
Molecular Mechanisms of the Cytotoxic Effect of Recombinant Selenoprotein SELENOM on Human Glioblastoma Cells

Abstract
:1. Introduction
2. Results
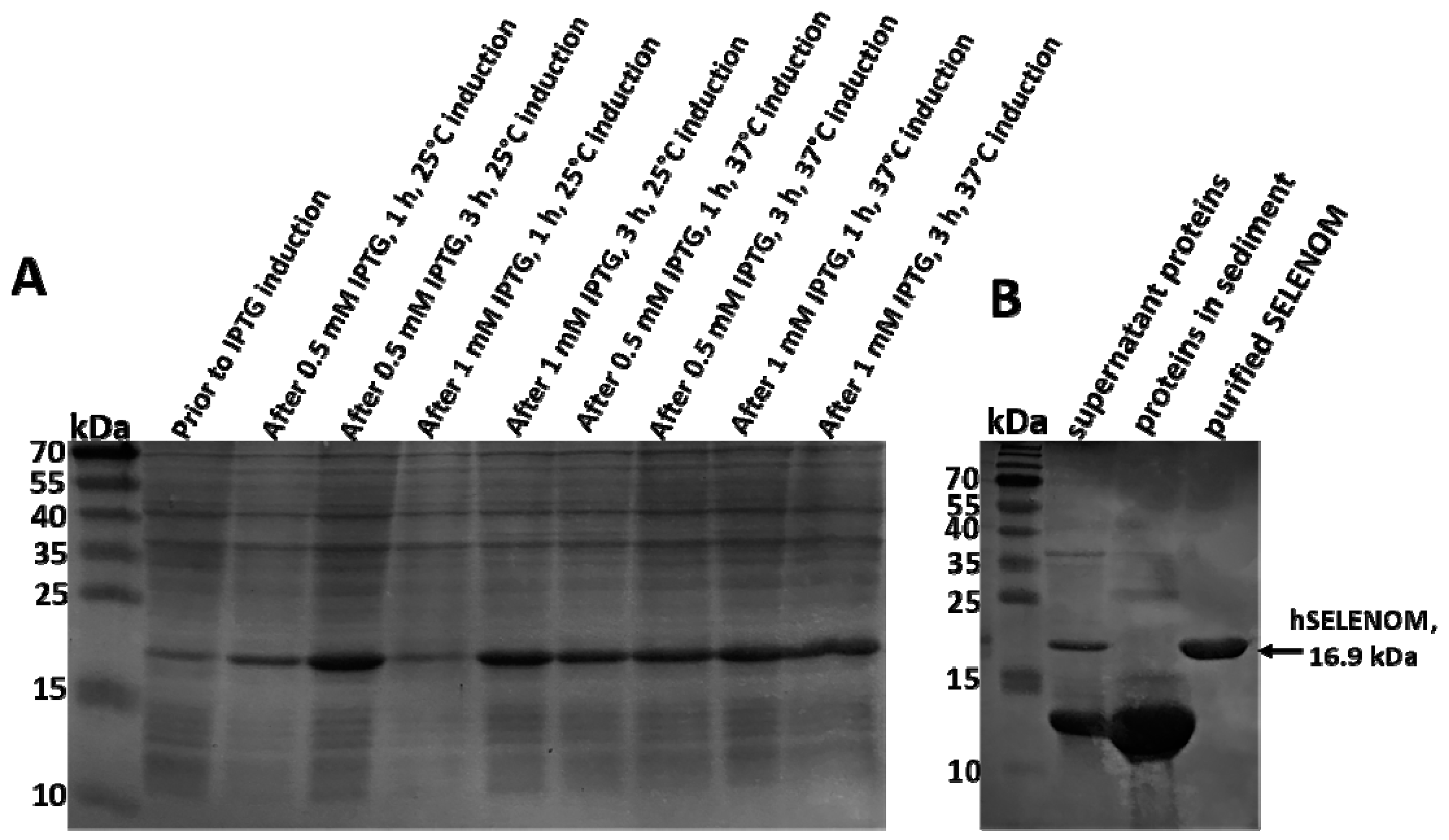
2.1. Optimal Conditions for the Synthesis of Human SELENOM in a Bacterial System Were Observed When Bacterial Cells Were Incubated with 0.5 MM IPTG for 3 h at 25 °C
2.2. Recombinant hSELENOM Reduced Proliferative Activity and Induced Apoptosis in Human Glioblastoma Cells
2.3. Recombinant hSELENOM Induced Increased mRNA Expression of Pro-Apoptotic Genes and the IRE1α Signaling Pathway UPR
2.4. Recombinant hSELENOM Depleted Endoplasmic Reticulum Ca2+ Pools
2.5. Activation of the Mechanism of Actin-Dependent Endocytosis under the Action of Recombinant hSELENOM on Human Glioblastoma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Bacterial Strains
4.2. Isolation of RNA and Reverse Transcription
4.3. Cloning and Site-Directed Mutagenesis
4.4. Isolation and Purification of Recombinant hSELENOM
4.5. Real-Time PCR
4.6. Western Blotting
4.7. Registration of Changes in Cytosolic Ca2+
4.8. MTT Analysis and Apoptosis Detection Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hatfield, D.L.; Gladyshev, V.N. How Selenium Has Altered Our Understanding of the Genetic Code. Mol. Cell. Biol. 2002, 22, 3565–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.-A.; Wu, Y.; Zappacosta, F.; Jeang, K.-T.; Lee, B.J.; Hatfield, D.L.; Gladyshev, V.N. Redox Regulation of Cell Signaling by Selenocysteine in Mammalian Thioredoxin Reductases. J. Biol. Chem. 1999, 274, 24522–24530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fomenko, D.E.; Marino, S.M.; Gladyshev, V.N. Functional diversity of cysteine residues in proteins and unique features of catalytic redox-active cysteines in thiol oxidoreductases. Mol. Cells. 2008, 26, 228–235. [Google Scholar] [PubMed]
- Zhou, J.-C.; Zhao, H.; Tang, J.-Y.; Li, J.-G.; Liu, X.-L.; Zhu, Y.-M. Molecular Cloning, Chromosomal Localization and Expres-sion Profiling of Porcine Selenoprotein M Gene. Genes Genom. 2011, 33, 529–534. [Google Scholar] [CrossRef]
- Dikiy, A.; Novoselov, S.V.; Fomenko, D.E.; Sengupta, A.; Carlson, B.A.; Cerny, R.L.; Ginalski, K.; Grishin, N.V.; Hatfield, D.L.; Gladyshev, V.N. SelT, SelW, SelH, and Rdx12: Genomics and Molecular Insights into the Functions of Selenoproteins of a Novel Thioredoxin-like Family. Biochemistry 2007, 46, 6871–6882. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-Q.; Ren, F.-Z.; Jiang, Y.-Y.; Lei, X. Characterization of Selenoprotein M and Its Response to Selenium Deficiency in Chicken Brain. Biol. Trace Element Res. 2015, 170, 449–458. [Google Scholar] [CrossRef]
