.; Nikbakht, F.; Bahmanpour, S.; Rastegar, K.; Namavar, R. Neuroprotective Effects of NMDA and Group I Metabotropic Glutamate Receptor Antagonists Against Neurodegeneration Induced by Homocysteine in Rat Hippocampus: In Vivo Study. J. Mol. Neurosci. 2013, 50, 551–557. [Google Scholar] [CrossRef]
- Pyrgioti, E.E.; Karakousis, N.D. B12 Levels and Frailty Syndrome. J. Frailty Sarcopenia Falls 2022, 7, 32–37. [Google Scholar] [CrossRef]
- Liu, L.; van Groen, T.; Kadish, I.; Tollefsbol, T.O. DNA Methylation Impacts on Learning and Memory in Aging. Neurobiol. Aging 2009, 30, 549–560. [Google Scholar] [CrossRef]
- Oliveira, A.M.; Hemstedt, T.J.; Bading, H. Rescue of Aging-Associated Decline in Dnmt3a2 Expression Restores Cognitive Abilities. Nat. Neurosci. 2012, 15, 1111–1113. [Google Scholar] [CrossRef]
- Saunderson, E.A.; Spiers, H.; Mifsud, K.R.; Gutierrez-Mecinas, M.; Trollope, A.F.; Shaikh, A.; Mill, J.; Reul, J.M.H.M. Stress-Induced Gene Expression and Behavior Are Controlled by DNA Methylation and Methyl Donor Availability in the Dentate Gyrus. Proc. Natl. Acad. Sci. USA 2016, 113, 4830–4835. [Google Scholar] [CrossRef]
- Loenen, W.A. S-Adenosylmethionine Metabolism and Aging. In Epigenetics of Aging and Longevity; Elsevier: Amsterdam, The Netherlands, 2018; pp. 59–93. [Google Scholar]
- Shen, L.; Ji, H.-F. Associations between Homocysteine, Folic Acid, Vitamin B12 and Alzheimer’s Disease: Insights from Meta-Analyses. J. Alzheimers Dis. 2015, 46, 777–790. [Google Scholar] [CrossRef]
- McCarter, S.J.; Teigen, L.M.; McCarter, A.R.; Benarroch, E.E.; St. Louis, E.K.; Savica, R. Low Vitamin B12 and Parkinson Disease. Mayo Clin. Proc. 2019, 94, 757–762. [Google Scholar] [CrossRef]
- Andrès, E.; Serraj, K.; Zhu, J.; Vermorken, A.J. The Pathophysiology of Elevated Vitamin B12 in Clinical Practice. QJM Int. J. Med. 2013, 106, 505–515. [Google Scholar] [CrossRef]
- Guest, J.; Bilgin, A.; Hokin, B.; Mori, T.A.; Croft, K.D.; Grant, R. Novel Relationships between B12, Folate and Markers of Inflammation, Oxidative Stress and NAD(H) Levels, Systemically and in the CNS of a Healthy Human Cohort. Nutr. Neurosci. 2015, 18, 355–364. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef]
- Tikhonov, S.; Batin, M.; Gladyshev, V.N.; Dmitriev, S.E.; Tyshkovskiy, A. AgeMeta: Quantitative Gene Expression Database of Mammalian Aging. Biochem. Mosc. 2024, 89, 313–321. [Google Scholar] [CrossRef]
- Argan, O.; Ural, D.; Karauzum, K.; Bozyel, S.; Aktaş, M.; Karauzum, I.; Kozdag, G.; Agacdiken Agir, A. Elevated Levels of Vitamin B12 in Chronic Stable Heart Failure: A Marker for Subclinical Liver Damage and Impaired Prognosis. Ther. Clin. Risk Manag. 2018, 14, 1067–1073. [Google Scholar] [CrossRef]
- Mendonça, N.; Jagger, C.; Granic, A.; Martin-Ruiz, C.; Mathers, J.C.; Seal, C.J.; Hill, T.R. Elevated Total Homocysteine in All Participants and Plasma Vitamin B12 Concentrations in Women Are Associated With All-Cause and Cardiovascular Mortality in the Very Old: The Newcastle 85+ Study. J. Gerontol. Ser. A 2018, 73, 1258–1264. [Google Scholar] [CrossRef]
- Sviri, S.; Khalaila, R.; Daher, S.; Bayya, A.; Linton, D.M.; Stav, I.; van Heerden, P.V. Increased Vitamin B12 Levels Are Associated with Mortality in Critically Ill Medical Patients. Clin. Nutr. 2012, 31, 53–59. [Google Scholar] [CrossRef]
- Ogrodnik, M. Cellular Aging beyond Cellular Senescence: Markers of Senescence Prior to Cell Cycle Arrest in Vitro and in Vivo. Aging Cell 2021, 20, e13338. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Wang, Z.; Liu, J.-P. Roles of Telomere Biology in Cell Senescence, Replicative and Chronological Ageing. Cells 2019, 8, 54. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- de Magalhães, J.P. Distinguishing between Driver and Passenger Mechanisms of Aging. Nat. Genet. 2024, 56, 204–211. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to Detect Senescence-Associated Beta-Galactosidase (SA-Βgal) Activity, a Biomarker of Senescent Cells in Culture and in Vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-Associated β-Galactosidase Is Lysosomal β-Galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Nobori, T.; Miura, K.; Wu, D.J.; Lois, A.; Takabayashi, K.; Carson, D.A. Deletions of the Cyclin-Dependent Kinase-4 Inhibitor Gene in Multiple Human Cancers. Nature 1994, 368, 753–756. [Google Scholar] [CrossRef]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of P16 INK4a in Peripheral Blood T-cells Is a Biomarker of Human Aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef]
