
Treatment with a Cholecystokinin Receptor Antagonist, Proglumide, Improves Efficacy of Immune Checkpoint Antibodies in Hepatocellular Carcinoma

 and
and
Abstract
:1. Introduction
2. Results
2.1. Receptor and Gastrin Expression in HCC Cells
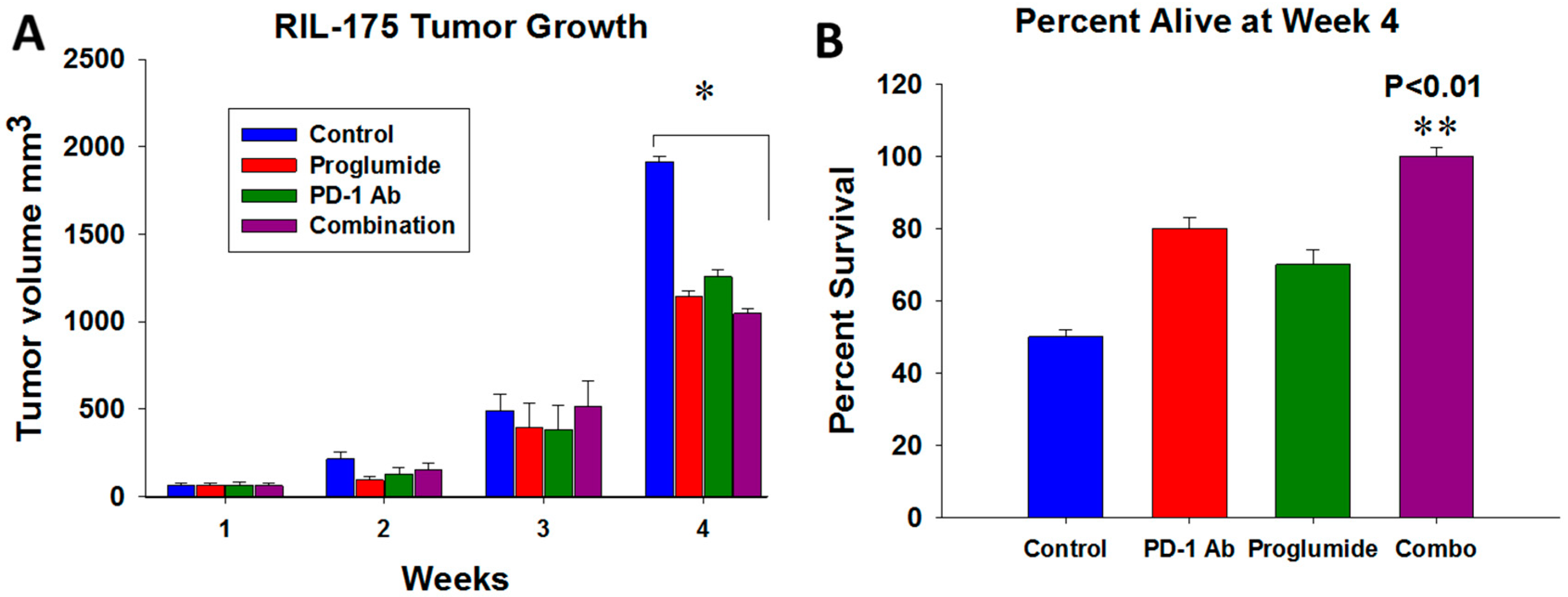
2.2. HCC Tumor Growth and Mouse Survival
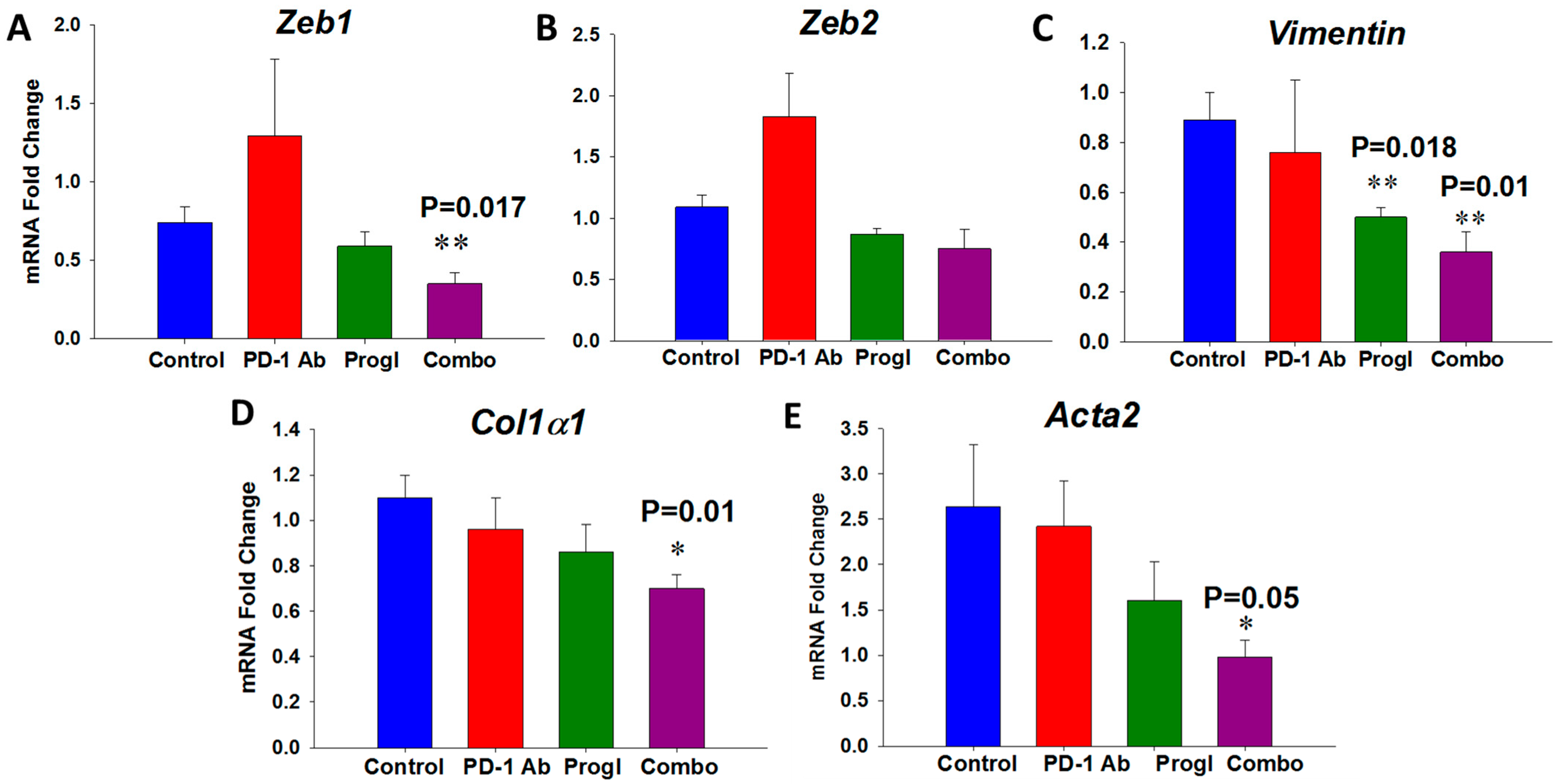
2.3. Proglumide Alters the Tumor Microenvironment by Decreasing Fibrosis and Increasing CD8+ T Cells
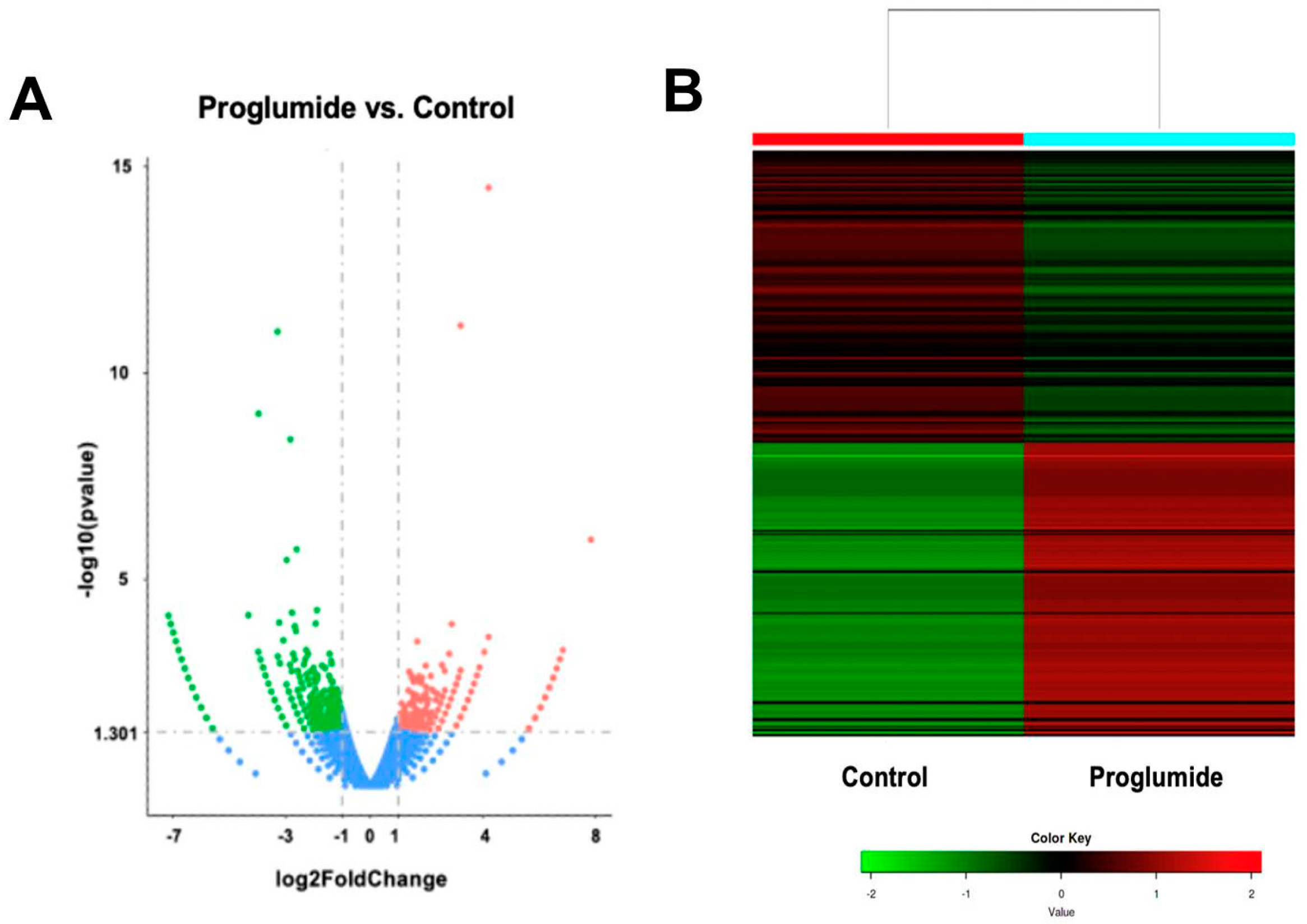
2.4. Differentially Expressed Genes in Tumors of Mice Treated with Proglumide and PD-1Ab
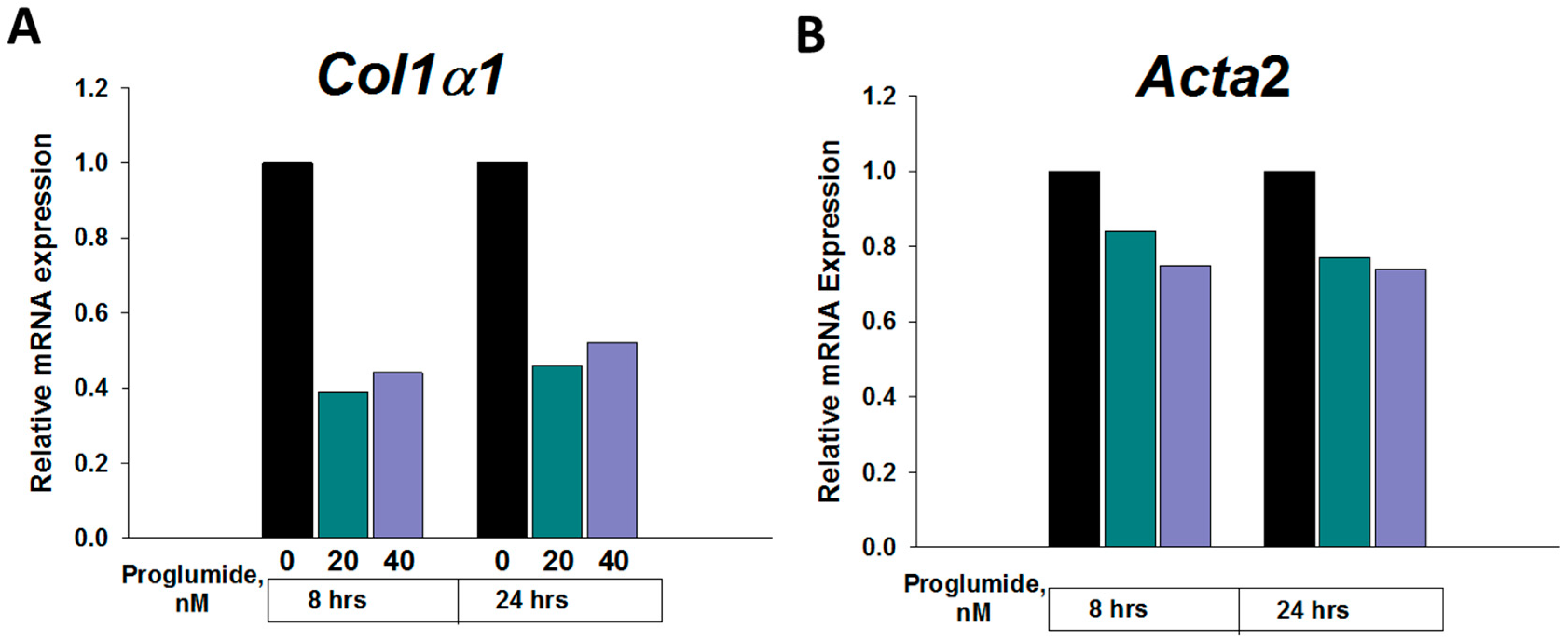
2.5. Proglumide Therapy Significantly Decreases Fibrosis-Associated Genes in Dt81Hepa1-6 Cells In Vitro
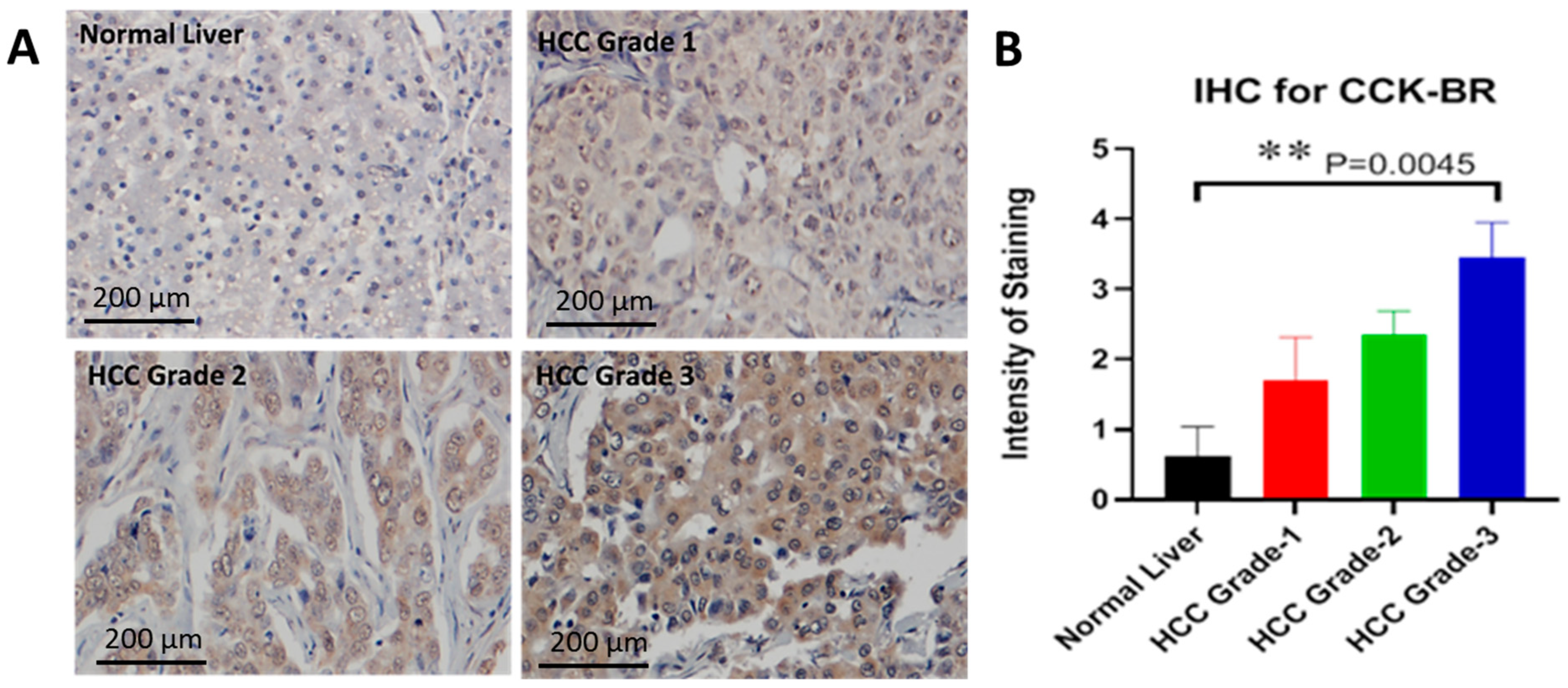
2.6. Characterization of the CCK-B Receptor in a Human Tissue Array
2.6.1. Upregulated Genes by Proglumide Using RNA Sequencing
2.6.2. Downregulated Genes by Proglumide in Human HCC Cells by RNA Sequencing
2.7. Differentially Expressed Genes in Human HCC Untreated or Treated with Proglumide by RNAseq
3. Discussion
4. Materials and Methods
4.1. HCC Cell Lines
4.2. Characterization of PD-L1 Receptor and Gastrin mRNA Expression in RIL-175 Cells
4.3. Study Design for the Treatment of HCC Murine Tumors with Proglumide and PD-1Ab
4.4. Analysis of the HCC Tumor Microenvironment for CD8+ T Cells and Fibrosis
4.5. Examination of Differentially Expressed Genes (DEGs) in Murine HCC Tumors and Cells
4.6. Immunohistochemistry for the CCK-BR in Human Liver Cancer
4.7. Differentially Expressed Genes by RNA Sequencing in Human HCC Cells Exposed to Proglumide
