
BGP-15 Protects against Doxorubicin-Induced Cell Toxicity via Enhanced Mitochondrial Function
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effects of BGP-15 Pretreatment on Cell Viability and LDH Release of DOX-Induced Cardiotoxicity
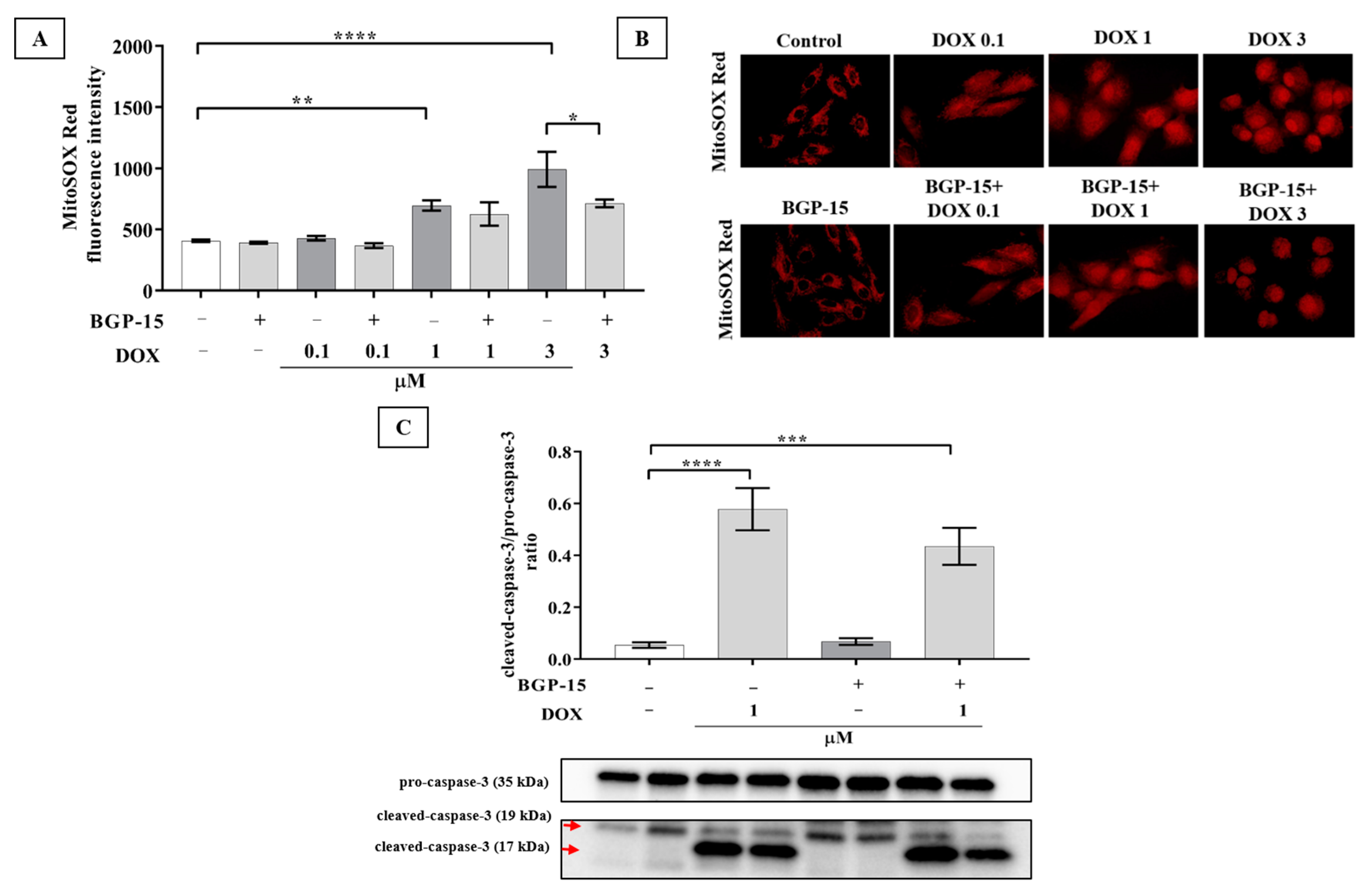
2.2. BGP-15 Attenuates the DOX-Induced Generation of Mitochondrial ROS and Slightly Diminishes the Activation of Caspase-3 Apoptosis Marker in H9c2 Cells
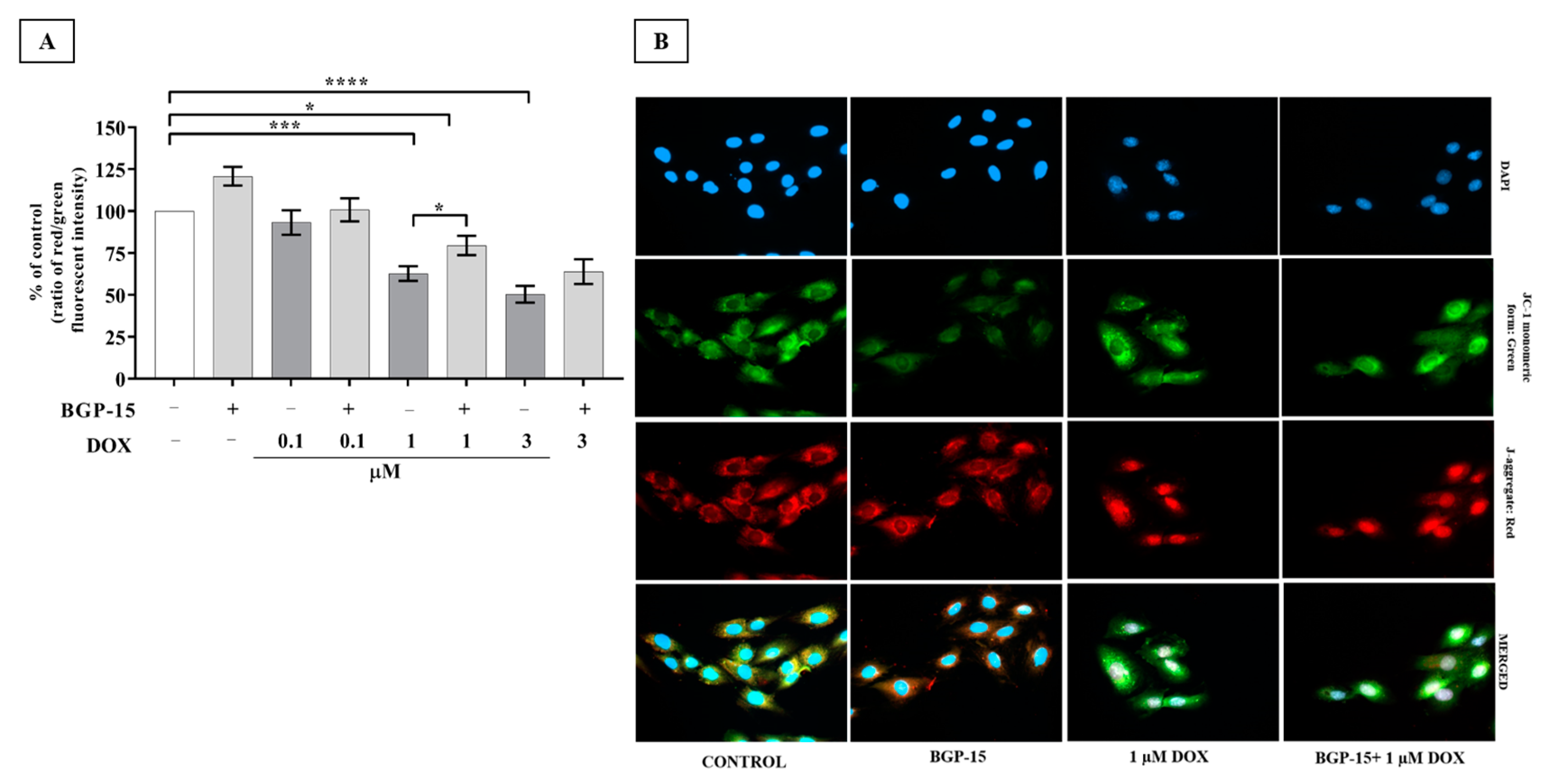
2.3. Effects of BGP-15 Pretreatment on Mitochondrial Depolarization of DOX-Exposed H9c2 Cells
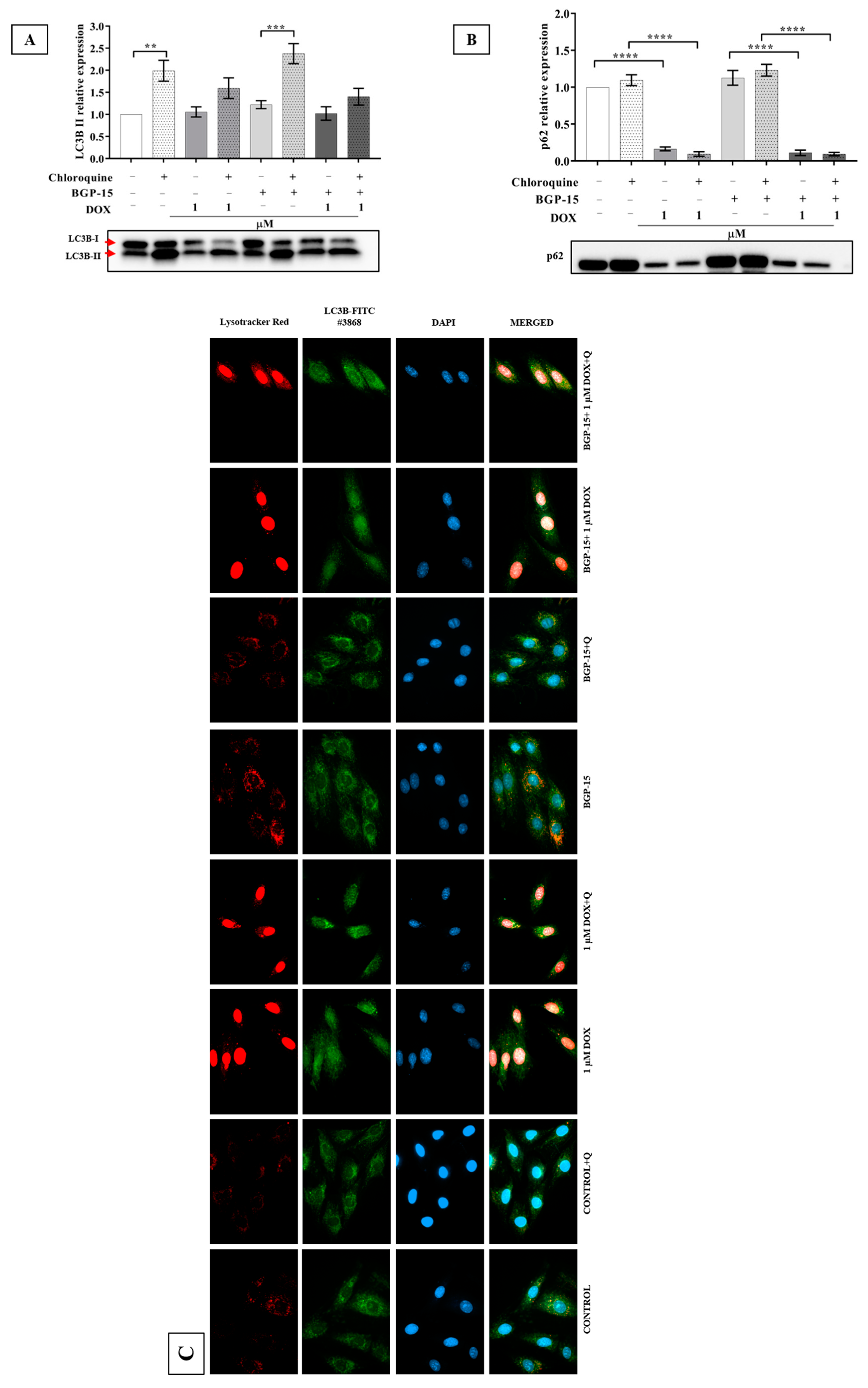
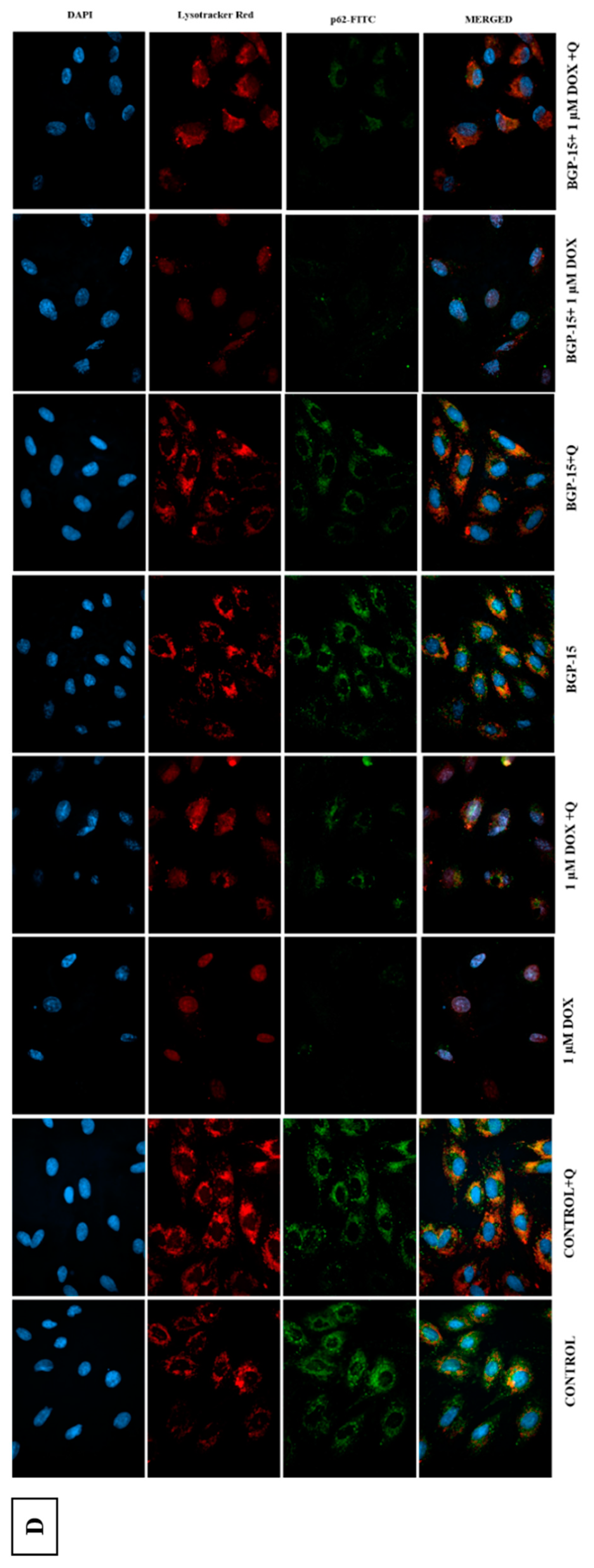
2.4. Effects of BGP-15 on Autophagy Flux in DOX-Induced Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Assay by MTT
4.4. Determination of Intracellular Reactive Oxygen Species Generation and Mitochondrial Function
4.5. Assessment of Mitochondrial Membrane Potential
4.6. LDH (Lactate Dehydrogenase) Release Assay
4.7. Autophagy Flux Determined by Fluorescent Microscopy
4.8. Protein Isolation
4.9. Western Blot Analysis
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Carvalho, F.S.; Burgeiro, A.; Garcia, R.; Moreno, A.J.; Carvalho, R.A.; Oliveira, P.J. Doxorubicin-induced cardiotoxicity: From bioenergetic failure and cell death to cardiomyopathy. Med. Res. Rev. 2014, 34, 106–135. