
Structures of MPND Reveal the Molecular Recognition of Nucleosomes

Abstract
:1. Introduction
2. Results
2.1. Crystal Structure Determination and Characterization of apo–MPND
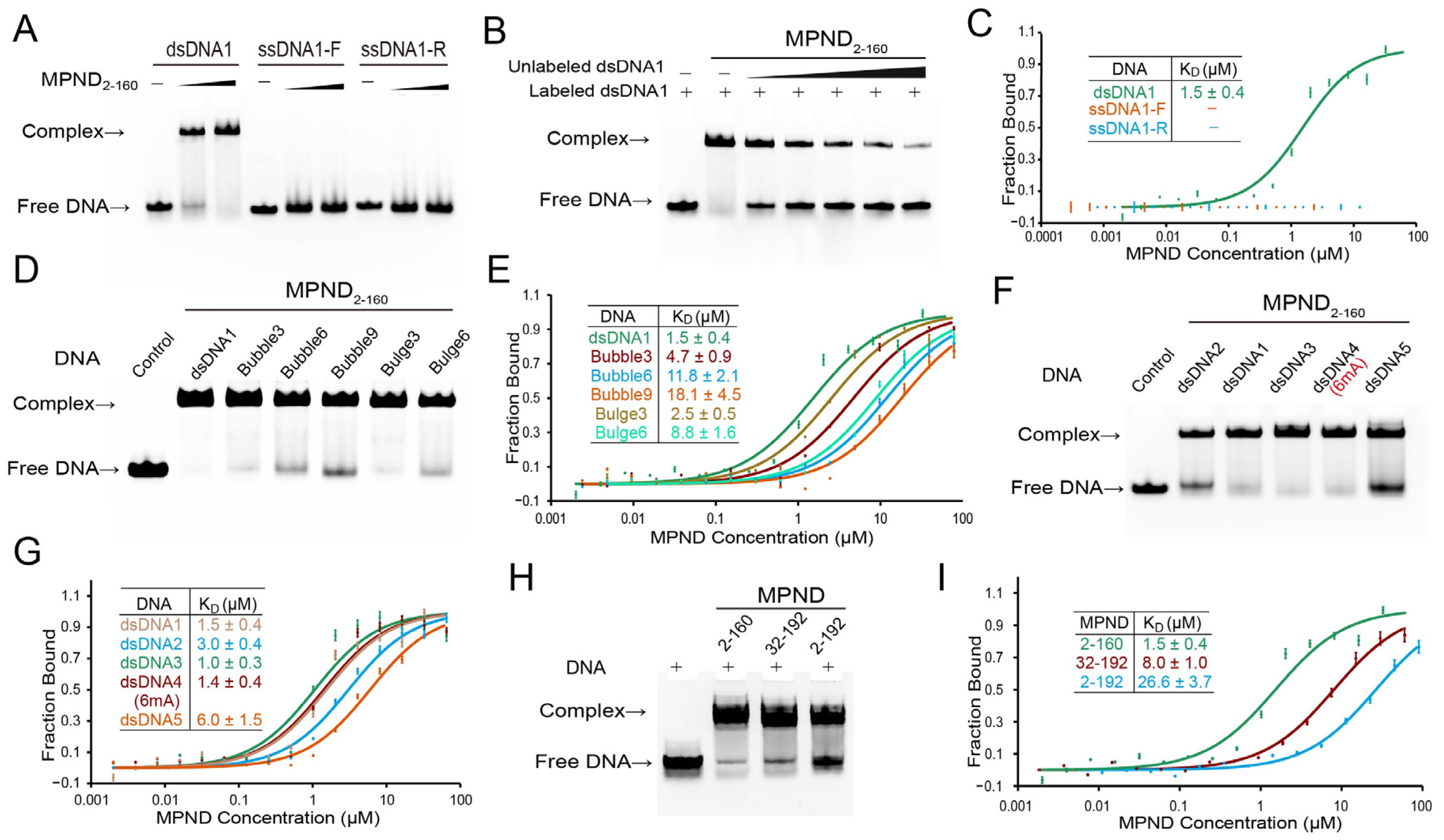
2.2. Specific Recognition and Binding of dsDNA by MPND
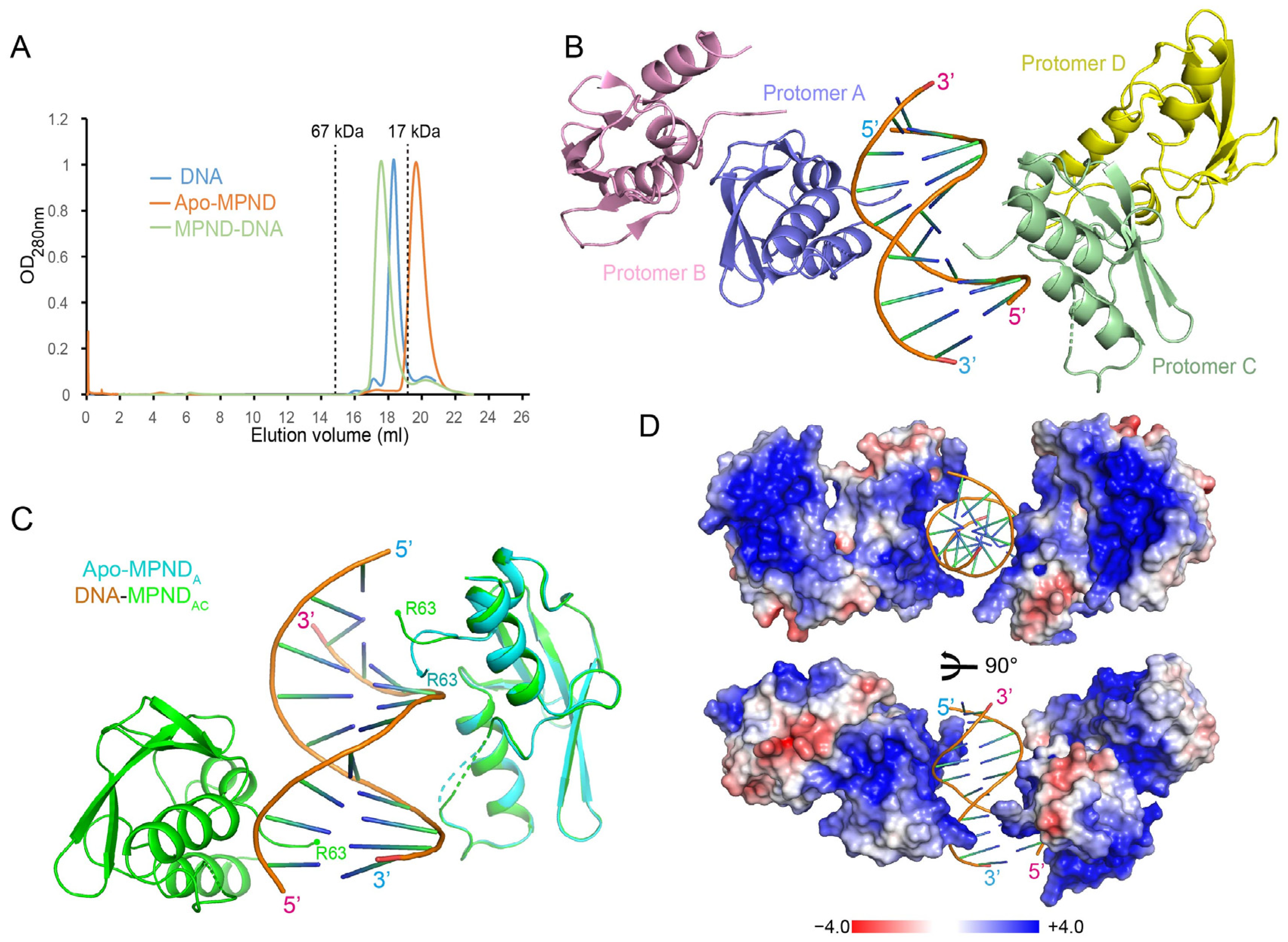
2.3. The Complex Structure of MPND with Double-Stranded DNA
2.4. Key Sites of Interaction between MPND and Nucleic Acid
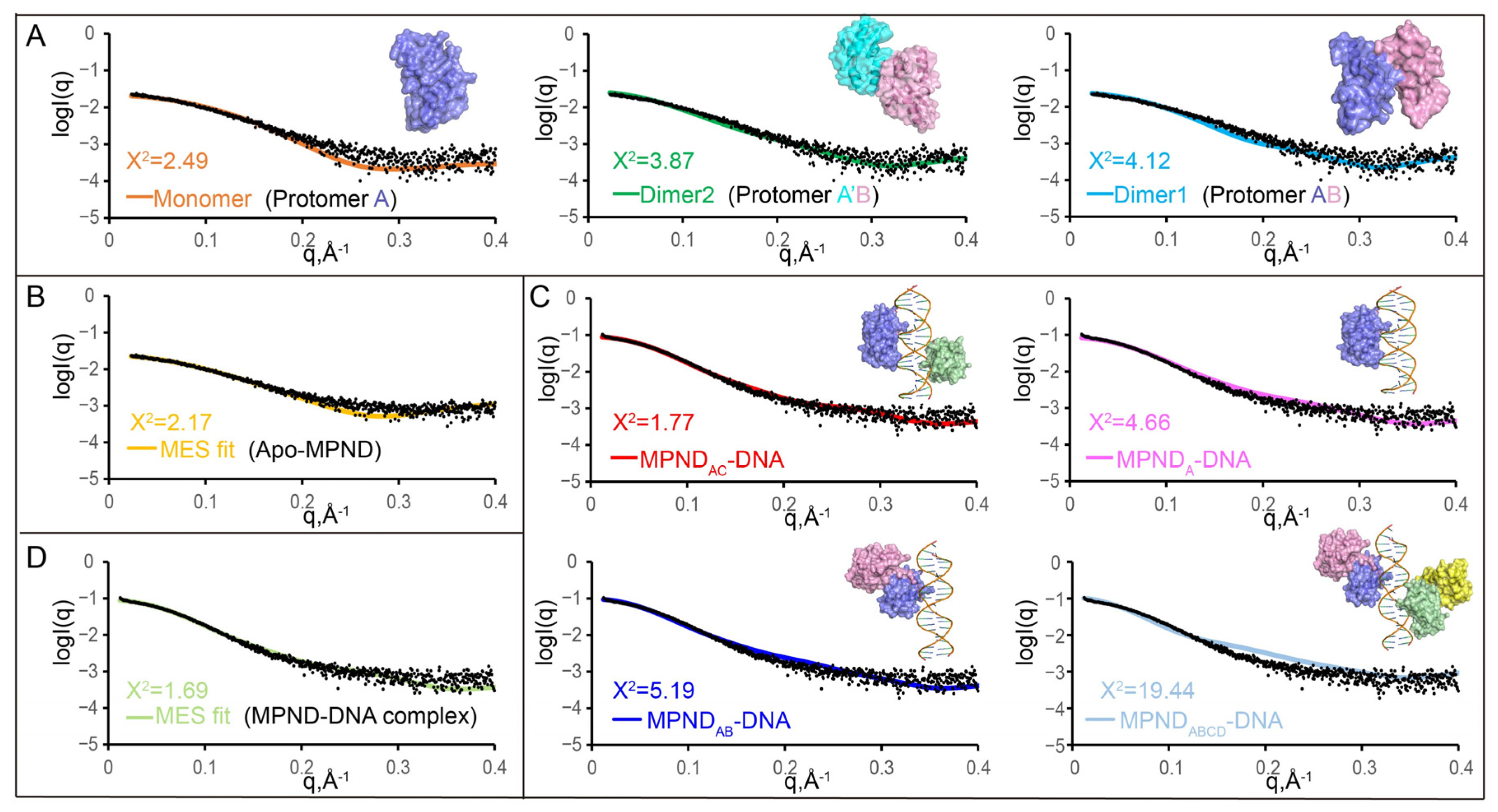
2.5. The Oligomeric State of apo–MPND and MPND–DNA Complexes in Solution
2.6. Direct Histone Binding by MPND and Cooperative Regulation by Acidic Regions and DNA
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification of Protein
4.2. Protein Crystallization and Data Collection
4.3. Structure Determination
4.4. Electrophoretic Mobility Shift Assays
4.5. Small-Angle X-ray Scattering
4.6. Microscale Thermophoresis (MST)
4.7. Pull-Down Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, I.A.; Clark, T.A.; Morgan, R.D.; Boitano, M.; Anton, B.P.; Luong, K.; Fomenkov, A.; Turner, S.W.; Korlach, J.; Roberts, R.J. The methylomes of six bacteria. Nucleic Acids Res. 2012, 40, 11450–11462. [Google Scholar] [CrossRef]
- Fang, G.; Munera, D.; Friedman, D.I.; Mandlik, A.; Chao, M.C.; Banerjee, O.; Feng, Z.; Losic, B.; Mahajan, M.C.; Jabado, O.J.; et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 2012, 30, 1232–1239. [Google Scholar] [PubMed]
- Casadesús, J.; Low, D. Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. 2006, 70, 830–856. [Google Scholar] [CrossRef]