4.8. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marisi, G.; Cucchetti, A.; Ulivi, P.; Canale, M.; Cabibbo, G.; Solaini, L.; Foschi, F.G.; De, M.S.; Ercolani, G.; Valgiusti, M.; et al. Ten Years of Sorafenib in Hepatocellular Carcinoma: Are There any Predictive and/or Prognostic Markers? World J. Gastroenterol. 2018, 24, 4152–4163. [Google Scholar] [CrossRef]
- Boland, P.; Wu, J. Systemic Therapy for Hepatocellular Carcinoma: Beyond Sorafenib. Chin. Clin. Oncol. 2018, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for Hepatocellular Carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.R.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus Sorafenib in Advanced Hepatocellular Carcinoma (CheckMate 459): A Randomised, Multicentre, Open-label, Phase 3 Trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib (KEYNOTE-224): A Non-randomised, Open-label Phase 2 Trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Kudo, M. Durvalumab plus Tremelimumab in Unresectable Hepatocellular Carcinoma. Hepatobiliary Surg. Nutr. 2022, 11, 592–596. [Google Scholar] [CrossRef]
- de Castria, T.B.; Khalil, D.N.; Harding, J.J.; O’Reilly, E.M.; Abou-Alfa, G.K. Tremelimumab and Durvalumab in the Treatment of Unresectable, Advanced Hepatocellular Carcinoma. Future Oncol. 2022, 18, 3769–3782. [Google Scholar] [CrossRef]
- Cheng, A.L.; Hsu, C.; Chan, S.L.; Choo, S.P.; Kudo, M. Challenges of Combination Therapy with Immune Checkpoint Inhibitors for Hepatocellular Carcinoma. J. Hepatol. 2020, 72, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Cusmai, A.; Gadaleta-Caldarola, G.; Palmiotti, G. Which Role for Predictors of Response to Immune Checkpoint Inhibitors in Hepatocellular Carcinoma? Expert Rev. Gastroenterol. Hepatol. 2022, 16, 333–339. [Google Scholar] [CrossRef]