- Reeves, M.A.; Bellinger, F.P.; Berry, M.J. The Neuroprotective Functions of Selenoprotein M and its Role in Cytosolic Calcium Regulation. Antioxid. Redox Signal. 2010, 12, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.Y.; Sin, J.S.; Kim, M.S.; Yim, S.Y.; Kim, Y.K.; Kim, C.K.; Kim, B.G.; Shim, S.B.; Jee, S.W.; Lee, S.H.; et al. Overexpression of human selenoprotein M differentially regulates the concentrations of antioxidants and H2O2, the activity of antioxidant enzymes, and the composition of white blood cells in a transgenic rat. Int. J. Mol. Med. 2008, 21, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Guerriero, E.; Accardo, M.; Capone, F.; Colonna, G.; Castello, G.; Costantini, S. Assessment of the Selenoprotein M (SELM) over-expression on human hepatocellular carcinoma tissues by immunohistochemistry. Eur. J. Histochem. 2014, 58, 2433. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Goltyaev, M.V.; Fesenko, E.E. Protein Partners of Selenoprotein SELM and the Role of Selenium Compounds in Regulation of Its Expression in Human Cancer Cells. Dokl. Biochem. Biophys. 2019, 488, 300–303. [Google Scholar] [CrossRef]
- Ferguson, A.D.; Labunskyy, V.; Fomenko, D.E.; Araç, D.; Chelliah, Y.; Amezcua, C.A.; Rizo, J.; Gladyshev, V.N.; Deisenhofer, J. NMR Structures of the Selenoproteins Sep15 and SelM Reveal Redox Activity of a New Thioredoxin-like Family. J. Biol. Chem. 2006, 281, 3536–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, J.; Jang, M.; Huh, J.H.; Na, J.-W.; Shim, M.; Carlson, B.A.; Tobe, R.; Tsuji, P.A.; Gladyshev, V.N.; Hatfield, D.L.; et al. Deficiency of the 15-kDa selenoprotein led to cytoskeleton remodeling and non-apoptotic membrane blebbing through a RhoA/ROCK pathway. Biochem. Biophys. Res. Commun. 2015, 456, 884–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varlamova, E.G.; Goltyaev, M.V.; Turovsky, E.A. The Role of Selenoproteins SELENOM and SELENOT in the Regulation of Apoptosis, ER Stress, and Calcium Homeostasis in the A-172 Human Glioblastoma Cell Line. Biology 2022, 11, 811. [Google Scholar] [CrossRef] [PubMed]
- Tujebajeva, R.M.; Copeland, P.; Xu, X.; Carlson, B.A.; Harney, J.W.; Driscoll, D.M.; Hatfield, D.L.; Berry, M.J. Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 2000, 1, 158–163. [Google Scholar] [CrossRef] [Green Version]
- Fagegaltier, D.; Hubert, N.; Yamada, K.; Mizutani, T.; Carbon, P.; Krol, A. Characterization of mSelB, a novel mammalian elongation factor for selenoprotein translation. EMBO J. 2000, 19, 4796–4805. [Google Scholar] [CrossRef] [Green Version]
- Varlamova, E.G. Protein–protein interactions of ER–resident selenoproteins with their physiological partners. Biochimie 2020, 171–172, 197–204. [Google Scholar] [CrossRef]
- Varlamova, E.G. Participation of selenoproteins localized in the ER in the processes occurring in this organelle and in the regulation of carcinogenesis-associated processes. J. Trace Elements Med. Biol. 2018, 48, 172–180. [Google Scholar] [CrossRef]