- Zhu, S.; Beaulaurier, J.; Deikus, G.; Wu, T.P.; Strahl, M.; Hao, Z.; Luo, G.; Gregory, J.A.; Chess, A.; He, C.; et al. Mapping and characterizing N6-methyladenine in eukaryotic genomes using single-molecule real-time sequencing. Genome Res. 2018, 28, 1067–1078. [Google Scholar] [PubMed]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Koziol, M.J.; Bradshaw, C.R.; Allen, G.E.; Costa, A.S.H.; Frezza, C.; Gurdon, J.B. Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat. Struct. Mol. Biol. 2016, 23, 24–30. [Google Scholar] [CrossRef]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizabal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA Methylation on N6-Adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar]
- Iyer, L.M.; Zhang, D.; Aravind, L. Adenine methylation in eukaryotes: Apprehending the complex evolutionary history and functional potential of an epigenetic modification. BioEssays 2016, 38, 27–40. [Google Scholar]
- Kweon, S.M.; Chen, Y.; Moon, E.; Kvederaviciutė, K.; Klimasauskas, S.; Feldman, D.E. An Adversarial DNA N(6)-Methyladenine-Sensor Network Preserves Polycomb Silencing. Mol. Cell. 2019, 74, 1138–1147. [Google Scholar] [CrossRef]
- Zhu, P.; Zhou, W.; Wang, J.; Puc, J.; Ohgi, K.A.; Erdjument-Bromage, H.; Tempst, P.; Glass, C.K.; Rosenfeld, M.G. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol. Cell. 2007, 27, 609–621. [Google Scholar] [CrossRef]
- Patterson-Fortin, J.; Shao, G.; Bretscher, H.; Messick, T.E.; Greenberg, R.A. Differential regulation of JAMM domain deubiquitinating enzyme activity within the RAP80 complex. J. Biol. Chem. 2010, 285, 30971–30981. [Google Scholar] [PubMed]
- Marchione, R.; Leibovitch, S.A.; Lenormand, J.L. The translational factor eIF3f: The ambivalent eIF3 subunit. Cell. Mol. Life Sci. 2013, 70, 3603–3616. [Google Scholar] [PubMed] [Green Version]