- Sasaki, M.; Kajiya, H.; Ozeki, S.; Okabe, K.; Ikebe, T. Reactive Oxygen Species Promotes Cellular Senescence in Normal Human Epidermal Keratinocytes through Epigenetic Regulation of P16INK4a. Biochem. Biophys. Res. Commun. 2014, 452, 622–628. [Google Scholar] [CrossRef]
- Warfel, N.A.; El-Deiry, W.S. P21WAF1 and Tumourigenesis: 20 Years After. Curr. Opin. Oncol. 2013, 25, 52–58. [Google Scholar] [CrossRef]
- Kamal, M.; Joanisse, S.; Parise, G. Bleomycin-Treated Myoblasts Undergo P21-Associated Cellular Senescence and Have Severely Impaired Differentiation. GeroScience 2023, 46, 1843–1859. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors P21 and P16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef]
- Shaw, P.H. The Role of P53 in Cell Cycle Regulation. Pathol.-Res. Pract. 1996, 192, 669–675. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Sine, C.C.; Jeganathan, K.B.; Zhang, C.; Fierro Velasco, R.O.; Baker, D.J.; Li, H.; van Deursen, J.M. Senescent Cells Limit P53 Activity via Multiple Mechanisms to Remain Viable. Nat. Commun. 2022, 13, 3722. [Google Scholar] [CrossRef]
- Beausejour, C.M. Reversal of Human Cellular Senescence: Roles of the P53 and P16 Pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (SASP) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Polsky, L.R.; Rentscher, K.E.; Carroll, J.E. Stress-Induced Biological Aging: A Review and Guide for Research Priorities. Brain. Behav. Immun. 2022, 104, 97–109. [Google Scholar] [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence Surveillance of Pre-Malignant Hepatocytes Limits Liver Cancer Development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Pribluda, A.; Elyada, E.; Wiener, Z.; Hamza, H.; Goldstein, R.E.; Biton, M.; Burstain, I.; Morgenstern, Y.; Brachya, G.; Billauer, H.; et al. A Senescence-Inflammatory Switch from Cancer-Inhibitory to Cancer-Promoting Mechanism. Cancer Cell 2013, 24, 242–256. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP Mediates Chemoresistance and Tumor-Initiating-Activity of Mesothelioma Cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Awad, P.; Campisi, J.; Desprez, P.-Y. Epithelial-Mesenchymal Transition Induced by Senescent Fibroblasts. Cancer Microenviron. 2012, 5, 39–44. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-Associated Reprogramming Promotes Cancer Stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Nacarelli, T.; Fukumoto, T.; Zundell, J.A.; Fatkhutdinov, N.; Jean, S.; Cadungog, M.G.; Borowsky, M.E.; Zhang, R. NAMPT Inhibition Suppresses Cancer Stem-like Cells Associated with Therapy-Induced Senescence in Ovarian Cancer. Cancer Res. 2020, 80, 890–900. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.-H.; Park, T.J. Senescent Tumor Cells Lead the Collective Invasion in Thyroid Cancer. Nat. Commun. 2017, 8, 15208. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Kauser, K.; Campisi, J.; Beauséjour, C.M. Secretion of Vascular Endothelial Growth Factor by Primary Human Fibroblasts at Senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef]
- Toste, P.A.; Nguyen, A.H.; Kadera, B.E.; Duong, M.; Wu, N.; Gawlas, I.; Tran, L.M.; Bikhchandani, M.; Li, L.; Patel, S.G.; et al. Chemotherapy-Induced Inflammatory Gene Signature and Protumorigenic Phenotype in Pancreatic CAFs via Stress-Associated MAPK. Mol. Cancer Res. 2016, 14, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.-H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal Senescence Establishes an Immunosuppressive Microenvironment That Drives Tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef] [PubMed]