- Varlamova, E.; Goltyaev, M.; Mal’Tseva, V.; Turovsky, E.; Sarimov, R.; Simakin, A.; Gudkov, S. Mechanisms of the Cytotoxic Effect of Selenium Nanoparticles in Different Human Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 7798. [Google Scholar] [CrossRef]
- Goltyaev, M.; Mal’Tseva, V.; Varlamova, E. Expression of ER-resident selenoproteins and activation of cancer cells apoptosis mechanisms under ER-stress conditions caused by methylseleninic acid. Gene 2020, 755, 144884. [Google Scholar] [CrossRef]
- Gliga, A.R.; Skoglund, S.; Wallinder, I.O.; Fadeel, B.; Karlsson, H.L. Size-dependent cytotoxicity of silver nanoparticles in human lung cells: The role of cellular uptake, agglomeration and Ag release. Part. Fibre Toxicol. 2014, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Varlamova, E.; Turovsky, E. The main cytotoxic effects of methylseleninic acid on various cancer cells. Int. J. Mol. Sci. 2021, 22, 6614. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Yan, R.; Li, R.; Zhang, X. Selenium as a pleiotropic agent for medical discovery and drug delivery. Int. J. Nanomed. 2018, 13, 7473–7490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, Y.P.; Goltyaev, M.V.; Gorbacheva, O.S.; Novoselov, S.V.; Varlamova, E.G.; Fesenko, E.E. Influence of Sodium Selenite on the mRNA Expression of the Mammalian Selenocysteine-Containing Protein Genes in Testicle and Prostate Cancer Cells. Dokl. Biochem. Biophys. 2018, 480, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Boylan, M.; Selvam, A.; Spallholz, J.E.; Björnstedt, M. Redox-Active Selenium Compounds—From Toxicity and Cell Death to Cancer Treatment. Nutrients 2015, 7, 3536–3556. [Google Scholar] [CrossRef] [Green Version]
- Zu, K.; Bihani, T.; Lin, A.; Park, Y.-M.; Mori, K.; Lp, C. Enhanced selenium effect on growth arrest by BiP/GRP78 knockdown in p53-null human prostate cancer cells. Oncogene 2005, 25, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Durrani, F.A.; Rustum, Y.M. Selective Modulation of the Therapeutic Efficacy of Anticancer Drugs by Selenium Containing Compounds against Human Tumor Xenografts. Clin. Cancer Res. 2004, 10, 2561–2569. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Chen, Y.; Zhao, G.; Sun, H.; Che, H.; Leng, X. Effect of molecular weight of chitosan and its oligosaccharides on antitumor activities of chitosan-selenium nanoparticles. Carbohydr. Polym. 2019, 231, 115689. [Google Scholar] [CrossRef]
- Menon, S.; Ks, S.D.; Santhiya, R.; Rajeshkumar, S.; Kumar, V. Selenium nanoparticles: A potent chemotherapeutic agent and an elucidation of its mechanism. Colloids Surf. B Biointerfaces 2018, 170, 280–292. [Google Scholar] [CrossRef]
