
An Update on Protein Kinases as Therapeutic Targets—Part I: Protein Kinase C Activation and Its Role in Cancer and Cardiovascular Diseases


Abstract
:1. Introduction
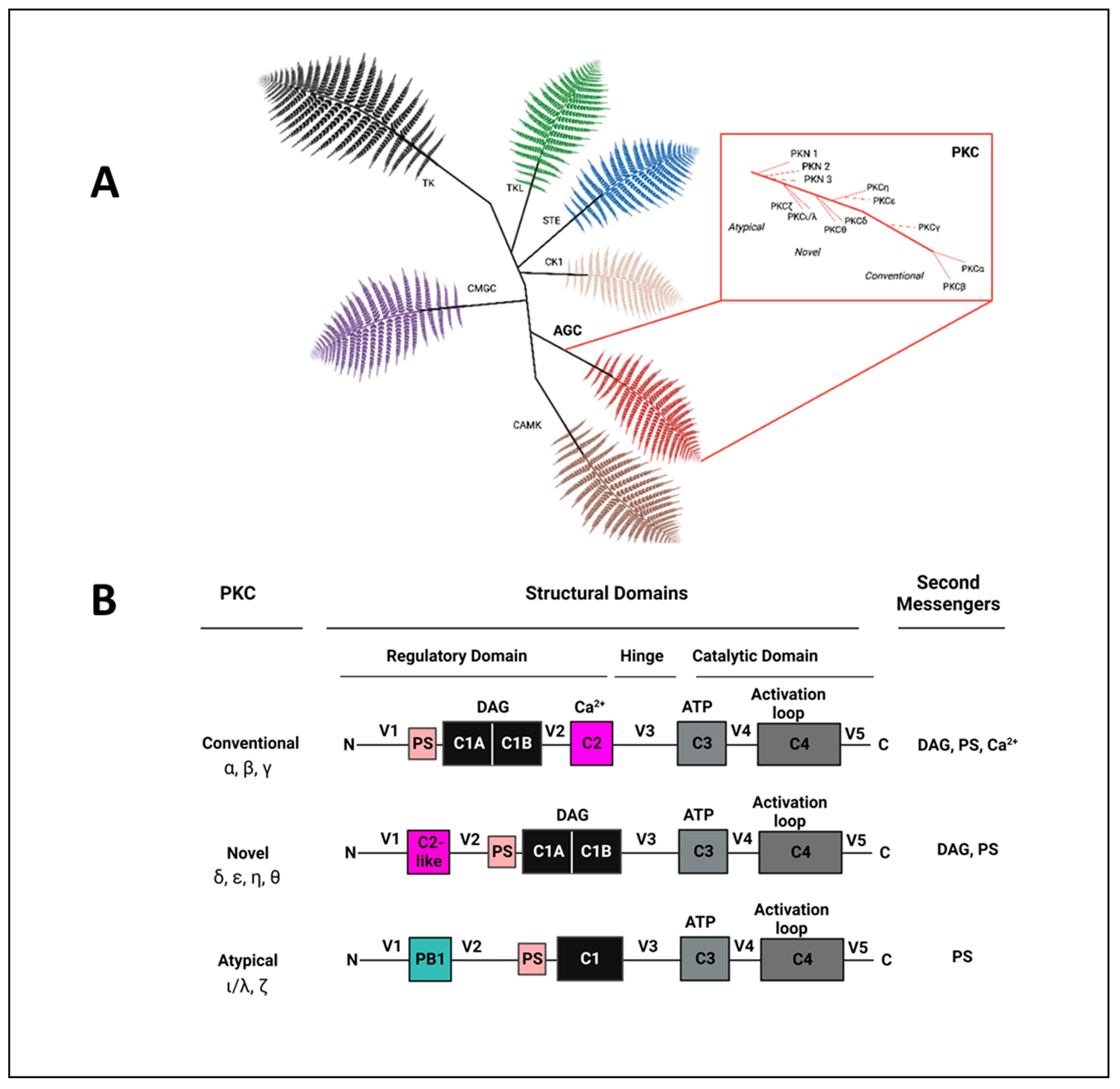
2. The Protein Kinase C (PKC) Family
2.1. Regulation by Lipid Second Messengers
2.2. Regulation by Scaffold Interactions
3. PKC in Cancer
4. PKC in Cardiovascular Diseases
5. PKC in Other Human Diseases
6. Targeting PKC
7. PKC Inhibitors in Clinical Trials

{kind=link}
{kind=link}
| Indication | Compound | Proposed Mechanism | Outcome | Regulatory Status | Refs. |
|---|---|---|---|---|---|
| Oncology | Bryostatin | Nonselective PKC activator | No benefit | Not approved | [430,431,432,433,434,435,436,437,438,439,440,441,442,443] |
| Aprinocarsen | PKCα inhibitor | No benefit | Not approved | [424,427,428,429,444,445] | |
| Enzastaurin (LY317615) | PKCβ inhibitor | No benefit | Not approved | [407,409,410,446,447,448,449,450] | |
| Tamoxifen | Nonselective PKC inhibitor at high doses, ER inhibitor | Used in the management of many breast and gynecologic cancers; failed trials for other malignancies | Approved | [451,452,453] | |
| Midostaurin | Nonselective PKC inhibitor, FLT3 inhibitor | Used in treatment of FLT3-mutated AML | Approved | [454] | |
| 7-Hydroxystaurosporine (UCN-01) | Nonselective PKC inhibitor, CHK1 inhibitor | No benefit | Not approved | [455,456,457] | |
| PMA | Nonspecific PKC activator | No benefit | Not approved | ||
| Safingol | PKCβI, PKCδ, and PKCε inhibitor; PI3K inhibitor | No benefit | Not approved | [458,459] | |
| 12-O-tetradecanoylphorbol-13-acetate | Nonselective PKC activator | No benefit, severe side effects | Not approved | [460] | |
| Diabetes mellitus | Ruboxistaurin(LY333531) | PKCβ inhibitor | Improved diabetic retinopathy but not nephropathy in early studies, minimal effect on neuropathy | Not approved | [461,462,463,464,465,466,467,468,469] |
| Cardiology | Delcasertib (KAI-9803) | PKCδ inhibitor | No benefit | Not approved | [470,471,472] |
| Flosequinan | Nonselective PKC inhibitor | Increased hospitalizations and HF mortality; early study termination | Withdrawn | [422,473] | |
| Volatile anesthetics | PKCε activator | Reduced troponin I, inotrope requirements, and length of hospitalization | Approved for other indications | [474,475,476,477,478] | |
| Adenosine | PKCε activator | Reduced MI, mortality, vasopressor requirements | Approved for other indications | [479,480,481] | |
| Acadesine | PKCε activator, AMPK activator | No reduction in death, MI, or stroke | Not approved | [482,483,484] | |
| Bipolar disorder | Endoxifen | Tamoxifen metabolite with four-fold increased PKC inhibition | Improved time to remission | Not approved | [485] |
| Nociception | KAI-1678 | Inhibits PKCε translocation | No benefit | Not approved | [486,487] |
| Inflammation | Sotrastaurin (AEB071) | Nonselective PKC inhibitor | Worse outcomes in transplant rejection, no benefit in malignancy or autoimmune trials | Not approved | [488,489,490,491] |
| Alzheimer’s Disease | Bryostatin | Nonselective PKC activator | Primary endpoint was not significant | Not approved | [492] |
8. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Johnson, J.L.; Yaron, T.M.; Huntsman, E.M.; Kerelsky, A.; Song, J.; Regev, A.; Lin, T.-Y.; Liberatore, K.; Cizin, D.M.; Cohen, B.M.; et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature 2023, 613, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Benn, C.L.; Dawson, L.A. Clinically Precedented Protein Kinases: Rationale for Their Use in Neurodegenerative Disease. Front. Aging Neurosci. 2020, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Patterson, H.; Nibbs, R.; McInnes, I.; Siebert, S. Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases. Clin. Exp. Immunol. 2014, 176, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Nussinov, R. The molecular basis of targeting protein kinases in cancer therapeutics. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Amin, F.; Ahmed, A.; Feroz, A.; Khaki, P.S.S.; Khan, M.S.; Tabrez, S.; Zaidi, S.K.; Abdulaal, W.H.; Shamsi, A.; Khan, W.; et al. An update on the association of protein kinases with cardiovascular diseases. Curr. Pharm. Des. 2019, 25, 174–183. [Google Scholar] [CrossRef]
- Krebs, E.G.; Fischer, E.H. The phosphorylase b to a converting enzyme of rabbit skeletal muscle. Biochim. Biophys. Acta 1956, 20, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Krebs, E.G.; Kent, A.B.; Fischer, E.H. The muscle phosphorylase b kinase reaction. J. Biol. Chem. 1958, 231, 73–83. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases-the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Eglen, R.; Reisine, T. Drug discovery and the human kinome: Recent trends. Pharmacol. Ther. 2011, 130, 144–156. [Google Scholar] [CrossRef]
- Arencibia, J.M.; Pastor-Flores, D.; Bauer, A.F.; Schulze, J.O.; Biondi, R.M. AGC protein kinases: From structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim. Biophys. Acta 2013, 1834, 1302–1321. [Google Scholar] [CrossRef]
- Kenakin, T.P. Chapter 6—Enzymes as Drug Targets. In Pharmacology in Drug Discovery and Development, 2nd ed.; Kenakin, T.P., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 131–156. [Google Scholar]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, L. Approval heralds new generation of kinase inhibitors? Nat. Biotechnol. 2001, 19, 599–600. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1–positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O. Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes. Cancer 2012, 3, 436–446. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; O’Brien, S.G.; Guilhot, F.; Goldman, J.M.; Hochhaus, A.; Hughes, T.P.; Radich, J.P.; Hatfield, A.K.; Mone, M.; Filian, J.; et al. International randomized study of interferon vs. STI571 (IRIS) 8-year follow up: Sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood 2009, 114, 1126. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2021. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2022, 27, 1075. [Google Scholar] [CrossRef]