- Angelini, P.D.; Fluck, M.F.Z.; Pedersen, K.; Parra-Palau, J.L.; Guiu, M.; Bernadó Morales, C.; Vicario, R.; Luque-García, A.; Navalpotro, N.P.; Giralt, J.; et al. Constitutive HER2 Signaling Promotes Breast Cancer Metastasis through Cellular Senescence. Cancer Res. 2013, 73, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Georgilis, A.; Klotz, S.; Hanley, C.J.; Herranz, N.; Weirich, B.; Morancho, B.; Leote, A.C.; D’Artista, L.; Gallage, S.; Seehawer, M.; et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-Tumorigenic Effects of Senescent Cells. Cancer Cell 2018, 34, 85–102.e9. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintás-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. P53-Mediated Senescence Impairs the Apoptotic Response to Chemotherapy and Clinical Outcome in Breast Cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-Induced Damage to the Tumor Microenvironment Promotes Prostate Cancer Therapy Resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M.; Yoshida, Y.; Hara, E.; Ohtani, N. The Role of Cellular Senescence and SASP in Tumour Microenvironment. FEBS J. 2023, 290, 1348–1361. [Google Scholar] [CrossRef]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for Cancer Therapy: Is All That Glitters Really Gold? Cancers 2021, 13, 723. [Google Scholar] [CrossRef]
- Khosla, S. Senescent Cells, Senolytics and Tissue Repair: The Devil May Be in the Dosing. Nat. Aging 2023, 3, 139–141. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Aït-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.-M.; De Vos, J.; et al. Rejuvenating Senescent and Centenarian Human Cells by Reprogramming through the Pluripotent State. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Grigorash, B.B.; van Essen, D.; Liang, G.; Grosse, L.; Emelyanov, A.; Kang, Z.; Korablev, A.; Kanzler, B.; Molina, C.; Lopez, E.; et al. P16High Senescence Restricts Cellular Plasticity during Somatic Cell Reprogramming. Nat. Cell Biol. 2023, 25, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Kovatcheva, M.; Melendez, E.; Chondronasiou, D.; Pietrocola, F.; Bernad, R.; Caballe, A.; Junza, A.; Capellades, J.; Holguín-Horcajo, A.; Prats, N.; et al. Vitamin B12 Is a Limiting Factor for Induced Cellular Plasticity and Tissue Repair. Nat. Metab. 2023, 5, 1911–1930. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Kirschner, K.; Thuret, J.-Y.; Pope, B.D.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of Repressive Histone Marks and Chromatin Compaction during Senescent Heterochromatic Layer Formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Narita, M. High-Order Chromatin Structure and the Epigenome in SAHFs. Nucleus 2013, 4, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Park, L.K.; Friso, S.; Choi, S.-W. Nutritional Influences on Epigenetics and Age-Related Disease. Proc. Nutr. Soc. 2012, 71, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between Oncogene-Induced DNA Damage Response and Heterochromatin in Senescence and Cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 2017, 8, 138. [Google Scholar] [CrossRef]
- Sarg, B.; Koutzamani, E.; Helliger, W.; Rundquist, I.; Lindner, H.H. Postsynthetic Trimethylation of Histone H4 at Lysine 20 in Mammalian Tissues Is Associated with Aging. J. Biol. Chem. 2002, 277, 39195–39201. [Google Scholar] [CrossRef] [PubMed]
- Ramponi, V.; Richart, L.; Kovatcheva, M.; Stephan-Otto Attolini, C.; Capellades, J.; Lord, A.E.; Yanes, O.; Ficz, G.; Serrano, M. Persister Cancer Cells Are Characterized by H4K20me3 Heterochromatin That Defines a Low Inflammatory Profile. bioRxiv 2024. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Z.; Zhang, L.; Ghosh, S.; Zheng, H.; Zhou, Z. Depleting the Methyltransferase Suv39h1 Improves DNA Repair and Extends Lifespan in a Progeria Mouse Model. Nat. Commun. 2013, 4, 1868. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Reversal of the Cellular Phenotype in the Premature Aging Disease Hutchinson-Gilford Progeria Syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant Nuclear Lamin A Leads to Progressive Alterations of Epigenetic Control in Premature Aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef]
- Pusceddu, I.; Herrmann, W.; Kleber, M.E.; Scharnagl, H.; März, W.; Herrmann, M. Telomere Length, Vitamin B12 and Mortality in Persons Undergoing Coronary Angiography: The Ludwigshafen Risk and Cardiovascular Health Study. Aging 2019, 11, 7083–7097. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Liu, D.; Songyang, Z. The Telosome/Shelterin Complex and Its Functions. Genome Biol. 2008, 9, 232. [Google Scholar] [CrossRef]
