
Obtaining Polyvinylpyrrolidone Fibers Using the Electroforming Method with the Inclusion of Microcrystalline High-Temperature Phosphates
 ,
,
Abstract
:1. Introduction
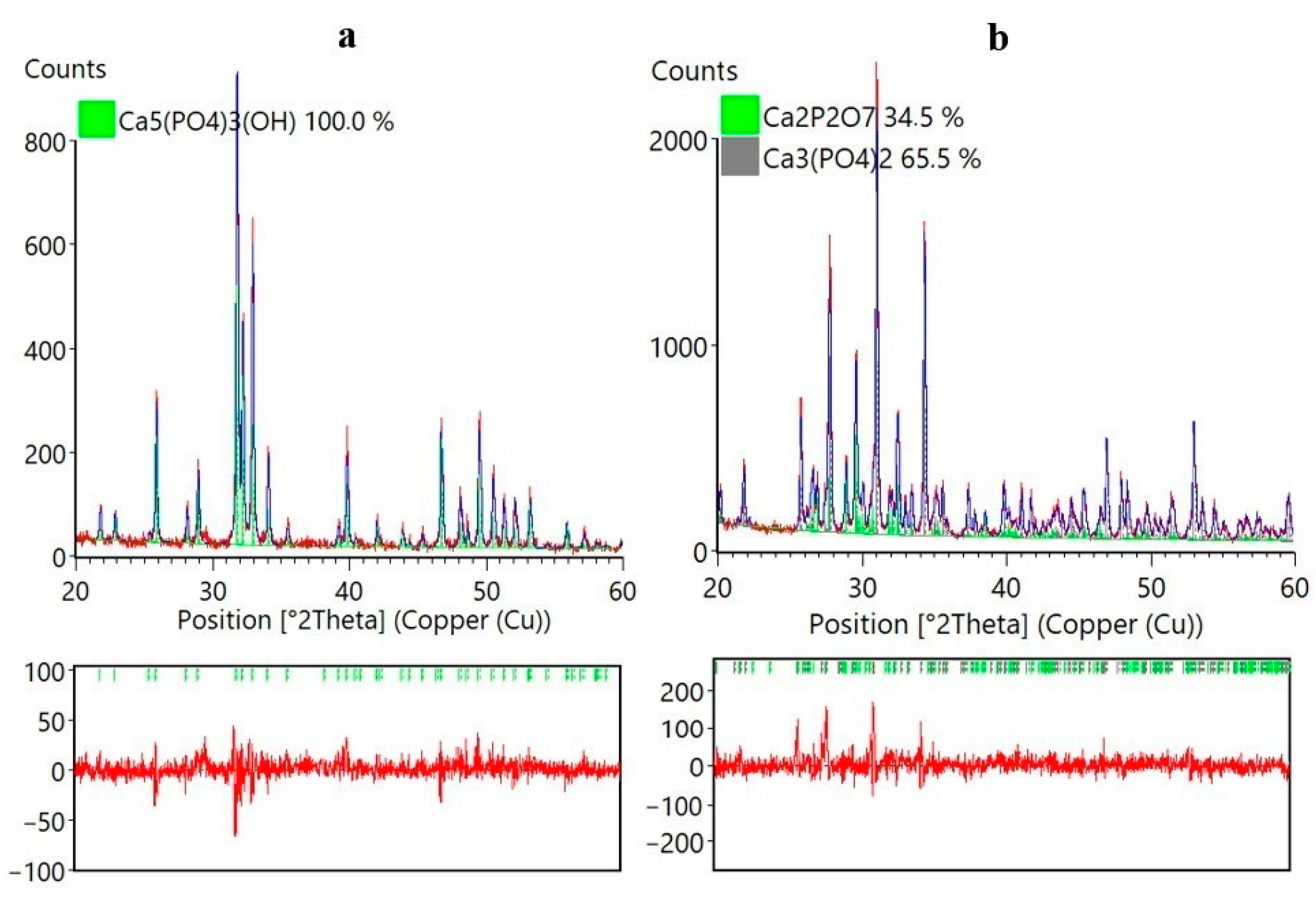
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Calcium Phosphate Synthesis
4.3. Electrospinning of Fibers
4.4. Experimental Method
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pańtak, P.; Czechowska, J.P.; Cichoń, E.; Zima, A. Novel double hybrid-type bone cements based on calcium phosphates, chitosan and citrus pectin. Int. J. Mol. Sci. 2023, 24, 13455. [Google Scholar] [CrossRef]
- Bakan, F.; Laçin, O.; Sarac, H. A novel low temperature sol–gel synthesis process for thermally stable nano crystalline hydroxyapatite. Powder Technol. 2013, 233, 295–302. [Google Scholar] [CrossRef]
- Chatakun, P.; Núñez-Toldrà, R.; López, E.J.D.; Gil-Recio, C.; Martínez-Sarrà, E.; Hernández-Alfaro, F.; Ferrés-Padró, E.; Giner-Tarrida, L.; Atari, M. The effect of five proteins on stem cells used for osteoblast differentiation and proliferation: A current review of the literature. Cell Mol. Life Sci. 2014, 71, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Leontiev, V.K. Biologically active synthetic calcium-phosphate-containing materials for dentistry. Stomatology 1996, 5, 4–6. [Google Scholar]
- Kovylin, R.S.; Aleinik, D.A.; Fedyushkin, I.L. Modern porous polymeric implants: Preparation, properties, and application. High-molecular compounds. Polym. Sci. Ser. C 2021, 63, 33–53. [Google Scholar] [CrossRef]
- Kołodziejska, B.; Figat, R.; Kolmas, J. Biomimetic apatite/natural polymer composite granules as multifunctional dental tissue regenerative material. Int. J. Mol. Sci. 2023, 24, 16751. [Google Scholar] [CrossRef] [PubMed]
- Zalewska, J.; Vivcharenko, V.; Belcarz, A. Gypsum-related Iipact on antibiotic-laded composite based on highly porous hydroxyapatite—Advantages and disadvantages. Int. J. Mol. Sci. 2023, 24, 17178. [Google Scholar] [CrossRef]
- Pisareva, E.V.; Vlasov, M.Y.; Volova, L.T. Indicators of bone tissue turnover in rabbits with the introduction of «allogeneic hydroxyapatite». News Samara Sci. Cent. Russ. Acad. Sci. 2015, 17, 908–912. [Google Scholar]
- Pisareva, E.V.; Volova, L.T.; Tikhonova, T.V.; Vlasov, M.Y.; Sokolovskaya, A.B.; Golub, Y.V. Application of nanostructured allogeneic hydroxyapatite for the correction of osteoresorption. In High Technologies, Fundamental and Applied Research in Physiology and Medicine; Polytechnic University: St. Petersburg, Russia, 2012; pp. 20–22. [Google Scholar]
- Sevastyanov, V.I.; Kirpichnikov, M.P. Biocompatible Materials; MIA: Moscow, Russia, 2011. [Google Scholar]
- Papezhuk, M.V.; Pilunova, E.; Ivanin, S.N.; Yakupov, R.P. Synthesis of microcrystalline hydroxyapatite and production of fibers using the electrospinning method based on it. Proc. Kola Sci. Cent. Russ. Acad. Sci. Ser. Tech. Sci. 2023, 14, 192–196. [Google Scholar]
- Radulescu, D.-E.; Vasile, O.R.; Andronescu, E.; Ficai, A. Latest research of doped hydroxyapatite for bone tissue engineering. Int. J. Mol. Sci. 2023, 24, 13157. [Google Scholar] [CrossRef]
