
The Blood–Brain Barrier Is Unaffected in the Ndufs4−/− Mouse Model of Leigh Syndrome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Biochemical and Anatomical Analysis of the BBB in Ndufs4−/− and Control Mice
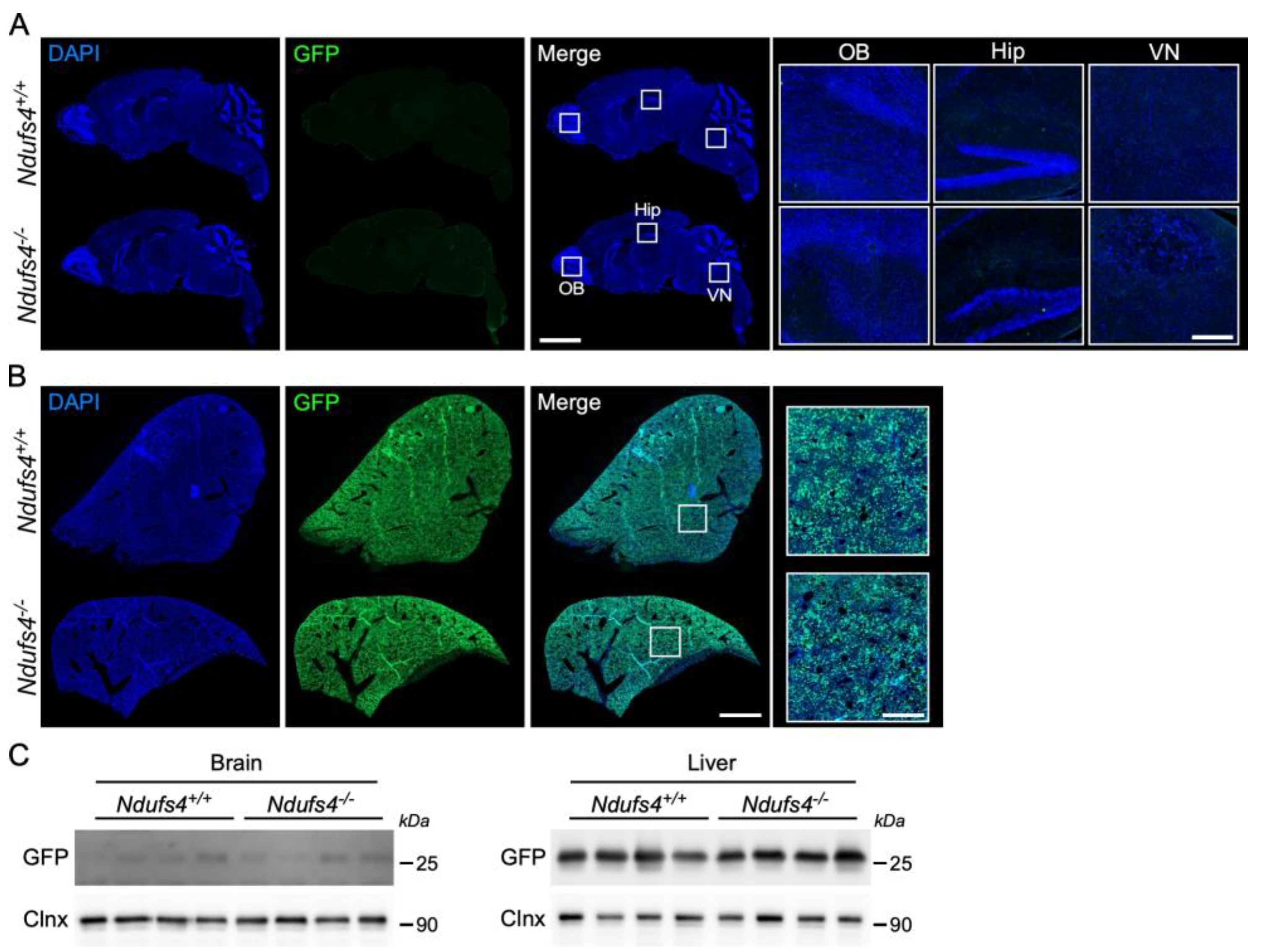
2.2. Analysis of AAV9-GFP Vector Distribution in Ndufs4−/− and Control Mice
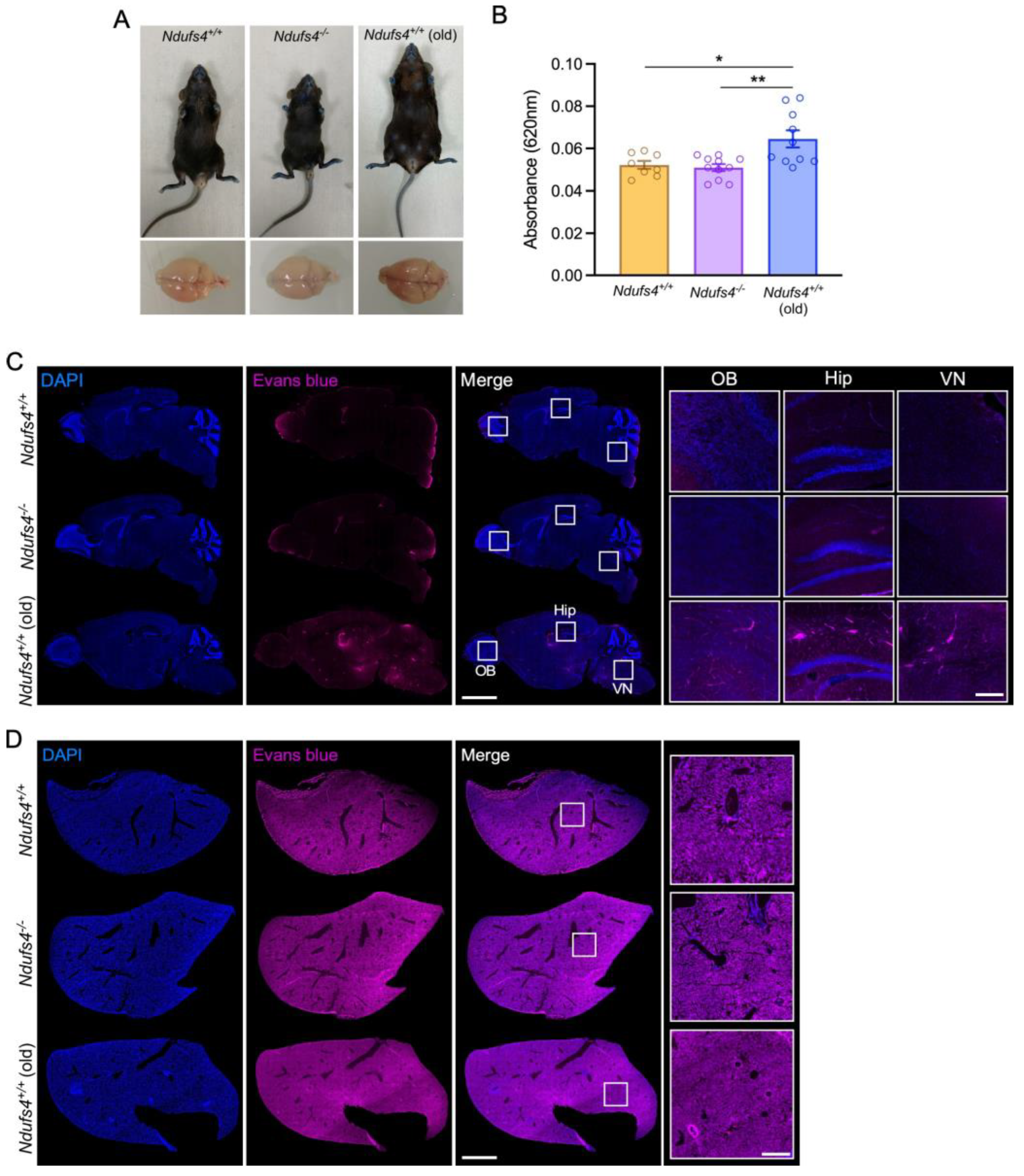
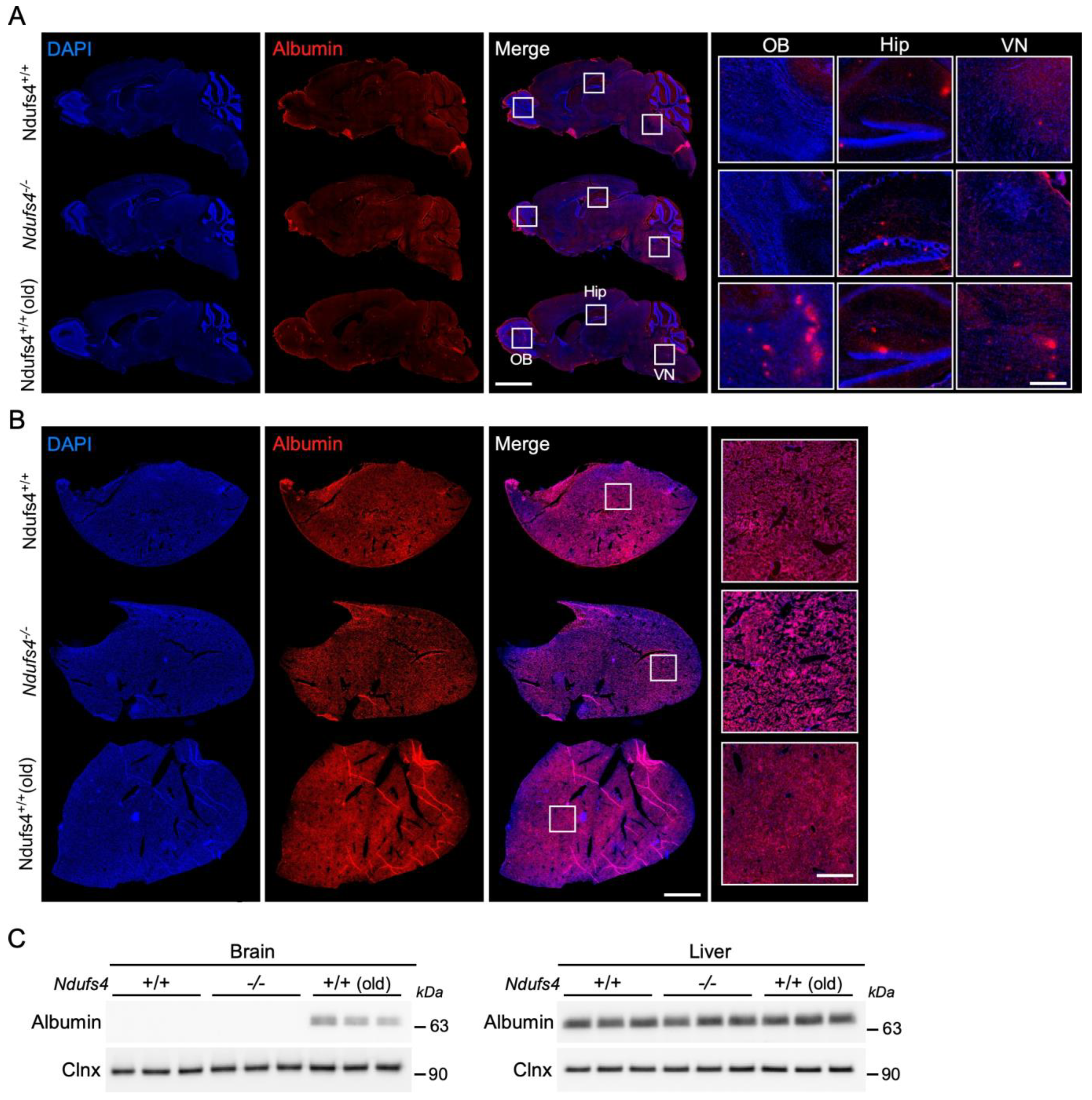
2.3. Evaluation of the Ndufs4−/− Mice BBB Permeability for Evans Blue and Albumin
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Three-Dimensional Mapping of the Brain Vasculature
4.3. Purification of Brain Vessels
4.4. Mitochondrial Complex I Activity Assay
4.5. Evans Blue Assay
4.6. Analysis of Evans Blue Content by Spectroscopy
4.7. Adeno-Associated Viral Vector Production, Titration and Injection
4.8. AAV Vector Injection
4.9. Tissue Staining and Microscopy
4.10. Western Blotting
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Bogush, M.; Heldt, N.A.; Persidsky, Y. Blood Brain Barrier Injury in Diabetes: Unrecognized Effects on Brain and Cognition. J. Neuroimmune Pharmacol. 2017, 12, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stahli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature 2020, 583, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Crockett, A.M.; Ryan, S.K.; Vasquez, A.H.; Canning, C.; Kanyuch, N.; Kebir, H.; Ceja, G.; Gesualdi, J.; Zackai, E.; McDonald-McGinn, D.; et al. Disruption of the blood-brain barrier in 22q11.2 deletion syndrome. Brain 2021, 144, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Di Pardo, A.; Amico, E.; Scalabri, F.; Pepe, G.; Castaldo, S.; Elifani, F.; Capocci, L.; De Sanctis, C.; Comerci, L.; Pompeo, F.; et al. Impairment of blood-brain barrier is an early event in R6/2 mouse model of Huntington Disease. Sci. Rep. 2017, 7, 41316. [Google Scholar] [CrossRef] [PubMed]