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Rabinovich-Nikitin, I.; Love, M.; Kirshenbaum, L.A. Inhibition of MMP prevents doxorubicin-induced cardiotoxicity by attenuating cardiac intracellular and extracellular matrix remodelling. Cardiovasc. Res. 2021, 117, 11–12. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Wu, W.; Yan, J.; Li, X.; Yu, H.; Yu, X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int. J. Cardiol. 2009, 134, 82–90. [Google Scholar] [CrossRef]
- Dirks-Naylor, A.J. The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sci 2013, 93, 913–916. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [Green Version]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Peto, A.; Kosa, D.; Feher, P.; Ujhelyi, Z.; Sinka, D.; Vecsernyes, M.; Szilvassy, Z.; Juhasz, B.; Csanadi, Z.; Vigh, L.; et al. Pharmacological Overview of the BGP-15 Chemical Agent as a New Drug Candidate for the Treatment of Symptoms of Metabolic Syndrome. Molecules 2020, 25, 429. [Google Scholar] [CrossRef] [Green Version]
- Nagy, G.; Szarka, A.; Lotz, G.; Doczi, J.; Wunderlich, L.; Kiss, A.; Jemnitz, K.; Veres, Z.; Banhegyi, G.; Schaff, Z.; et al. BGP-15 inhibits caspase-independent programmed cell death in acetaminophen-induced liver injury. Toxicol. Appl. Pharmacol. 2010, 243, 96–103. [Google Scholar] [CrossRef]