- Fischer, P.M. Approved and Experimental Small-Molecule Oncology Kinase Inhibitor Drugs: A Mid-2016 Overview. Med. Res. Rev. 2017, 37, 314–367. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Yang, X.; Duan, Y.; Han, J.; Liao, C. Small-molecule kinase inhibitors for the treatment of nononcologic diseases. J. Med. Chem. 2021, 64, 1283–1345. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P. Ripasudil: First Global Approval. Drugs 2014, 74, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Poh, A.L.; Tang, W.H.W. Novel Insights and Treatment Strategies for Right Heart Failure. Curr. Heart Fail Rep. 2018, 15, 141–155. [Google Scholar] [CrossRef]
- Lovly, C.M.; Shaw, A.T. Molecular pathways: Resistance to kinase inhibitors and implications for therapeutic strategies. Clin. Cancer Res. 2014, 20, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Djikic, T.; Gagic, Z.; Nikolic, K. Design and Discovery of Kinase Inhibitors Using Docking Studies. In Molecular Docking for Computer-Aided Drug Design; Elsevier: Amsterdam, The Netherlands, 2021; pp. 337–365. [Google Scholar]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Gild, M.L.; Bullock, M.; Robinson, B.G.; Clifton-Bligh, R. Multikinase inhibitors: A new option for the treatment of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 617–624. [Google Scholar] [CrossRef]
- Gild, M.L.; Tsang, V.H.M.; Clifton-Bligh, R.J.; Robinson, B.G. Multikinase inhibitors in thyroid cancer: Timing of targeted therapy. Nat. Rev. Endocrinol. 2021, 17, 225–234. [Google Scholar] [CrossRef]
- Smolinski, M.P.; Bu, Y.; Clements, J.; Gelman, I.H.; Hegab, T.; Cutler, D.L.; Fang, J.W.; Fetterly, G.; Kwan, R.; Barnett, A.; et al. Discovery of Novel Dual Mechanism of Action Src Signaling and Tubulin Polymerization Inhibitors (KX2-391 and KX2-361). J. Med. Chem. 2018, 61, 4704–4719. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Cai, M.; Shao, L.; Zhang, J. Targeting Protein Kinases Degradation by PROTACs. Front. Chem. 2021, 9, 679120. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.-M.; Dong, J.; Xu, Z.-Y.; Cheng, X.-D.; Zhang, W.-D.; Qin, J.-J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef] [PubMed]
- Goulet, D.R.; Atkins, W.M. Considerations for the design of antibody-based therapeutics. J. Pharm. Sci. 2020, 109, 74–103. [Google Scholar] [CrossRef]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.-J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef]
- Gharwan, H.; Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.M.; Hu, X.Q.; Liu, X.X.; Ruan, B.F.; Xu, J.; Liao, C. From monoclonal antibodies to small molecules: The development of inhibitors targeting the PD-1/PD-L1 pathway. Drug Discov. Today 2016, 21, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef]
- Takai, Y.; Kishimoto, A.; Inoue, M.; Nishizuka, Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. I. Purification and characterization of an active enzyme from bovine cerebellum. J. Biol. Chem. 1977, 252, 7603–7609. [Google Scholar] [CrossRef]
- Takai, Y.; Kishimoto, A.; Iwasa, Y.; Kawahara, Y.; Mori, T.; Nishizuka, Y. Calcium-dependent activation of a multifunctional protein kinase by membrane phospholipids. J. Biol. Chem. 1979, 254, 3692–3695. [Google Scholar] [CrossRef]
- Hagiwara, M. Alternative splicing: A new drug target of the post-genome era. Biochim. Et Biophys. Acta (BBA) Proteins Proteom. 2005, 1754, 324–331. [Google Scholar] [CrossRef]