- Cope, G.A.; Suh, G.S.; Aravind, L.; Schwarz, S.E.; Zipursky, S.L.; Koonin, E.V.; Deshaies, R.J. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 2002, 298, 608–611. [Google Scholar] [PubMed]
- Meng, C.; Xia, S.; He, Y.; Tang, X.; Zhang, G.; Zhou, T. Discovery of Prognostic Signature Genes for Overall Survival Prediction in Gastric Cancer. Comput. Math. Methods Med. 2020, 2020, 5479279. [Google Scholar] [PubMed]
- Zhang, J.; Huang, J.Y.; Chen, Y.N.; Yuan, F.; Zhang, H.; Yan, F.H.; Wang, M.J.; Wang, G.; Su, M.; Lu, G.; et al. Whole genome and transcriptome sequencing of matched primary and peritoneal metastatic gastric carcinoma. Sci. Rep. 2015, 5, 13750. [Google Scholar]
- Long, F.; Vagin, A.A.; Young, P.; Murshudov, G.N. BALBES: A molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 125–132. [Google Scholar] [CrossRef]
- Horton, J.R.; Woodcock, C.B.; Opot, S.B.; Reich, N.O.; Zhang, X.; Cheng, X. The cell cycle-regulated DNA adenine methyltransferase CcrM opens a bubble at its DNA recognition site. Nat. Commun. 2019, 10, 4600. [Google Scholar] [CrossRef]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Pelikan, M.; Hura, G.L.; Hammel, M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 2009, 28, 174–189. [Google Scholar] [PubMed]
- Tian, Z.; Li, X.; Li, M.; Wu, W.; Zhang, M.; Tang, C.; Li, Z.; Liu, Y.; Chen, Z.; Yang, M.; et al. Crystal structures of REF6 and its complex with DNA reveal diverse recognition mechanisms. Cell Discov. 2020, 6, 17. [Google Scholar]
- Gao, Y.; Zhang, Q.; Lang, Y.; Liu, Y.; Dong, X.; Chen, Z.; Tian, W.; Tang, J.; Wu, W.; Tong, Y.; et al. Human apo-SRP72 and SRP68/72 complex structures reveal the molecular basis of protein translocation. J. Mol. Cell Biol. 2017, 9, 220–230. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol. 1997, 276, 307–326. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [PubMed]