- Viscardi, G.; Tralongo, A.C.; Massari, F.; Lambertini, M.; Mollica, V.; Rizzo, A.; Comito, F.; Di, L.R.; Alfieri, S.; Imbimbo, M.; et al. Comparative Assessment of Early Mortality Risk upon Immune Checkpoint Inhibitors Alone or in Combination with Other Agents Across Solid Malignancies: A Systematic Review and Meta-analysis. Eur. J. Cancer 2022, 177, 175–185. [Google Scholar] [CrossRef]
- Gay, M.D.; Safronenka, A.; Cao, H.; Liu, F.H.; Malchiodi, Z.X.; Tucker, R.D.; Kroemer, A.; Shivapurkar, N.; Smith, J.P. Targeting the Cholecystokinin Receptor: A Novel Approach for Treatment and Prevention of Hepatocellular Cancer. Cancer Prev. Res. 2021, 14, 17–29. [Google Scholar] [CrossRef]
- Zender, L.; Xue, W.; Cordon-Cardo, C.; Hannon, G.J.; Lucito, R.; Powers, S.; Flemming, P.; Spector, M.S.; Lowe, S.W. Generation and Analysis of Genetically Defined Liver Carcinomas Derived from Bipotential Liver Progenitors. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, B.; Raymond, V.A.; Cassim, S.; Lapierre, P.; Bilodeau, M. Highly Tumorigenic Hepatocellular Carcinoma Cell Line with Cancer Stem Cell-Like Properties. PLoS ONE 2017, 12, e0171215. [Google Scholar] [CrossRef]
- Forte, J.G.; Wolsin, J.M. HCl Secretion by the Gastric Oxyntic Cell; Johnson, L., Ed.; Physiology of the Gastrointestinal Tract; Raven: New York, NY, USA, 1987; pp. 853–863. [Google Scholar]
- Smith, J.P.; Shih, A.; Wu, Y.; McLaughlin, P.J.; Zagon, I.S. Gastrin Regulates Growth of Human Pancreatic Cancer in a Tonic and Autocrine Fashion. Am. J. Physiol. 1996, 270, R1078–R1084. [Google Scholar] [CrossRef] [PubMed]