- Duarte Lobo, D.; Nobre, R.J.; Oliveira Miranda, C.; Pereira, D.; Castelhano, J.; Sereno, J.; Koeppen, A.; Castelo-Branco, M.; Pereira de Almeida, L. The blood-brain barrier is disrupted in Machado-Joseph disease/spinocerebellar ataxia type 3: Evidence from transgenic mice and human post-mortem samples. Acta Neuropathol. Commun. 2020, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.A.A.; Seo, H.S.; Armien, A.G.; Bates, F.S.; Tolar, J.; Azarin, S.M. Modeling and rescue of defective blood-brain barrier function of induced brain microvascular endothelial cells from childhood cerebral adrenoleukodystrophy patients. Fluids Barriers CNS 2018, 15, 9. [Google Scholar] [CrossRef]
- Alelwani, W.; Elmorsy, E.; Kattan, S.W.; Babteen, N.A.; Alnajeebi, A.M.; Al-Ghafari, A.; Carter, W.G. Carbamazepine induces a bioenergetics disruption to microvascular endothelial cells from the blood-brain barrier. Toxicol. Lett. 2020, 333, 184–191. [Google Scholar] [CrossRef]
- Doll, D.N.; Hu, H.; Sun, J.; Lewis, S.E.; Simpkins, J.W.; Ren, X. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke 2015, 46, 1681–1689. [Google Scholar] [CrossRef]
- Haileselassie, B.; Joshi, A.U.; Minhas, P.S.; Mukherjee, R.; Andreasson, K.I.; Mochly-Rosen, D. Mitochondrial dysfunction mediated through dynamin-related protein 1 (Drp1) propagates impairment in blood brain barrier in septic encephalopathy. J. Neuroinflamm. 2020, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, L.; Hargreaves, I.P.; Georgian, A.R.; Turner, C.; Dalton, R.N.; Abbott, N.J.; Heales, S.J.R.; Preston, J.E. CoQ(10) Deficient Endothelial Cell Culture Model for the Investigation of CoQ(10) Blood-Brain Barrier Transport. J. Clin. Med. 2020, 9, 3236. [Google Scholar] [CrossRef] [PubMed]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhu, M.; Zhang, Y.; Diao, Y. Crossing the blood-brain barrier