- Xu, Q.; Parks, C.G.; DeRoo, L.A.; Cawthon, R.M.; Sandler, D.P.; Chen, H. Multivitamin Use and Telomere Length in Women. Am. J. Clin. Nutr. 2009, 89, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA Methylation: A Review of Molecular Mechanisms and the Evidence for Folate’s Role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef]
- Rzepka, Z.; Rok, J.; Kowalska, J.; Banach, K.; Wrześniok, D. Cobalamin Deficiency May Induce Astrosenescence—An In Vitro Study. Cells 2022, 11, 3408. [Google Scholar] [CrossRef]
- Kwan, D.N.; Rocha, J.T.Q.; Niero-Melo, L.; Custódio, M.A.; Oliveira, C.C. P53 and P21 Expression in Bone Marrow Clots of Megaloblastic Anemia Patients. Int. J. Clin. Exp. Pathol. 2020, 13, 1829–1833. [Google Scholar] [PubMed]
- Bhanothu, V.; Venkatesan, V.; Kondapi, A.K.; Ajumeera, R. Restrictions and Supplementations Effects of Vitamins B6, B9 and B12 on Growth, Vasculogenesis and Senescence of BG01V Human Embryonic Stem Cell Derived Embryoid Bodies. Nutr. Clin. Métab. 2021, 35, 297–316. [Google Scholar] [CrossRef]
- Praveen, G.; Sivaprasad, M.; Reddy, G.B. Chapter Eleven—Telomere Length and Vitamin B12. In Vitamins and Hormones; Litwack, G., Ed.; Vitamin B12; Academic Press: Cambridge, MA, USA, 2022; Volume 119, pp. 299–324. [Google Scholar]
- Zhang, R.; Bons, J.; Scheidemantle, G.; Liu, X.; Bielska, O.; Carrico, C.; Rose, J.; Heckenbach, I.; Scheibye-Knudsen, M.; Schilling, B.; et al. Histone Malonylation Is Regulated by SIRT5 and KAT2A. iScience 2023, 26, 106193. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Pooya, S.; Lorentz, S.; Gauchotte, G.; Arnold, C.; Gueant, J.-L.; Battaglia-Hsu, S.-F. Decreased Vitamin B12 Availability Induces ER Stress through Impaired SIRT1-Deacetylation of HSF1. Cell Death Dis. 2013, 4, e553. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Verdura, S.; Llorach-Parés, L.; Fernández-Arroyo, S.; Joven, J.; Martin-Castillo, B.; Bosch-Barrera, J.; Brunet, J.; Nonell-Canals, A.; Sanchez-Martinez, M.; et al. Metformin Is a Direct SIRT1-Activating Compound: Computational Modeling and Experimental Validation. Front. Endocrinol. 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-J.; Defossez, P.-A.; Guarente, L. Requirement of NAD and SIR2 for Life-Span Extension by Calorie Restriction in Saccharomyces cerevisiae. Science 2000, 289, 2126–2128. [Google Scholar] [CrossRef]
- Gomes, A.P.; Ilter, D.; Low, V.; Endress, J.E.; Fernández-García, J.; Rosenzweig, A.; Schild, T.; Broekaert, D.; Ahmed, A.; Planque, M.; et al. Age-Induced Accumulation of Methylmalonic Acid Promotes Tumour Progression. Nature 2020, 585, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Fu, Q.; Sun, Y.; Li, Q. Epigenetic Clock: A Promising Biomarker and Practical Tool in Aging. Ageing Res. Rev. 2022, 81, 101743. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M. Loss of Epigenetic Information as a Cause of Mammalian Aging. Cell 2023, 186, 305–326. [Google Scholar] [CrossRef]
- Sen, P.; Donahue, G.; Li, C.; Egervari, G.; Yang, N.; Lan, Y.; Robertson, N.; Shah, P.P.; Kerkhoven, E.; Schultz, D.C. Spurious Intragenic Transcription Is a Feature of Mammalian Cellular Senescence and Tissue Aging. Nat. Aging 2023, 3, 402–417. [Google Scholar] [CrossRef]
- Vanyushin, B.F.; Nemirovsky, L.E.; Klimenko, V.V.; Vasiliev, V.K.; Belozersky, A.N. The 5-Methylcytosine in DNA of Rats. Gerontology 1973, 19, 138–152. [Google Scholar] [CrossRef]
- Wilson, V.L.; Smith, R.A.; Ma, S.; Cutler, R.G. Genomic 5-Methyldeoxycytidine Decreases with Age. J. Biol. Chem. 1987, 262, 9948–9951. [Google Scholar] [CrossRef]
- Drinkwater, R.D.; Blake, T.J.; Morley, A.A.; Turner, D.R. Human Lymphocytes Aged in Vivo Have Reduced Levels of Methylation in Transcriptionally Active and Inactive DNA. Mutat. Res. 1989, 219, 29–37. [Google Scholar] [CrossRef]