- Szabados, E.; Literati-Nagy, P.; Farkas, B.; Sumegi, B. BGP-15, a nicotinic amidoxime derivate protecting heart from ischemia reperfusion injury through modulation of poly(ADP-ribose) polymerase. Biochem. Pharmacol. 2000, 59, 937–945. [Google Scholar] [CrossRef]
- Bombicz, M.; Priksz, D.; Gesztelyi, R.; Kiss, R.; Hollos, N.; Varga, B.; Nemeth, J.; Toth, A.; Papp, Z.; Szilvassy, Z.; et al. The Drug Candidate BGP-15 Delays the Onset of Diastolic Dysfunction in the Goto-Kakizaki Rat Model of Diabetic Cardiomyopathy. Molecules 2019, 24, 586. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Wallace, K.B.; Sardao, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Sapra, G.; Tham, Y.K.; Cemerlang, N.; Matsumoto, A.; Kiriazis, H.; Bernardo, B.C.; Henstridge, D.C.; Ooi, J.Y.; Pretorius, L.; Boey, E.J.; et al. The small-molecule BGP-15 protects against heart failure and atrial fibrillation in mice. Nat. Commun. 2014, 5, 5705. [Google Scholar] [CrossRef] [Green Version]
- Horvath, O.; Ordog, K.; Bruszt, K.; Deres, L.; Gallyas, F.; Sumegi, B.; Toth, K.; Halmosi, R. BGP-15 Protects against Heart Failure by Enhanced Mitochondrial Biogenesis and Decreased Fibrotic Remodelling in Spontaneously Hypertensive Rats. Oxidative Med. Cell. Longev. 2021, 2021, 1250858. [Google Scholar] [CrossRef]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Munoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol.-Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [Green Version]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxidative Med. Cell. Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Montaigne, D.; Marechal, X.; Preau, S.; Baccouch, R.; Modine, T.; Fayad, G.; Lancel, S.; Neviere, R. Doxorubicin induces mitochondrial permeability transition and contractile dysfunction in the human myocardium. Mitochondrion 2011, 11, 22–26. [Google Scholar] [CrossRef]
- Nordgren, K.K.; Wallace, K.B. Keap1 redox-dependent regulation of doxorubicin-induced oxidative stress response in cardiac myoblasts. Toxicol. Appl. Pharmacol. 2014, 274, 107–116. [Google Scholar] [CrossRef]
- Sumegi, K.; Fekete, K.; Antus, C.; Debreceni, B.; Hocsak, E.; Gallyas, F., Jr.; Sumegi, B.; Szabo, A. BGP-15 Protects against Oxidative Stress- or Lipopolysaccharide-Induced Mitochondrial Destabilization and Reduces Mitochondrial Production of Reactive Oxygen Species. PLoS ONE 2017, 12, e0169372. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Zilinyi, R.; Czompa, A.; Czegledi, A.; Gajtko, A.; Pituk, D.; Lekli, I.; Tosaki, A. The Cardioprotective Effect of Metformin in Doxorubicin-Induced Cardiotoxicity: The Role of Autophagy. Molecules 2018, 23, 1184. [Google Scholar] [CrossRef] [Green Version]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [Green Version]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007, 3, 154–157. [Google Scholar] [CrossRef] [Green Version]