- Geoffrion, L.D.; Hesabizadeh, T.; Medina-Cruz, D.; Kusper, M.; Taylor, P.; Vernet-Crua, A.; Chen, J.; Ajo, A.; Webster, T.J.; Guisbiers, G. Naked Selenium Nanoparticles for Antibacterial and Anticancer Treatments. ACS Omega 2020, 5, 2660–2669. [Google Scholar] [CrossRef]
- Turovsky, E.; Varlamova, E. Mechanism of Ca2+-Dependent Pro-Apoptotic Action of Selenium Nanoparticles, Mediated by Activation of Cx43 Hemichannels. Biology 2021, 10, 743. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Turovsky, E.A.; Blinova, E.V. Therapeutic Potential and Main Methods of Obtaining Selenium Nanoparticles. Int. J. Mol. Sci. 2021, 22, 10808. [Google Scholar] [CrossRef] [PubMed]
- Varlamova, E.G.; Goltyaev, M.V.; Simakin, A.V.; Gudkov, S.V.; Turovsky, E.A. Comparative Analysis of the Cytotoxic Effect of a Complex of Selenium Nanoparticles Doped with Sorafenib, “Naked” Selenium Nanoparticles, and Sorafenib on Human Hepatocyte Carcinoma HepG2 Cells. Int. J. Mol. Sci. 2022, 23, 6641. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of Mammalian Selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varlamova, E.G.; Goltyaev, M.V.; Novoselov, V.I.; Fesenko, E.E. Cloning, intracellular localization, and expression of the mammalian selenocysteine-containing protein SELENOI (SelI) in tumor cell lines. Dokl. Biochem. Biophys. 2017, 476, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Shchedrina, V.A.; Everley, R.A.; Zhang, Y.; Gygi, S.P.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein K Binds Multiprotein Complexes and Is Involved in the Regulation of Endoplasmic Reticulum Homeostasis. J. Biol. Chem. 2011, 286, 42937–42948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Park, K.J.; Jang, J.K.; Jeon, Y.H.; Ko, K.Y.; Kwon, J.H.; Lee, S.-R.; Kim, I.Y. Selenoprotein S-dependent Selenoprotein K Binding to p97(VCP) Protein Is Essential for Endoplasmic Reticulum-associated Degradation. J. Biol. Chem. 2015, 290, 29941–29952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Z.-Q.; Zhang, X.; Fan, H.-H.; Wang, X.-S.; Wu, H.-M.; Zhang, L.; Cheng, W.-H.; Zhu, J.-H. Selenoprotein T Promotes Proliferation and G1-to-S Transition in SK-N-SH Cells: Implications in Parkinson’s Disease. J. Nutr. 2019, 149, 2110–2119. [Google Scholar] [CrossRef]
- Sengupta, A.; Carlson, B.A.; Labunskyy, V.M.; Gladyshev, V.N.; Hatfield, D.L. Selenoprotein T deficiency alters cell adhesion and elevates selenoprotein W expression in murine fibroblast cells. Biochem. Cell Biol. 2009, 87, 953–961. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Goltyaev, M.V.; Kuznetsova, Y.P. Effect of Sodium Selenite on Gene Expression of SELF, SELW, and TGR Selenoproteins in Adenocarcinoma Cells of the Human Prostate. Mol. Biol. 2018, 52, 519–526. [Google Scholar] [CrossRef]