- Kim, J.D.; Seo, K.W.; Lee, E.A.; Quang, N.N.; Cho, H.R.; Kwon, B. A novel mouse PKCδ splice variant, PKCδIX, inhibits etoposide-induced apoptosis. Biochem. Biophys. Res. Commun. 2011, 410, 177–182. [Google Scholar] [CrossRef]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.; Boeckeler, K.; Parker, P.J. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 2010, 11, 103–112. [Google Scholar] [CrossRef]
- Kikkawa, U. The story of PKC: A discovery marked by unexpected twists and turns. IUBMB Life 2019, 71, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Faria, A.; Persaud, S.J. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol. Ther. 2017, 172, 50–62. [Google Scholar] [CrossRef]
- Spitaler, M.; Cantrell, D.A. Protein kinase C and beyond. Nat. Immunol. 2004, 5, 785–790. [Google Scholar] [CrossRef]
- Zanin-Zhorov, A.; Dustin, M.L.; Blazar, B.R. PKC-theta function at the immunological synapse: Prospects for therapeutic targeting. Trends Immunol. 2011, 32, 358–363. [Google Scholar] [CrossRef]
- Altman, A.; Kong, K.F. Protein kinase C inhibitors for immune disorders. Drug Discov. Today 2014, 19, 1217–1221. [Google Scholar] [CrossRef]
- Sipka, S.; Bíró, T.; Czifra, G.; Griger, Z.; Gergely, P.; Brugós, B.; Tarr, T. The role of protein kinase C isoenzymes in the pathogenesis of human autoimmune diseases. Clin. Immunol. 2022, 241, 109071. [Google Scholar] [CrossRef]
- Deka, S.J.; Trivedi, V. Potentials of PKC in cancer progression and anticancer drug development. Curr. Drug Discov. Technol. 2019, 16, 135–147. [Google Scholar] [CrossRef]
- Simonis, G.; Braun, M.U.; Kirrstetter, M.; Schön, S.P.; Strasser, R.H. Mechanisms of myocardial remodeling: Ramiprilat blocks the expressional upregulation of protein kinase C-epsilon in the surviving myocardium early after infarction. J. Cardiovasc. Pharmacol. 2003, 41, 780–787. [Google Scholar] [CrossRef]
- Koyanagi, T.; Noguchi, K.; Ootani, A.; Inagaki, K.; Robbins, R.C.; Mochly-Rosen, D. Pharmacological inhibition of epsilon PKC suppresses chronic inflammation in murine cardiac transplantation model. J. Mol. Cell Cardiol. 2007, 43, 517–522. [Google Scholar] [CrossRef]
- Marrocco, V.; Bogomolovas, J.; Ehler, E.; Dos Remedios, C.G.; Yu, J.; Gao, C.; Lange, S. PKC and PKN in heart disease. J. Mol. Cell. Cardiol. 2019, 128, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, K.; Churchill, E.; Mochly-Rosen, D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc. Res. 2006, 70, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Brum, P.C.; Mochly-Rosen, D. betaIIPKC and epsilonPKC isozymes as potential pharmacological targets in cardiac hypertrophy and heart failure. J. Mol. Cell Cardiol. 2011, 51, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Koyanagi, T.; Palaniyandi, S.S.; Fajardo, G.; Churchill, E.N.; Budas, G.; Disatnik, M.H.; Bernstein, D.; Brum, P.C.; Mochly-Rosen, D. Pharmacological inhibition of betaIIPKC is cardioprotective in late-stage hypertrophy. J. Mol. Cell. Cardiol. 2011, 51, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Hardman, C.; Ho, S.; Shimizu, A.; Luu-Nguyen, Q.; Sloane, J.L.; Soliman, M.S.A.; Marsden, M.D.; Zack, J.A.; Wender, P.A. Synthesis and evaluation of designed PKC modulators for enhanced cancer immunotherapy. Nat. Commun. 2020, 11, 1879. [Google Scholar] [CrossRef] [PubMed]