with AAV vectors. Metab. Brain Dis. 2021, 36, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Grange, R.M.H.; Sharma, R.; Shah, H.; Reinstadler, B.; Goldberger, O.; Cooper, M.K.; Nakagawa, A.; Miyazaki, Y.; Hindle, A.G.; Batten, A.J.; et al. Hypoxia ameliorates brain hyperoxia and NAD(+) deficiency in a murine model of Leigh syndrome. Mol. Genet. Metab. 2021, 133, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef]
- Perry, E.A.; Bennett, C.F.; Luo, C.; Balsa, E.; Jedrychowski, M.; O’Malley, K.E.; Latorre-Muro, P.; Ladley, R.P.; Reda, K.; Wright, P.M.; et al. Tetracyclines promote survival and fitness in mitochondrial disease models. Nat. Metab. 2021, 3, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; Grillo, A.S.; Ito, T.K.; Stanaway, I.B.; Nguyen, B.M.G.; Ying, K.; Tung, H.; Smith, K.; Tran, N.; Velikanje, G.; et al. Acarbose suppresses symptoms of mitochondrial disease in a mouse model of Leigh syndrome. Nat. Metab. 2023, 5, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Corra, S.; Cerutti, R.; Balmaceda, V.; Viscomi, C.; Zeviani, M. Double administration of self-complementary AAV9NDUFS4 prevents Leigh disease in Ndufs4−/− mice. Brain 2022, 145, 3405–3414. [Google Scholar] [CrossRef]
- Di Meo, I.; Marchet, S.; Lamperti, C.; Zeviani, M.; Viscomi, C. AAV9-based gene therapy partially ameliorates the clinical phenotype of a mouse model of Leigh syndrome. Gene Ther. 2017, 24, 661–667. [Google Scholar] [CrossRef]
- Quintana, A.; Zanella, S.; Koch, H.; Kruse, S.E.; Lee, D.; Ramirez, J.M.; Palmiter, R.D. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J. Clin. Investig. 