- Kovalevskiy, O.; Nicholls, R.A.; Long, F.; Carlon, A.; Murshudov, G.N. Overview of refinement procedures within REFMAC5: Utilizing data from different sources. Acta Crystallogr. D Struct. Biol. 2018, 74, 215–227. [Google Scholar]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Alexander, N.; Woetzel, N.; Meiler, J. BCL Cluster: A method for clustering biological molecules coupled with visualization in the Pymol Molecular Graphics System. IEEE Int. Conf. Comput. Adv. Biol. Med. Sci. 2011, 2011, 13–18. [Google Scholar]
- Nielsen, S.S.; Toft, K.N.; Snakenborg, D.; Jeppesen, M.G.; Jacobsen, J.K.; Vestergaard, B.; Kutter, J.P.; Arleth, L. BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. J. Appl. Crystallogr. 2009, 42, 959–964. [Google Scholar]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013, 105, 962–974. [Google Scholar] [CrossRef]
- Trewhella, J.; Duff, A.P.; Durand, D.; Gabel, F.; Guss, J.M.; Hendrickson, W.A.; Hura, G.L.; Jacques, D.A.; Kirby, N.M.; Kwan, A.H.; et al. 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: An update. Acta Crystallogr. D Struct. Biol. 2017, 73, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Bond, C.S.A.W. Schüttelkopf. ALINE: A WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr. D 2009, 65, 510–512. [Google Scholar] [CrossRef]
- Mooers, B.H.M. Shortcuts for faster image creation in PyMOL. Protein Sci. 2020, 29, 268–276. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apo–MPND | MPND–DNA Complex | |
|---|---|---|
| (PDB ID: 7YDT) | (PDB ID: 7YDW) | |
| Data collection | ||
| Space group | P212121 | P1 |
| Cell dimensions | ||
| a,b,c (Å) | 37.68, 62.84, 112.6 | 37.78, 62.92, 69.09 |
| α,β,γ (˚) | 90, 90, 90 | 72.58, 89.88, 89.98 |