- Ducheyne, P.; Radin, S.; King, L. The effect of calcium phosphate ceramic composition and structure on in vitro behavior. I. Dissolution. J. Biomed. Mater. Res. 1993, 27, 25–34. [Google Scholar] [CrossRef]
- Moreno, E.; Gregory, T.; Brown, W. Preparation and Solubility of Hydroxyapatite. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1968, 72, 773–782. [Google Scholar] [CrossRef]
- Putlyaev, V.I.; Safronova, T.V. New generation of calcium phosphate materials: The role of phase and chemical composition. Glass Ceram. 2005, 3, 30–33. [Google Scholar]
- Gregory, T.M.; Moreno, E.C.; Patel, J.M.; Brown, W.E. Solubility of β-Ca3(PO4)2 in the system Ca(OH)2-H3PO4-H2O at 5, 15, 25, and 37 °C. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1974, 78, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, G.; Lemaitre, S. Calculation of the solubility diagrams in the system Ca(OH)2- H3PO4-KOH-HNO3-CO2-H2O. J. Cryst. Growth 1990, 104, 820–832. [Google Scholar] [CrossRef]
- Fulmer, M.T.; Ison, I.C.; Hankermayer, C.R.; Constantz, B.R.; Ross, J. Measurements of the solubilities and dissolution rates of several hydroxyapatites. Biomaterials 2002, 23, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Vamze, J.; Pilmane, M.; Skagers, A. Biocompatibility of pure and mixed hydroxyapatite and α-tricalcium phosphate implanted in rabbit bone. J. Mater. Sci. Mater. Med. 2015, 26, 73. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhao, G.; Liu, H. Histological evaluation of collagen-hydroxyapatite composite as osseous implants in the repair of mandibular defect. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 1998, 12, 74–76. [Google Scholar] [PubMed]
- Dhand, V.; Rhee, K.Y.; Park, S.J. The facile and low temperature synthesis of nanophase hydroxyapatite crystals using wet chemistry. Mater. Sci. Eng. C 2014, 36, 152–159. [Google Scholar] [CrossRef]
- McKay, W.F. Ceramic Fusion Implants and Compositions. U.S. Patent US6037519A, 14 March 2000. [Google Scholar]
- Kumar, S.S.D.; Abrahamse, H. Advancement of nanobiomaterials to deliver natural compounds for tissue engineering applications. Int. J. Mol. Sci. 2020, 21, 6752. [Google Scholar] [CrossRef]
- Torres-Martínez, E.J.; Bravo, J.M.C.; Medina, A.S.; González, G.L.P.; Gómez, L.J.V.A. Summary of electrospun nanofibers as drug delivery system: Drugs loaded and biopolymers used as matrices. Curr. Drug Deliv. 2018, 15, 1360–1374. [Google Scholar] [CrossRef]
- Xue, J.; Wu, T.; Dai, Y.; Xia, Y. Electrospinning and electrospun nanofibers: Methods, materials, and applications. Chem. Rev. 2019, 119, 5298–5415. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Polymers as biomaterials for tissue engineering and controlled drug delivery. In Tissue Engineering I; Springer: Berlin/Heidelberg, Germany, 2006; Volume 102, pp. 47–90. [Google Scholar] [CrossRef]
- Maitz, M.F. Applications of synthetic polymers in clinical medicine. Biosurf. Biotribol. 2015, 1, 161–176. [Google Scholar] [CrossRef]