- Malchiodi, Z.X.; Cao, H.; Gay, M.D.; Safronenka, A.; Bansal, S.; Tucker, R.D.; Weinberg, B.A.; Cheema, A.; Shivapurkar, N.; Smith, J.P. Cholecystokinin Receptor Antagonist Improves Efficacy of Chemotherapy in Murine Models of Pancreatic Cancer by Altering the Tumor Microenvironment. Cancers 2021, 13, 4949. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Putt, K.S.; Coppola, D.; Leon, M.E.; Khalil, F.K.; Centeno, B.A.; Clark, N.; Stark, V.E.; Morse, D.L.; Low, P.S. Assessment of Cholecystokinin 2 Receptor (CCK2R) in Neoplastic Tissue. Oncotarget 2016, 7, 14605–14615. [Google Scholar] [CrossRef]
- McLaughlin, J.; Grazia, L.M.; Jones, M.N.; D’Amato, M.; Dockray, G.J.; Thompson, D.G. Fatty Acid Chain Length Determines Cholecystokinin Secretion and Effect on Human Gastric Motility. Gastroenterology 1999, 116, 46–53. [Google Scholar] [CrossRef]
- Caplin, M.; Khan, K.; Savage, K.; Rode, J.; Varro, A.; Michaeli, D.; Grimes, S.; Brett, B.; Pounder, R.; Dhillon, A. Expression and Processing of Gastrin in Hepatocellular Carcinoma, Fibrolamellar Carcinoma and Cholangiocarcinoma. J. Hepatol. 1999, 30, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Dudzinski, S.; Beckermann, K.E.; Young, K.; McKinley, E.; McIntyre, J.; Rathmell, J.C.; Xu, J.; Gore, J.C. MRI of Tumor T Cell Infiltration in Response to Checkpoint Inhibitor Therapy. J. Immunother. Cancer. 2020, 8, e000328. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, K.; Datta, M.; Hato, T.; Kitahara, S.; Chen, I.X.; Matsui, A.; Kikuchi, H.; Mamessier, E.; Aoki, S.; Ramjiawan, R.R.; et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology 2020, 71, 1247–1261. [Google Scholar] [CrossRef]