- Fernàndez-Roig, S.; Lai, S.-C.; Murphy, M.M.; Fernandez-Ballart, J.; Quadros, E.V. Vitamin B12 Deficiency in the Brain Leads to DNA Hypomethylation in the TCblR/CD320 Knockout Mouse. Nutr. Metab. 2012, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Amenyah, S.D.; Hughes, C.F.; Ward, M.; Rosborough, S.; Deane, J.; Thursby, S.-J.; Walsh, C.P.; Kok, D.E.; Strain, J.J.; McNulty, H.; et al. Influence of Nutrients Involved in One-Carbon Metabolism on DNA Methylation in Adults—A Systematic Review and Meta-Analysis. Nutr. Rev. 2020, 78, 647–666. [Google Scholar] [CrossRef]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of Blood Can Be Tracked by DNA Methylation Changes at Just Three CpG Sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Hübner, U.; Bodis, M.; Graeber, S.; Geisel, J. Effect of Adding B-Vitamins to Vitamin D and Calcium Supplementation on CpG Methylation of Epigenetic Aging Markers. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 411–417. [Google Scholar] [CrossRef]
- Sae-Lee, C.; Corsi, S.; Barrow, T.M.; Kuhnle, G.G.C.; Bollati, V.; Mathers, J.C.; Byun, H. Dietary Intervention Modifies DNA Methylation Age Assessed by the Epigenetic Clock. Mol. Nutr. Food Res. 2018, 62, 1800092. [Google Scholar] [CrossRef]
- Lee, Y.-T.; Savini, M.; Chen, T.; Yang, J.; Zhao, Q.; Ding, L.; Gao, S.M.; Senturk, M.; Sowa, J.N.; Wang, J.D.; et al. Mitochondrial GTP Metabolism Controls Reproductive Aging in C. Elegans. Dev. Cell 2023, 58, 2718–2731.e7. [Google Scholar] [CrossRef] [PubMed]
- Monasso, G.S.; Küpers, L.K.; Jaddoe, V.W.V.; Heil, S.G.; Felix, J.F. Associations of Circulating Folate, Vitamin B12 and Homocysteine Concentrations in Early Pregnancy and Cord Blood with Epigenetic Gestational Age: The Generation R Study. Clin. Epigenet. 2021, 13, 95. [Google Scholar] [CrossRef]
- Hirata, T.; Arai, Y.; Yuasa, S.; Abe, Y.; Takayama, M.; Sasaki, T.; Kunitomi, A.; Inagaki, H.; Endo, M.; Morinaga, J.; et al. Associations of Cardiovascular Biomarkers and Plasma Albumin with Exceptional Survival to the Highest Ages. Nat. Commun. 2020, 11, 3820. [Google Scholar] [CrossRef]
- Bogdanova, D.A.; Kolosova, E.D.; Pukhalskaia, T.V.; Levchuk, K.A.; Demidov, O.N.; Belotserkovskaya, E.V. The Differential Effect of Senolytics on SASP Cytokine Secretion and Regulation of EMT by CAFs. Int. J. Mol. Sci. 2024, 25, 4031. [Google Scholar] [CrossRef]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.-P. Senescence-Associated IL-6 and IL-8 Cytokines Induce a Self- and Cross-Reinforced Senescence/Inflammatory Milieu Strengthening Tumorigenic Capabilities in the MCF-7 Breast Cancer Cell Line. Cell Commun. Signal. 2017, 15, 17. [Google Scholar] [CrossRef]
- Christen, W.G.; Cook, N.R.; Van Denburgh, M.; Zaharris, E.; Albert, C.M.; Manson, J.E. Effect of Combined Treatment With Folic Acid, Vitamin B 6, and Vitamin B12 on Plasma Biomarkers of Inflammation and Endothelial Dysfunction in Women. J. Am. Heart Assoc. 2018, 7, e008517. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, C. The Return of the Scarlet Pimpernel: Cobalamin in Inflammation II—Cobalamins Can Both Selectively Promote All Three Nitric Oxide Synthases (NOS), Particularly INOS and ENOS, and, as Needed, Selectively Inhibit INOS and NNOS. J. Nutr. Environ. Med. 2007, 16, 181–211. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Wang, M.-Y.; Lin, M.-C.; Lin, P.-T. Associations between Vitamin B-12 Status and Oxidative Stress and Inflammation in Diabetic Vegetarians and Omnivores. Nutrients 2016, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Gubernatorova, E.O.; Polinova, A.I.; Petropavlovskiy, M.M.; Namakanova, O.A.; Medvedovskaya, A.D.; Zvartsev, R.V.; Telegin, G.B.; Drutskaya, M.S.; Nedospasov, S.A. Dual Role of TNF and LTα in Carcinogenesis as Implicated by Studies in Mice. Cancers 2021, 13, 1775. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, S.P.; Quadros, E.V.; Regec, A. Transcobalamin II. Chem. Biochem. Vitam. B 1999, 12, 441–473. [Google Scholar]
- Scalabrino, G.; Carpo, M.; Bamonti, F.; Pizzinelli, S.; D’Avino, C.; Bresolin, N.; Meucci, G.; Martinelli, V.; Comi, G.C.; Peracchi, M. High Tumor Necrosis Factor-α in Levels in Cerebrospinal Fluid of Cobalamin-deficient Patients. Ann. Neurol. 2004, 56, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Ahuja, R.; Mutti, E.; Veber, D.; Seetharam, S.; Scalabrino, G.; Seetharam, B. Cobalamin-Mediated Regulation of Transcobalamin Receptor Levels in Rat Organs. Arch. Biochem. Biophys. 2007, 463, 128–132. [Google Scholar] [CrossRef]