2012, 122, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Reynaud-Dulaurier, R.; Benegiamo, G.; Marrocco, E.; Al-Tannir, R.; Surace, E.M.; Auwerx, J.; Decressac, M. Gene replacement therapy provides benefit in an adult mouse model of Leigh syndrome. Brain 2020, 143, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pinheiro, P.; Cerutti, R.; Luna-Sanchez, M.; Zeviani, M.; Viscomi, C. A Single Intravenous Injection of AAV-PHP.B-hNDUFS4 Ameliorates the Phenotype of Ndufs4−/− Mice. Mol. Ther. Methods Clin. Dev. 2020, 17, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Kruse, S.E.; Watt, W.C.; Marcinek, D.J.; Kapur, R.P.; Schenkman, K.A.; Palmiter, R.D. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008, 7, 312–320. [Google Scholar] [CrossRef] [PubMed]
- van de Wal, M.A.E.; Adjobo-Hermans, M.J.W.; Keijer, J.; Schirris, T.J.J.; Homberg, J.R.; Wieckowski, M.R.; Grefte, S.; van Schothorst, E.M.; van Karnebeek, C.; Quintana, A.; et al. Ndufs4 knockout mouse models of Leigh syndrome: Pathophysiology and intervention. Brain 2022, 145, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Farmer, B.C.; Walsh, A.E.; Kluemper, J.C.; Johnson, L.A. Lipid Droplets in Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 742. [Google Scholar] [CrossRef]
- Quintana, A.; Kruse, S.E.; Kapur, R.P.; Sanz, E.; Palmiter, R.D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 10996–11001. [Google Scholar] [CrossRef]
- Wick, M.J.; Harral, J.W.; Loomis, Z.L.; Dempsey, E.C. An Optimized Evans Blue Protocol to Assess Vascular Leak in the Mouse. J. Vis. Exp. 2018, 139, e57037. [Google Scholar]
- Senatorov, V.V., Jr.; Friedman, A.R.; Milikovsky, D.Z.; Ofer, J.; Saar-Ashkenazy, R.; Charbash, A.; Jahan, N.; Chin, G.; Mihaly, E.; Lin, J.M.; et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFbeta signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 2019, 11, eaaw8283. [Google Scholar] [CrossRef]
- Rahman, S. Leigh syndrome. Handb. Clin. Neurol. 2023, 194, 43–63. [Google Scholar]