- Buzko, V.; Ivanin, S.; Goryachko, A.; Shutkin, I.; Pushankina, P.; Petriev, I. Magnesium spinel ferrites development for FDM 3D-printing material for microwave absorption. Processes 2023, 11, 60. [Google Scholar] [CrossRef]
- Velasco Barraza, R.; Alvarez Suarez, A.S.; Villarreal Gomez, L.J.; Paz González, J.A.; Iglesias, A.L.; Vera Graziano, R. Designing a low-cost electrospinning device for practical learning in a bioengineering biomaterials course. Rev. Mex. Ing. Bioméd. 2016, 37, 7–16. [Google Scholar] [CrossRef]
- Stafin, K.; Śliwa, P.; Piątkowski, M. Towards polycaprolactone-based scaffolds for alveolar bone tissue engineering: A biomimetic approach in a 3D printing technique. Int. J. Mol. Sci. 2023, 24, 16180. [Google Scholar] [CrossRef]
- Prokopchuk, N.R.; Shashok, J.S.; Prishchepenko, D.V.; Melamed, V.D. (Review). Electrospinning of nanofibers from chitosan solution. Polym. Mater. Technol. 2015, 1, 36–56. [Google Scholar]
- Shin, Y.M.; Hohman, M.M.; Brenner, M.P.; Rutledge, G.C. Electrospinning: A whipping fluid jet generates submicron polymer fibers. Appl. Phys. Lett. 2001, 78, 1149–1151. [Google Scholar] [CrossRef]
- Prahasti, G.; Zulfiand, A.; Munir, M.M. Needleless electrospinning system with wire spinneret: Analternative way to control morphology, size, and productivity ofnanofibers. Nano Express 2020, 1, 010046. [Google Scholar] [CrossRef]
- Brydson, J.A. Plastics Materials; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Anunziata, O.A.; Maria, L.; Beltramone, M.R. Hydroxyapatite/MCM-41 and SBA-15 nano-composites: Preparation, characterization and applications. J. Mater. 2009, 2, 1508–1519. [Google Scholar] [CrossRef]
- Kaimonov, M.R.; Safronova, T.V.; Filippov, Y.Y.; Shatalova, T.B.; Preobrazhenskii, I.I. Calcium phosphate powder for obtaining of composite bioceramics. Inorg. Mater. Appl. Res. 2021, 12, 34–39. [Google Scholar] [CrossRef]
- Ivanin, S.N.; Buz’ko, V.Y.; Sokolov, M.E.; Magomadova, M.A.; Panyushkin, V.T. Structure and properties of the mixed-ligand complex compound of gadolinium stearate with benzoyltrifluoroacetone. J. Struct. Chem. 2021, 62, 563–570. [Google Scholar] [CrossRef]
- Ivanin, S.N.; Panyushkin, V.T.; Buzko, V.Y.; Selivantev, Y.M.; Kostyrina, T.V. Synthesis, investigation, and molecular simulation of possible structures of a heteroligand complex of gadolinium stearate with acetylacetone. J. Struct. Chem. 2021, 62, 19–28. [Google Scholar] [CrossRef]
- Berzina-Cimdina, L.; Borodajenko, N. Research of calcium phosphates using fourier transform infrared spectroscopy. In Infrared Spectroscopy—Materials Science, Engineering and Technology; InTech: Rijeka, Croatia, 2012; Volume 6, pp. 124–148. [Google Scholar]
- Binitha, M.P.; Pradyumnan, P.P. Dielectric property studies of biologically compatible brushite single crystals used as bone graft substitute. J. Biomater. Nanobiotechnol. 2013, 4, 119–122. [Google Scholar] [CrossRef]
- Boanini, E.; Gazzano, M.; Bigi, A. Ionic substitutions in calcium phosphates synthesized at low temperature. Acta Biomater. 2010, 6, 1882–1894. [Google Scholar] [CrossRef]