- Veber, D.; Mutti, E.; Tacchini, L.; Gammella, E.; Tredici, G.; Scalabrino, G. Indirect Down-regulation of Nuclear NF-κB Levels by Cobalamin in the Spinal Cord and Liver of the Rat. J. Neurosci. Res. 2008, 86, 1380–1387. [Google Scholar] [CrossRef]
- Homann, L.; Rentschler, M.; Brenner, E.; Böhm, K.; Röcken, M.; Wieder, T. IFN-γ and TNF Induce Senescence and a Distinct Senescence-Associated Secretory Phenotype in Melanoma. Cells 2022, 11, 1514. [Google Scholar] [CrossRef] [PubMed]
- Kandhaya-Pillai, R.; Yang, X.; Tchkonia, T.; Martin, G.M.; Kirkland, J.L.; Oshima, J. TNF -α/ IFN -γ Synergy Amplifies Senescence-associated Inflammation and SARS-CoV -2 Receptor Expression via Hyper-activated JAK / STAT1. Aging Cell 2022, 21, e13646. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-Responsive Gene 1 Protein Links Metabolism to Immunity by Catalyzing Itaconic Acid Production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Campanello, G.C.; Flicker, D.; Grabarek, Z.; Hu, J.; Luo, C.; Banerjee, R.; Mootha, V.K. The Human Knockout Gene CLYBL Connects Itaconate to Vitamin B12. Cell 2017, 171, 771–782.e11. [Google Scholar] [CrossRef] [PubMed]
- Ruetz, M.; Campanello, G.C.; Purchal, M.; Shen, H.; McDevitt, L.; Gouda, H.; Wakabayashi, S.; Zhu, J.; Rubin, E.J.; Warncke, K.; et al. Itaconyl-CoA Forms a Stable Biradical in Methylmalonyl-CoA Mutase and Derails Its Activity and Repair. Science 2019, 366, 589–593. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, S.; Zhao, L.; Cheng, P.; Liu, J.; Guo, F.; Xiao, J.; Zhu, W.; Chen, A. Aging Relevant Metabolite Itaconate Inhibits Inflammatory Bone Loss. Front. Endocrinol. 2022, 13, 885879. [Google Scholar] [CrossRef] [PubMed]
- Martucci, M.; Conte, M.; Bucci, L.; Giampieri, E.; Fabbri, C.; Palmas, M.; Izzi, M.; Salvioli, S.; Zambrini, A.; Orsi, C.; et al. Twelve-Week Daily Consumption of Ad Hoc Fortified Milk with ω-3, D, and Group B Vitamins Has a Positive Impact on Inflammaging Parameters: A Randomized Cross-Over Trial. Nutrients 2020, 12, 3580. [Google Scholar] [CrossRef]
- Ghosh, T.S. Toward an Improved Definition of a Healthy Microbiome for Healthy Aging. Nat. Aging 2022, 2, 1054–1069. [Google Scholar] [CrossRef]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut Microbiota Composition Correlates with Diet and Health in the Elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Lu, W.; Wu, T.; Yuan, W.; Zhu, J.; Lee, Y.K.; Zhao, J.; Zhang, H.; Chen, W. Human Gut Microbiome Aging Clocks Based on Taxonomic and Functional Signatures through Multi-View Learning. Gut Microbes 2022, 14, 2025016. [Google Scholar] [CrossRef]
- Berlin, H.; Berlin, R.; Brante, G. Oral treatment of pernicious anemia with high doses of vitamin b12 without intrinsic factor. Acta Med. Scand. 1968, 184, 247–258. [Google Scholar] [CrossRef]
- Kuzminski, A.M.; Del Giacco, E.J.; Allen, R.H.; Stabler, S.P.; Lindenbaum, J. Effective Treatment of Cobalamin Deficiency With Oral Cobalamin. Blood 1998, 92, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Seetharam, B.; Alpers, D.H. Absorption and Transport of Cobalamin (Vitamin B12). Annu. Rev. Nutr. 1982, 2, 343–369. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.H.; Stabler, S.P. Identification and Quantitation of Cobalamin and Cobalamin Analogues in Human Feces. Am. J. Clin. Nutr. 2008, 87, 1324–1335. [Google Scholar] [CrossRef]
- Bárcena, C.; Valdés-Mas, R.; Mayoral, P.; Garabaya, C.; Durand, S.; Rodríguez, F.; Fernández-García, M.T.; Salazar, N.; Nogacka, A.M.; Garatachea, N.; et al. Healthspan and Lifespan Extension by Fecal Microbiota Transplantation into Progeroid Mice. Nat. Med. 2019, 25, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Romano, S.; Ansorge, R.; Aboelnour, A.; Le Gall, G.; Savva, G.M.; Pontifex, M.G.; Telatin, A.; Baker, D.; Jones, E.; et al. Fecal Microbiota Transfer between Young and Aged Mice Reverses Hallmarks of the Aging Gut, Eye, and Brain. Microbiome 2022, 10, 68. [Google Scholar] [CrossRef]