- Bruix, J.; Reig, M.; Sherman, M. Evidence-Based Diagnosis, Staging, and Treatment of Patients with Hepatocellular Carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-Associated Fibrosis as a Regulator of Tumor Immunity and Response to Immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1037–1048. [Google Scholar] [CrossRef]
- Singh, P.; Owlia, A.; Espeijo, R.; Dai, B. Novel Gastrin Receptors Mediate Mitogenic Effects of Gastrin and Processing Intermediates of Gastrin on Swiss 3T3 Fibroblasts. Absence of Detectable Cholecystokinin (CCK)-A and CCK-B Receptors. J. Biol. Chem. 1995, 270, 8429–8438. [Google Scholar] [CrossRef]
- Berna, M.J.; Seiz, O.; Nast, J.F.; Benten, D.; Blaker, M.; Koch, J.; Lohse, A.W.; Pace, A. CCK1 and CCK2 Receptors are Expressed on Pancreatic Stellate Cells and Induce Collagen Production. J. Biol. Chem. 2010, 285, 38905–38914. [Google Scholar] [CrossRef]
- Tucker, R.D.; Ciofoaia, V.; Nadella, S.; Gay, M.D.; Cao, H.; Huber, M.; Safronenka, A.; Shivapurkar, N.; Kallakury, B.; Kruger, A.J.; et al. A Cholecystokinin Receptor Antagonist Halts Nonalcoholic Steatohepatitis and Prevents Hepatocellular Carcinoma. Dig. Dis. Sci. 2020, 65, 189–203. [Google Scholar] [CrossRef]
- Rabiee, A.; Gay, M.D.; Shivapurkar, N.; Cao, H.; Nadella, S.; Smith, C.I.; Lewis, J.; Bansal, S.; Cheema, A.; Smith, J.P. Safety and Dosing Study of a Cholecystokinin Receptor Antagonist in Non-alcoholic Steatohepatitis (NASH). Clin. Pharmacol. Ther. 2022, 112, 1271–1279. [Google Scholar] [CrossRef]
- Kurzepa, J.; Madro, A.; Czechowska, G.; Kurzepa, J.; Celinski, K.; Kazmierak, W.; Slomka, M. Role of MMP-2 and MMP-9 and Their Natural Inhibitors in Liver Fibrosis, Chronic Pancreatitis and Non-Specific Inflammatory Bowel Diseases. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 570–579. [Google Scholar] [CrossRef]