- Gyongyosi, A.; Szoke, K.; Fenyvesi, F.; Fejes, Z.; Debreceni, I.B.; Nagy, B., Jr.; Tosaki, A.; Lekli, I. Inhibited autophagy may contribute to heme toxicity in cardiomyoblast cells. Biochem. Biophys. Res. Commun. 2019, 511, 732–738. [Google Scholar] [CrossRef]
- Gurtler, A.; Kunz, N.; Gomolka, M.; Hornhardt, S.; Friedl, A.A.; McDonald, K.; Kohn, J.E.; Posch, A. Stain-Free technology as a normalization tool in Western blot analysis. Anal. Biochem. 2013, 433, 105–111. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyongyosi, A.; Csaki, N.; Peto, A.; Szoke, K.; Fenyvesi, F.; Bacskay, I.; Lekli, I. BGP-15 Protects against Doxorubicin-Induced Cell Toxicity via Enhanced Mitochondrial Function. Int. J. Mol. Sci. 2023, 24, 5269. https://doi.org/10.3390/ijms24065269
Gyongyosi A, Csaki N, Peto A, Szoke K, Fenyvesi F, Bacskay I, Lekli I. BGP-15 Protects against Doxorubicin-Induced Cell Toxicity via Enhanced Mitochondrial Function. International Journal of Molecular Sciences. 2023; 24(6):5269. https://doi.org/10.3390/ijms24065269
Chicago/Turabian StyleGyongyosi, Alexandra, Nikolett Csaki, Agota Peto, Kitti Szoke, Ferenc Fenyvesi, Ildiko Bacskay, and Istvan Lekli. 2023. "BGP-15 Protects against Doxorubicin-Induced Cell Toxicity via Enhanced Mitochondrial Function" International Journal of Molecular Sciences 24, no. 6: 5269. https://doi.org/10.3390/ijms24065269