- Diamond, A.M. Selenoproteins of the Human Prostate: Unusual Properties and Role in Cancer Etiology. Biol. Trace Elem. Res. 2019, 192, 51–59. [Google Scholar] [CrossRef]
- Björnstedt, M.; Kumar, S.; Björkhem, L.; Spyrou, G.; Holmgren, A. Selenium and the thioredoxin and glutaredoxin systems. Biomed. Environ. Sci. 1997, 10, 271–279. [Google Scholar] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Yoshida, H.; Akazawa, R.; Negishi, M.; Mori, K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002, 366, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.-H.; Chu, G.C.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005, 24, 4368–4380. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Chapman, J.S.; Barnett, D.W.; Curley, R.W., Jr.; Clagett-Dame, M. The Unhydrolyzable Fenretinide Analogue 4-Hydroxybenzylretinone Induces the Proapoptotic Genes GADD153 (CHOP) and Bcl-2–Binding Component 3 (PUMA) and Apoptosis that Is Caspase- Dependent and Independent of the Retinoic Acid Receptor. Cancer Res. 2007, 67, 6270–6277. [Google Scholar] [CrossRef] [Green Version]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.-O.; Aw, T.-Y.; Holbrook, N.J. Gadd153 Sensitizes Cells to Endoplasmic Reticulum Stress by Down-Regulating Bcl2 and Perturbing the Cellular Redox State. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of Caspase-12, an Endoplastic Reticulum (ER) Resident Caspase, through Tumor Necrosis Factor Receptor-associated Factor 2-dependent Mechanism in Response to the ER Stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Yuan, J. Cross-Talk between Two Cysteine Protease Families. Activation of Caspase-12 by Calpain in Apoptosis. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Hermel, E.; Castro-Obregon, S.; del Rio, G.; Ellerby, L.M.; Ellerby, H.M.; Bredesen, D.E. Coupling Endoplasmic Reticulum Stress to the Cell Death Program. Mechanism of Caspase Activation. J. Biol. Chem. 2001, 276, 33869–33874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK Regulation of Endoplasmic Reticulum Ca2+: A Control Point for Apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Oberley, T.D. Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res 2001, 61, 7071–7078. [Google Scholar]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Ryan, P.M.; Bedard, K.; Breining, T.; Cribb, A.E. Disruption of the endoplasmic reticulum by cytotoxins in LLC-PK1 cells. Toxicol. Lett. 2005, 159, 154–163. [Google Scholar] [CrossRef]
- Tan, Y.; Dourdin, N.; Wu, C.; De Veyra, T.; Elce, J.S.; Greer, P.A. Ubiquitous Calpains Promote Caspase-12 and JNK Activation during Endoplasmic Reticulum Stress-induced Apoptosis. J. Biol. Chem. 2006, 281, 16016–16024. [Google Scholar] [CrossRef] [Green Version]