- Rodriguez-Aznar, E.; Wiesmuller, L.; Sainz, B., Jr.; Hermann, P.C. EMT and Stemness-Key Players in Pancreatic Cancer Stem Cells. Cancers 2019, 11, 1136. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the Crossroads of Epithelial-Mesenchymal Transition, Metastasis and Therapy Resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Li, S. Vimentin in Cancer and its Potential as a Molecular Target for Cancer Therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New Insights into the Role of EMT in Tumor Immune Escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed]
- Firrincieli, D.; Boissan, M.; Chignard, N. Epithelial-Mesenchymal Transition in the Liver. Gastroenterol. Clin. Biol. 2010, 34, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef]
- Kapanadze, T.; Gamrekelashvili, J.; Ma, C.; Chan, C.; Zhao, F.; Hewitt, S.; Zender, L.; Kapoor, V.; Felsher, D.W.; Manns, M.P.; et al. Regulation of Accumulation and Function of Myeloid Derived Suppressor Cells in Different Murine Models of Hepatocellular Carcinoma. J. Hepatol. 2013, 59, 1007–1013. [Google Scholar] [CrossRef]
- Lopez-Terrada, D.; Cheung, S.W.; Finegold, M.J.; Knowles, B.B. Hep G2 is a hepatoblastoma-derived cell line. Hum. Pathol. 2009, 40, 1512–1515. [Google Scholar] [CrossRef]
- Smith, J.P.; Solomon, T.E. Effects of Gastrin, Proglumide, and Somatostatin on Growth of Human Colon Cancer. Gastroenterology 1988, 95, 1541–1548. [Google Scholar] [CrossRef]