- Chen, L.; Cui, Y.; Jiang, D.; Ma, C.Y.; Tse, H.F.; Hwu, W.L.; Lian, Q. Management of Leigh syndrome: Current status and new insights. Clin. Genet. 2018, 93, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Ahishali, B.; Kaya, M. Evaluation of Blood-Brain Barrier Integrity Using Vascular Permeability Markers: Evans Blue, Sodium Fluorescein, Albumin-Alexa Fluor Conjugates, and Horseradish Peroxidase. Methods Mol. Biol. 2021, 2367, 87–103. [Google Scholar] [PubMed]
- Saunders, N.R.; Dziegielewska, K.M.; Mollgard, K.; Habgood, M.D. Markers for blood-brain barrier integrity: How appropriate is Evans blue in the twenty-first century and what are the alternatives? Front. Neurosci. 2015, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Salmina, A.B.; Kuvacheva, N.V.; Morgun, A.V.; Komleva, Y.K.; Pozhilenkova, E.A.; Lopatina, O.L.; Gorina, Y.V.; Taranushenko, T.E.; Petrova, L.L. Glycolysis-mediated control of blood-brain barrier development and function. Int. J. Biochem. Cell Biol. 2015, 64, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Dell’agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Torraco, A.; Peralta, S.; Iommarini, L.; Diaz, F. Mitochondrial Diseases Part I: Mouse models of OXPHOS deficiencies caused by defects in respiratory complex subunits or assembly factors. Mitochondrion 2015, 21, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Hanaford, A.R.; Khanna, A.; Truong, V.; James, K.; Chen, Y.; Mulholland, M.; Kayser, B.; Liao, R.W.; Sedensky, M.; Morgan, P.; et al. Peripheral macrophages drive CNS disease in the Ndufs4−/− model of Leigh syndrome. Brain Pathol. 2023, 33, e13192. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Cerda, J., 3rd; Kirk, J.M.; Turner, W.D.; Rasmussen, T.L.; Flores Suarez, C.P.; Dickinson, M.E.; Wythe, J.D. EZ Clear for simple, rapid, and robust mouse whole organ clearing. Elife 2022, 11, e77419. [Google Scholar] [CrossRef]