- Buzko, V.; Babushkin, M.; Ivanin, S.; Goryachko, A.; Petriev, I. Study of electromagnetic shielding properties of composites based on glass fiber metallized with metal films. Coatings 2022, 12, 1173. [Google Scholar] [CrossRef]
- Petriev, I.; Baryshev, M.G.; Voronin, K.A.; Lutsenko, I.S.; Pushankina, P.D.; Kopytov, G.F. Gas Transmission properties of Pd–Ag membranes coated with modifying layer. Russ. Phys. J. 2020, 63, 457–461. [Google Scholar] [CrossRef]
- Buz’ko, V.Y.; Ivanin, S.N.; Shutkin, I.Y.; Goryachko, A.I.; Udodov, S.A.; Ozolin, A.V. Atomic composition, microstructure, and electromagnetic properties of schungite micropowder. Condens. Matter Interphases 2023, 25, 3–13. [Google Scholar] [CrossRef]
- Petriev, I.; Pushankina, P.; Andreev, G.; Ivanin, S.; Dzhimak, S. High-performance hydrogen-selective Pd-Ag membranes modified with Pd-Pt nanoparticles for use in steam reforming membrane reactors. Int. J. Mol. Sci. 2023, 24, 17403. [Google Scholar] [CrossRef]
- Schwarzenbach, G.; Flashka, G. Complexometric Titration; M.: Chemistry; U.S. Department of Energy: London, UK, 1970; p. 360. [Google Scholar]
- Carrodeguas, R.G.; Alonso, L.M.; García-Menocal, J.A.D.; Alonso, L.M.; Molins, M.P.G.; Manent, S.M.; Gil Mur, J.; Pérez, J.T.; Estany, J.A.P. Hydrothermal method for preparing calcium phosphate monoliths. Mater. Res. 2003, 6, 395–401. [Google Scholar] [CrossRef]
- Kannan, S.; Goetz-Neunhoeffer, F.; Neubauer, J.; Ferreira, J.M.F. Ionic substitutions in biphasic hydroxyapatite and b-tricalcium phosphate mixtures: Structural analysis by rietveld refinement. J. Am. Ceram. Soc. 2008, 91, 1–12. [Google Scholar] [CrossRef]
- Hughes, J.M.; Cameron, M.; Crowley, K.D. Structural variations in natural F, OH, and Cl apatites. Am. Mineral. 1989, 74, 870–876. [Google Scholar]
- Kamiyama, T.; Hoshikawa, A.; Yashima, M.; Sakai, A. Crystal structure analysis of beta-tricalcium phosphate Ca3(PO4)2 by neutron powder diffraction. J. Solid State Chem. 2003, 175, 272–277. [Google Scholar] [CrossRef]
- Boudin, S.; Grandin, A.; Borel, M.M.; Leclaire, A.; Raveau, B. Redetermination of the β-Ca2P2O7 structure. Acta Crystallogr. Sect. C 1993, 49, 2062–2064. [Google Scholar] [CrossRef]
- Bian, J.; Kim, D.W.; Hong, K.S. Microwave dielectric properties of Ca2P2O7. J. Eur. Ceram. Soc. 2003, 23, 2589–2592. [Google Scholar] [CrossRef]
- Parodi, J.A.; Hickok, R.L.; Segelken, W.G.; Cooper, J.R. Electronic paramagnetic resonance study of the thermal decomposition of dibasic calcium orthophosphate. J. Electrochem. Soc. 1965, 112, 688–692. [Google Scholar] [CrossRef]
- Griesiute, D.; Garskaite, E.; Antuzevics, A.; Klimavicius, V.; Balevicius, V.; Zarkov, A.; Katelnikovas, A.; Sandberg, D.; Kareiva, A. Synthesis, structural and luminescent properties of Mn doped calcium pyrophosphate (Ca2P2O7) polymorphs. Sci. Rep. 2022, 12, 7116. [Google Scholar] [CrossRef]
- Tazibt, N.; Kaci, M.; Dehouche, N.; Ragoubi, M.; Atanase, L.I. Effect of filler content on the morphology and physical properties of poly(lactic acid)-hydroxyapatite. Compos. Mater. 2023, 16, 809. [Google Scholar] [CrossRef]