- Smith, J.P.; Wang, S.; Nadella, S.; Jablonski, S.A.; Weiner, L.M. Cholecystokinin Receptor Antagonist Alters Pancreatic Cancer Microenvironment and Increases Efficacy of Immune Checkpoint Antibody Therapy in Mice. Cancer Immunol. Immunother. 2018, 67, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Function | Expression in Combination Therapy | PubMed |
|---|---|---|---|---|
| Igfbp3 | insulin-like growth factor binding protein 3 | Metastases suppression gene | Up | 21697285 |
| Ptgs2 | prostaglandin-endo-peroxide synthase 2 | Downstream marker of ferroptosis, cell death | Up | 30925886 |
| Tert | Telomerase reverse transcriptase | Promotes immortalization by protecting telomeres | Up | 30447097 |
| Col1αα1 | Collagen 1-alpha | Makes collagen/fibrosis/promotes metastases | Down | 31181620 |
| Zeb2 | Zinc Finger E-Box Binding Homeobox 2 | Regulator of EMT | Down | 30157906 |
| Zeb1 | Zinc Finger E-Box Binding Homeobox 1 | Regulator of EMT | Down | 25607528 |
| Vimentin | fibroblast intermediate filament, | Promotes EMT | Down | 25965826 |
| Lef1 | Lymphoid enhancer binding factor 1 | Promotes EMT & WNT signaling | Down | 28670499 |
| Acta2 | Alpha-smooth muscle actin | EMT and fibrosis | Down | 23128109 |
| Sfrp2 | Secreted frizzled-related protein 2 | Promotes EMT | Down | 28218291 |
| Pathway | p-Value |
|---|---|
| Phagosome Formation | 5.88 × 10−3 |
| Pyroptosis Signaling Pathway | 1.55 × 10−2 |
| Macrophage migration inhibitory factor (MIF)-mediated Glucocorticoid Regulation | 1.74 × 10−2 |
| G-Protein-Coupled Receptor Signaling | 1.78 × 10−2 |
| Inhibition of Matrix Metalloproteases | 2.02 × 10−2 |
| mGene | 5′-Forward Primer-3′ | 5′-Reverse Primer-3′ |
|---|---|---|
| Cckar | CTTTTCTGCCTGGATCAACCT | ACCGTGATAACCAGCGTGTTC |
| Cckbr | GATGGCTGCTACGTGCAACT | CGCACCACCCGCTTCTTAG |