- Kosmidis, S.; Negrean, A.; Dranovsky, A.; Losonczy, A.; Kandel, E.R. A fast, aqueous, reversible three-day tissue clearing method for adult and embryonic mouse brain and whole body. Cell Rep. Methods 2021, 1, 100090. [Google Scholar] [CrossRef]
- Boulay, A.C.; Saubamea, B.; Decleves, X.; Cohen-Salmon, M. Purification of Mouse Brain Vessels. J. Vis. Exp. 2015, 105, e53208. [Google Scholar]
- Quintana, A.; Morgan, P.G.; Kruse, S.E.; Palmiter, R.D.; Sedensky, M.M. Altered anesthetic sensitivity of mice lacking Ndufs4, a subunit of mitochondrial complex I. PLoS ONE 2012, 7, e42904. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.A.; Mulholland, M.; Snell, J.; Howe, M.; James, K.; Hanaford, A.R.; Morgan, P.G.; Sedensky, M.; Johnson, S.C. Volatile anaesthetic toxicity in the genetic mitochondrial disease Leigh syndrome. Br. J. Anaesth. 2023, 131, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Lai, T.W. Optimization of Evans blue quantitation in limited rat tissue samples. Sci. Rep. 2014, 4, 6588. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reynaud-Dulaurier, R.; Clément, R.; Yjjou, S.; Cresson, C.; Saoudi, Y.; Faideau, M.; Decressac, M. The Blood–Brain Barrier Is Unaffected in the Ndufs4−/− Mouse Model of Leigh Syndrome. Int. J. Mol. Sci. 2024, 25, 4828. https://doi.org/10.3390/ijms25094828
Reynaud-Dulaurier R, Clément R, Yjjou S, Cresson C, Saoudi Y, Faideau M, Decressac M. The Blood–Brain Barrier Is Unaffected in the Ndufs4−/− Mouse Model of Leigh Syndrome. International Journal of Molecular Sciences. 2024; 25(9):4828. https://doi.org/10.3390/ijms25094828
Chicago/Turabian StyleReynaud-Dulaurier, Robin, Romain Clément, Sara Yjjou, Cassandra Cresson, Yasmina Saoudi, Mathilde Faideau, and Michael Decressac. 2024. "The Blood–Brain Barrier Is Unaffected in the Ndufs4−/− Mouse Model of Leigh Syndrome" International Journal of Molecular Sciences 25, no. 9: 4828. https://doi.org/10.3390/ijms25094828