- Biagi, E.; Franceschi, C.; Rampelli, S.; Severgnini, M.; Ostan, R.; Turroni, S.; Consolandi, C.; Quercia, S.; Scurti, M.; Monti, D.; et al. Gut Microbiota and Extreme Longevity. Curr. Biol. 2016, 26, 1480–1485. [Google Scholar] [CrossRef]
- Sato, Y.; Atarashi, K.; Plichta, D.R.; Arai, Y.; Sasajima, S.; Kearney, S.M.; Suda, W.; Takeshita, K.; Sasaki, T.; Okamoto, S.; et al. Novel Bile Acid Biosynthetic Pathways Are Enriched in the Microbiome of Centenarians. Nature 2021, 599, 458–464. [Google Scholar] [CrossRef]
- Kawamoto, S.; Uemura, K.; Hori, N.; Takayasu, L.; Konishi, Y.; Katoh, K.; Matsumoto, T.; Suzuki, M.; Sakai, Y.; Matsudaira, T.; et al. Bacterial Induction of B Cell Senescence Promotes Age-Related Changes in the Gut Microbiota. Nat. Cell Biol. 2023, 25, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.-F.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin Regulates Global DNA Methylation via Mitochondrial One-Carbon Metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Aroda, V.R.; Edelstein, S.L.; Goldberg, R.B.; Knowler, W.C.; Marcovina, S.M.; Orchard, T.J.; Bray, G.A.; Schade, D.S.; Temprosa, M.G.; White, N.H.; et al. Long-Term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J. Clin. Endocrinol. Metab. 2016, 101, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Verheijen, B.M.; Navapanich, Z.; McGann, E.G.; Shemtov, S.; Lai, G.-J.; Arora, P.; Towheed, A.; Haroon, S.; Holczbauer, A.; et al. Evolutionary Conservation of the Fidelity of Transcription. Nat. Commun. 2023, 14, 1547. [Google Scholar] [CrossRef] [PubMed]
- Debès, C.; Papadakis, A.; Grönke, S.; Karalay, Ö.; Tain, L.S.; Mizi, A.; Nakamura, S.; Hahn, O.; Weigelt, C.; Josipovic, N.; et al. Ageing-Associated Changes in Transcriptional Elongation Influence Longevity. Nature 2023, 616, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Mohsin, M.; Iqrar, S.; Manzoor, O.; Siddiqi, T.O.; Ahmad, A. Live Cell Imaging of Vitamin B12 Dynamics by Genetically Encoded Fluorescent Nanosensor. Sens. Actuators B Chem. 2018, 257, 866–874. [Google Scholar] [CrossRef]
- Soleja, N.; Agrawal, N.; Nazir, R.; Ahmad, M.; Mohsin, M. Enhanced Sensitivity and Detection Range of FRET-Based Vitamin B12 Nanosensor. 3 Biotech 2020, 10, 87. [Google Scholar] [CrossRef]
Figure 1.
Vitamin B12 transport and cobalamin structural forms.

Figure 2.
MMUT function in mitochondria. Propionate formed from the catabolism of branched amino acids and odd-chain fatty acids is converted to D-methylmalonyl-CoA. Methylmanolyl-CoA epimerase isomerizes D-methylmalonyl-CoA to the L-form. L-methylmalonyl-CoA is the substrate for MMUT, converting it to succinyl-CoA, which enters the TCA cycle. Abbreviations: MMA—methylmalonic acid, TCA cycle—the tricarboxylic acid cycle.
Figure 2.
MMUT function in mitochondria. Propionate formed from the catabolism of branched amino acids and odd-chain fatty acids is converted to D-methylmalonyl-CoA. Methylmanolyl-CoA epimerase isomerizes D-methylmalonyl-CoA to the L-form. L-methylmalonyl-CoA is the substrate for MMUT, converting it to succinyl-CoA, which enters the TCA cycle. Abbreviations: MMA—methylmalonic acid, TCA cycle—the tricarboxylic acid cycle.

Figure 3.
The MS catalytic cycle and its connection to cellular metabolism. Red arrows indicate the pathway of a methyl radical. Abbreviations: MS—methionine synthase, THF—[5,6,7,8]-tetrahydrofolate, DHF—[7,8]-dihydrofolate, Hcy—L-homocysteine, SAH—S-adenosyl-L-homocysteine, SAM—S-adenosyl-L-methionine, GSH—reduced glutathione, MetCbl—MeCbl, Cbl—cobalamin, CH3˙—methyl radical.
Figure 3.
The MS catalytic cycle and its connection to cellular metabolism. Red arrows indicate the pathway of a methyl radical. Abbreviations: MS—methionine synthase, THF—[5,6,7,8]-tetrahydrofolate, DHF—[7,8]-dihydrofolate, Hcy—L-homocysteine, SAH—S-adenosyl-L-homocysteine, SAM—S-adenosyl-L-methionine, GSH—reduced glutathione, MetCbl—MeCbl, Cbl—cobalamin, CH3˙—methyl radical.

Figure 4.
Consequences of vitamin B12 deficiency at cellular, tissue, and organismal levels. Arrows indicate direction of change: ↑—increase, ↓—decrease.
Figure 4.
Consequences of vitamin B12 deficiency at cellular, tissue, and organismal levels. Arrows indicate direction of change: ↑—increase, ↓—decrease.