| Resolution (Å) a | 50.0–2.06 | 50.0–2.47 |
| (2.10–2.06) | (2.51–2.47) | |
| Rmerge | 11.5% (49.6%) | 10.5% (32.5%) |
| I/σ | 13.3 (2.0) | 9.5 (2.0) |
| Completeness (%) | 97.6 (83.7) | 96.0 (80.8) |
| Total no. of reflections | 97,574 | 71,276 |
| Unique reflections | 17,365 | 21,839 |
| Redundancy | 5.8 (5.1) | 3.4 (3.1) |
| CC1/2 | 0.950 (0.883) | 0.968 (0.894) |
| Refinement | ||
| Resolution (Å) | 50.0–2.06 (2.11–2.06) | 50.0–2.47 (2.53–2.47) |
| No. of reflections | 16,061 | 19,835 |
| Rwork/Rfree (%) | 23.0/24.9 | 24.5/25.4 |
| No. of atoms | ||
| Protein | 1427 | 2841 |
| Ligand/ions | 0 | 409 |
| Water | 62 | 161 |
| Average B-factors (Å2) | ||
| Protein | 37.94 | 50.42 |
| Ligand/ion | 0.00 | 63.17 |
| Water | 39.13 | 52.33 |
| rms deviations b | ||
| Bond lengths (Å) | 0.002 | 0.003 |
| Bond angles, ° | 1.149 | 1.181 |
| Ramachandran plot, % c | 94.9/5.1/0 | 94.1/5.9/0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Li, X.; Tian, Z.; Ma, L.; Ma, J.; Liu, Y.; Shang, G.; Liang, A.; Wu, W.; Chen, Z. Structures of MPND Reveal the Molecular Recognition of Nucleosomes. Int. J. Mol. Sci. 2023, 24, 3368. https://doi.org/10.3390/ijms24043368
Yang M, Li X, Tian Z, Ma L, Ma J, Liu Y, Shang G, Liang A, Wu W, Chen Z. Structures of MPND Reveal the Molecular Recognition of Nucleosomes. International Journal of Molecular Sciences. 2023; 24(4):3368. https://doi.org/10.3390/ijms24043368
Chicago/Turabian StyleYang, Meiting, Xiaorong Li, Zizi Tian, Lulu Ma, Jun Ma, Yunlong Liu, Guohui Shang, Ailing Liang, Wei Wu, and Zhongzhou Chen. 2023. "Structures of MPND Reveal the Molecular Recognition of Nucleosomes" International Journal of Molecular Sciences 24, no. 4: 3368. https://doi.org/10.3390/ijms24043368