- Klee, W.E.; Engel, G. Infrared spectra of the phosphate ions in various apatite. J. Inorg. Nucl. Chem. 1970, 32, 1837–1843. [Google Scholar] [CrossRef]
- Ivanin, S.N.; Buzko, V.Y.; Goryachko, A.I.; Panyushkin, V.T. Optical and magnetic properties of a heteroligand complexof gadolinium stearate with acetylacetone. Russ. J. Phys. Chem. A 2021, 95, 326–331. [Google Scholar] [CrossRef]
- Papezhuk, M.V.; Volynkin, V.A.; Stroganova, T.A.; Usacheva, T.R.; Thi, L.P. Theoretical and experimental study of inclusion complex formation of β-cyclodextrin with some 1,4-diazepine derivatives. Macroheterocycles 2020, 13, 64–73. [Google Scholar] [CrossRef]
- Pavlova, T.V.; Bavykina, T.Y. Comparative evaluation of the mineral composition and ultramicrostructure of dental tissues in norm and with caries. Mod. Sci.-Intensive Technol. 2009, 12, 15–18. [Google Scholar]
- Teotia, A.K.; Raina, D.B.; Singh, C.; Sinha, N.; Isaksson, H.; Tägil, M.; Lidgren, L.; Kumar, A. Nano-hydroxyapatite bone substitute functionalized with bone active molecules for enhanced cranial bone regeneration. ACS Appl. Mater. Interfaces 2017, 9, 6816–6828. [Google Scholar] [CrossRef]
- Safronova, T.V.; Putlyaev, V.I.; Shekhirev, M.A.; Kuztsov, A.V. Composite ceramics containing a bioresorbable phase. Glass Ceram. 2007, 3, 31–35. [Google Scholar] [CrossRef]
- Kuczumow, A.; Chałas, R.; Nowak, J.; Smułek, W.; Jarzębski, M. Novel approach to tooth chemistry: Quantification of human enamel apatite in context for new biomaterials and nanomaterials development. Int. J. Mol. Sci. 2021, 22, 279. [Google Scholar] [CrossRef]
- Veresov, A.G.; Putlyaev, V.I.; Tretyakov, Y.D. Chemistry of inorganic biomaterials based on calcium phosphates. Russian Chem. J. 2004, 48, 52–64. [Google Scholar]
- Kokubo, T.; Kim, H.M.; Kawashita, M. Novel bioactive materials with different mechanical properties. Biomaterials 2003, 24, 2161–2175. [Google Scholar] [CrossRef]
- Puleo, D.A.; Nanci, A. Understanding and controlling the bone–implant interface. Biomaterials 1999, 20, 2311–2321. [Google Scholar] [CrossRef]
- Collins, A.M. Physical Techniques. In Nanotechnology Cookbook; Elsevier: Oxford, UK, 2012; pp. 205–253. [Google Scholar] [CrossRef]
- Leonova, L.A.; Guzeeva, T.I.; Guzeev, V.V. Study of the synthesis process of hydroxyapatite. Chem. Sustain. Dev. 2010, 18, 107–110. [Google Scholar]
- Liu, D.M.; Troczynski, T.; Tseng, W.J. Water-based sol-gel synthesis of hydroxyapatite: Process development. Biomaterials 2001, 13, 1721–1730. [Google Scholar] [CrossRef]
- Goloshchapov, D.L.; Kashkarov, V.M.; Rumyantseva, N.A.; Seredin, P.V.; Len’shin, A.S.; Agapov, B.L.; Domashevskaya, E.P. Obtaining nanocrystalline hydroxyapatite by chemical precipitation using a biogenic source of calcium. Condens. Media Interphase 2011, 13, 427–441. [Google Scholar]
- ISO 10993-5-2023; Medical Devices. Assessment of the Biological Effects of Medical Devices. Part 5. Cytotoxicity Studies Using In Vitro Methods. ISO: Geneva, Switzerland, 2023.