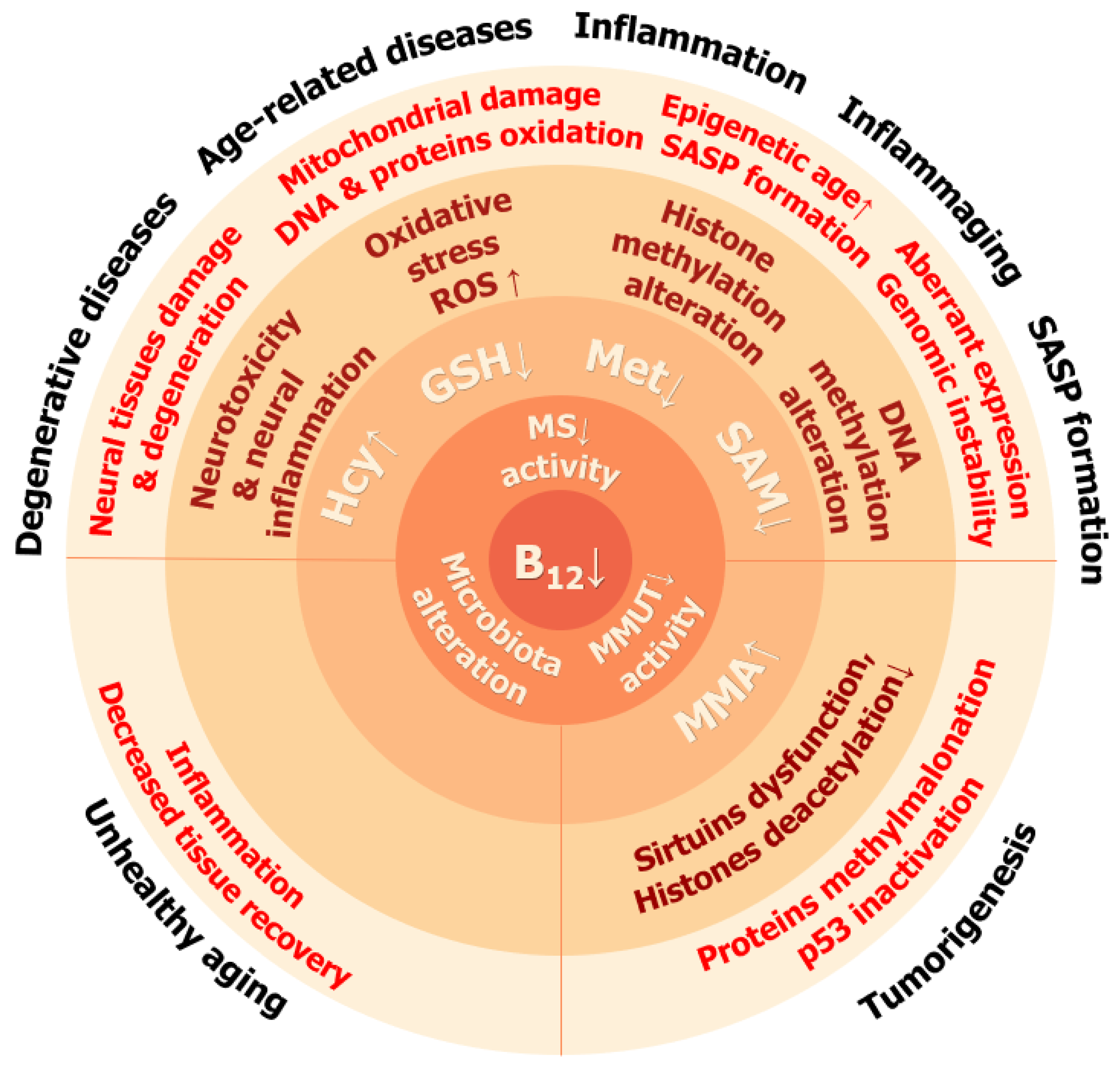

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Functions of histone post-translational modifications.
| Modification | Role | Reference |
|---|---|---|
| H3K9me2 H3K27me2 H3K9me3 H3K27me3 H3K79me3 H4K20me3 H2A/H4R3me2 | Repression of gene expression | [83,84,90] |
| H3K4me1 H3K4me3 H2BK5me1 H3K9me1 H3K27me1 H4K20me1 H3K79me1 H3K79me2 H3K9ac H3K14ac H3K27ac H3K63ac H3K122ac | Increase in gene expression | [83,84,91,92,93,94] |
| H3K36me3 | Prevention of transcription initiation outside promoters | [88] |
| H3K4 methylation | Prevention of DNA methylation | [89] |
Table 2.
Assessment of vitamin B12 in human blood plasma.
| Analyte | Reference Interval | B12 Deficiency | Advantages and Disadvantages |
|---|---|---|---|
| Total B12, pM | 200–600 | <148 | Requires digestion of proteins |
| Holotranscobalamin, pM | 40–100 | <35 | Highly automated |
| Homocysteine, μM | 8–15 | >15 | Several confounders: B9 and B6 levels, thyroid function, glomerular filtration rate, sex, and age |
| 2-methylmalonic acid, nM | 0.04–0.37 | >0.37 | Results may be unreliable in cases of kidney deficiency or infection |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Simonenko, S.Y.; Bogdanova, D.A.; Kuldyushev, N.A. Emerging Roles of Vitamin B12 in Aging and Inflammation. Int. J. Mol. Sci. 2024, 25, 5044. https://doi.org/10.3390/ijms25095044
AMA Style
Simonenko SY, Bogdanova DA, Kuldyushev NA. Emerging Roles of Vitamin B12 in Aging and Inflammation. International Journal of Molecular Sciences. 2024; 25(9):5044. https://doi.org/10.3390/ijms25095044
Chicago/Turabian StyleSimonenko, Sergey Yu., Daria A. Bogdanova, and Nikita A. Kuldyushev. 2024. "Emerging Roles of Vitamin B12 in Aging and Inflammation" International Journal of Molecular Sciences 25, no. 9: 5044. https://doi.org/10.3390/ijms25095044
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.