| Cd274 | TGCGGACTACAAGCGAATCACG | CTCAGCTTCTGGATAACCCTCG |
| Gast | CCCAGGGTCCTCAACACTTC | GCCAAAGTCCATCCATCCGT |
| Hprt | TCCTCCTCAGACCGCTTT | TTTTCCAAATCCTCGGCATAATG |
| mGene | 5′-Forward Primer-3′ | 5′-Reverse Primer-3′ |
|---|---|---|
| Zeb1 | GCTGGCAAGACAACGTGAAAG | GCCTCAGGATAAATGACGGC |
| Zeb2 | GGGACAGATCAGCACCAAAT | GACCCAGAATGAGACAAGCG |
| Vimentin | TCCACACGCACCTACAGTCT | CCGAGGACCGGGTCACATA |
| Col1α1 | CGCCATCAAGGTCTACTG | ACGGGAATCCATCGGTC |
| Acta2 | TGCCGAGCGTGAGATTGT | CCCGTCAGGCAGTTCGTAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shivapurkar, N.; Gay, M.D.; He, A.; Chen, W.; Golnazar, S.; Cao, H.; Duka, T.; Kallakury, B.; Vasudevan, S.; Smith, J.P. Treatment with a Cholecystokinin Receptor Antagonist, Proglumide, Improves Efficacy of Immune Checkpoint Antibodies in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 3625. https://doi.org/10.3390/ijms24043625
Shivapurkar N, Gay MD, He A, Chen W, Golnazar S, Cao H, Duka T, Kallakury B, Vasudevan S, Smith JP. Treatment with a Cholecystokinin Receptor Antagonist, Proglumide, Improves Efficacy of Immune Checkpoint Antibodies in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2023; 24(4):3625. https://doi.org/10.3390/ijms24043625
Chicago/Turabian StyleShivapurkar, Narayan, Martha D. Gay, Aiwu (Ruth) He, Wenqiang Chen, Shermineh Golnazar, Hong Cao, Tetyana Duka, Bhaskar Kallakury, Sona Vasudevan, and Jill P. Smith. 2023. "Treatment with a Cholecystokinin Receptor Antagonist, Proglumide, Improves Efficacy of Immune Checkpoint Antibodies in Hepatocellular Carcinoma" International Journal of Molecular Sciences 24, no. 4: 3625. https://doi.org/10.3390/ijms24043625