- Hammadi, M.; Oulidi, A.; Gackière, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S.; et al. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leakviatranslocon during unfolded protein response: Involvement of GRP78. FASEB J. 2012, 27, 1600–1609. [Google Scholar] [CrossRef]
- Sukumaran, P.; Da Conceicao, V.N.; Sun, Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium Signaling Regulates Autophagy and Apoptosis. Cells 2021, 10, 2125. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer 5′– > 3′ | Reverse Primer 5′– > 3′ |
|---|---|---|
| TGA– > TGT | GTAGAGACCTGCGGGGGATGTCAGCTGAACCGCC | GCTGACATCCCCCGCAGGTCTCTACCCG |
| Cloning primers | TACAGAATTCATGAGCCTCCTGTTGCCTCCG | CGTCGACCTACAGGTCAGCGTGGTCCGAAG |
| Gene Name | Forward Primer 5′– > 3′ | Reverse Primer 5′– > 3′ |
|---|---|---|
| GAPDH | ACATCGCTCAGACACCATG | GCCAGTGAGCTTCCCGTT |
| SELENOT | TCTCCTAGTGGCGGCGTC | GTCTATATATTGGTTGAGGGAGG |
| SELENOM | AGCCTCCTGTTGCCTCCGC | AGGTCAGCGTGGTCCGAAG |
| SELENOF | TACGGTTGTTGTTGGCGAC | CAAATTGTGCTTCCTCCTGAC |
| SELENOK | TTTACATCTCGAACGGACAAG | CAGCCTTCCACTTCTTGATG |
| SELENOS | TGGGACAGCATGCAAGAAG | GCGTCCAGGTCTCCAGG |
| SELENON | TGATCTGCCTGCCCAATG | TCAGGAACTGCATGTAGGTGG |
| DIO2 | AGCTTCCTCCTCGATGCC | AAAGGAGGTCAAGTGGCTG |
| CHOP | GCTCTGATTGACCGAATGG | TCTGGGAAAGGTGGGTAGTG |
| GADD34 | CTCCGAGAAGGTCACTGTCC | GACGAGCGGGAAGGTGTGG |
| PUMA | CAGATATGCGCCCAGAGAT | CCATTCGTGGGTGGTCTTC |
| BIM | GGACGACCTCAACGCACAGTACGAG | GTAAGGGCAGGAGTCCCA |
| CAS-3 | GCATTGAGACAGACAGTGGTG | AATAGAGTTCTTTTGTGAGCATG |
| CAS-4 | CACGCCTGGCTCTCATCATA | TAGCAAATGCCCTCAGCG |
| BAX | GGGCTGGACATTGGACTTC | AACACAGTCCAAGGCAGCTG |
| BAK | GAGAGTGGCATCAATTGGGG | CAGCCACCCCTCTGTGCAATCCA |
| BCL-2 | GGTGAACTGGGGGAGGATTG | AGCCAGGAGAAATCAAACAGAG |
| ATF-4 | GTGTTCTCTGTGGGTCTGCC | GACCCTTTTCTTCCCCCTTG |
| ATF-6 | AACCCTAGTGTGAGCCCTGC | GTTCAGAGCACCCTGAAGA |
| XBPu | ACTCAGACTACGTGCACCTC | GTCAATACCGCCAGAATCC |
| XBPs | CTGAGTCCGCAGCGGTGCAGG | GGTCCAAGTTGTCCAGAATG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogachev, V.V.; Goltyaev, M.V.; Varlamova, E.G.; Turovsky, E.A. Molecular Mechanisms of the Cytotoxic Effect of Recombinant Selenoprotein SELENOM on Human Glioblastoma Cells. Int. J. Mol. Sci. 2023, 24, 6469. https://doi.org/10.3390/ijms24076469
Rogachev VV, Goltyaev MV, Varlamova EG, Turovsky EA. Molecular Mechanisms of the Cytotoxic Effect of Recombinant Selenoprotein SELENOM on Human Glioblastoma Cells. International Journal of Molecular Sciences. 2023; 24(7):6469. https://doi.org/10.3390/ijms24076469
Chicago/Turabian StyleRogachev, Vladimir V., Michael V. Goltyaev, Elena G. Varlamova, and Egor A. Turovsky. 2023. "Molecular Mechanisms of the Cytotoxic Effect of Recombinant Selenoprotein SELENOM on Human Glioblastoma Cells" International Journal of Molecular Sciences 24, no. 7: 6469. https://doi.org/10.3390/ijms24076469