- Safronova, T.V.; Putlyaev, V.I. Medical inorganic materials science in Russia: Calcium phosphate materials. Nanosyst. Phys. Chem. Math. 2013, 4, 24–47. [Google Scholar]
- Gurin, A.N.; Komlev, V.S.; Fadeeva, I.V.; Barinov, S.M. Octacalcium phosphate—A precursor of biological mineralization, a promising osteoplastic material. Dentistry. 2010, 4, 65–72. [Google Scholar]
- Tiwari, G.; Rai, A.K.; Tiwari, R. Cyclodextrins in delivery systems: Applications. J. Pharm. Bioallied Sci. 2010, 2, 72–79. [Google Scholar] [CrossRef]
- Hrib, J.; Sirc, J.; Hobzova, R.; Hampejsova, Z.; Bosakova, Z.; Munzarova, M.; Michalek, J. Nanofibers for drug delivery—Incorporation and release of model molecules, influence of molecular weight and polymer structure. Beilstein J. Nanotechnol. 2015, 6, 1939–1945. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Formula | Crystal System | Space Group | a, Å | b, Å | c, Å | Alpha, ° | Beta, ° | Gamma, ° |
|---|---|---|---|---|---|---|---|---|
| Ca3(PO4)2 | Rhombohedral | R-3c | 10.4290 | 10.4290 | 37.3800 | 90 | 90 | 120 |
| Ca2P2O7 | Tetragonal | P41 | 6.6840 | 6.6840 | 24.1450 | 90 | 90 | 90 |
| Ca5(PO4)3(OH) | Hexagonal | P63/m | 9.4320 | 9.4320 | 6.8810 | 90 | 90 | 120 |
| Samples | ||
|---|---|---|
| CPs | HAp | |
| Ca | 19.22 | 23.04 |
| P | 15.17 | 16.45 |
| O | 65.62 | 60.51 |
| Ca/P | 1.27 | 1.4 |
| Samples | Concentration of Ca2+, mol/L | |
|---|---|---|
| 20 °C | 37 °C | |
| HAp | 1·10−3 | 1.5·10−3 |
| CPs | 2.5·10−4 | 3.1·10−4 |
| Fibers (CPs) | 1·10−4 | 1.2·10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papezhuk, M.V.; Ivanin, S.N.; Yakupov, R.P.; Buz’ko, V.Y.; Sukhno, I.V.; Gneush, A.N.; Petriev, I.S. Obtaining Polyvinylpyrrolidone Fibers Using the Electroforming Method with the Inclusion of Microcrystalline High-Temperature Phosphates. Int. J. Mol. Sci. 2024, 25, 2298. https://doi.org/10.3390/ijms25042298
Papezhuk MV, Ivanin SN, Yakupov RP, Buz’ko VY, Sukhno IV, Gneush AN, Petriev IS. Obtaining Polyvinylpyrrolidone Fibers Using the Electroforming Method with the Inclusion of Microcrystalline High-Temperature Phosphates. International Journal of Molecular Sciences. 2024; 25(4):2298. https://doi.org/10.3390/ijms25042298
Chicago/Turabian StylePapezhuk, Marina Vladimirovna, Sergei Nikolaevich Ivanin, Roman Pavlovich Yakupov, Vladimir Yurievich Buz’ko, Igor Vladimirovich Sukhno, Anna Nikolaevna Gneush, and Iliya Sergeevich Petriev. 2024. "Obtaining Polyvinylpyrrolidone Fibers Using the Electroforming Method with the Inclusion of Microcrystalline High-Temperature Phosphates" International Journal of Molecular Sciences 25, no. 4: 2298. https://doi.org/10.3390/ijms25042298