- Kanev, G.K.; de Graaf, C.; de Esch, I.J.; Leurs, R.; Würdinger, T.; Westerman, B.A.; Kooistra, A.J. The Landscape of Atypical and Eukaryotic Protein Kinases. Trends Pharmacol. Sci. 2019, 40, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Ghosh, D.; Williams, R.O., 3rd. Just how prevalent are peptide therapeutic products? A critical review. Int. J. Pharm. 2020, 587, 119491. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef]
- Wang, B.; Wu, H.; Hu, C.; Wang, H.; Liu, J.; Wang, W.; Liu, Q. An overview of kinase downregulators and recent advances in discovery approaches. Signal Transduct. Target. Ther. 2021, 6, 423. [Google Scholar] [CrossRef]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.E.; Jura, N. Prospects for pharmacological targeting of pseudokinases. Nat. Rev. Drug Discov. 2019, 18, 501–526. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef] [PubMed]
- Szilveszter, K.P.; Németh, T.; Mócsai, A. Tyrosine Kinases in Autoimmune and Inflammatory Skin Diseases. Front. Immunol. 2019, 10, 1862. [Google Scholar] [CrossRef] [PubMed]
- Zarrin, A.A.; Bao, K.; Lupardus, P.; Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 2021, 20, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef]
- Ayala-Aguilera, C.C.; Valero, T.; Lorente-Macías, Á.; Baillache, D.J.; Croke, S.; Unciti-Broceta, A. Small Molecule Kinase Inhibitor Drugs (1995–2021): Medical Indication, Pharmacology, and Synthesis. J. Med. Chem. 2022, 65, 1047–1131. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef]
- Gould, C.M.; Newton, A.C. The life and death of protein kinase C. Curr. Drug Targets 2008, 9, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.E.; Fields, F.; Kunisawa, R.; Bishop, J.; Thorner, J. A candidate protein kinase C gene, PKC1, is required for the S. cerevisiae cell cycle. Cell 1990, 62, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Chen, C.; Levin, D. Saccharomyces cerevisiae PKC1 encodes a protein kinase C (PKC) homolog with a substrate specificity similar to that of mammalian PKC. J. Biol. Chem. 1994, 269, 16829–16836. [Google Scholar] [CrossRef] [PubMed]
- Giorgione, J.R.; Lin, J.H.; McCammon, J.A.; Newton, A.C. Increased membrane affinity of the C1 domain of protein kinase Cdelta compensates for the lack of involvement of its C2 domain in membrane recruitment. J. Biol. Chem. 2006, 281, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Dries, D.R.; Gallegos, L.L.; Newton, A.C. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J. Biol. Chem. 2007, 282, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef] [PubMed]
- eranen, L.M.; Dutil, E.M.; Newton, A.C. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr. Biol. 1995, 5, 1394–1403. [Google Scholar] [CrossRef]
- Tsutakawa, S.E.; Medzihradszky, K.F.; Flint, A.J.; Burlingame, A.L.; Koshland, D.E., Jr. Determination of in vivo phosphorylation sites in protein kinase C. J. Biol. Chem. 1995, 270, 26807–26812. [Google Scholar] [CrossRef]
- Gould, C.M.; Kannan, N.; Taylor, S.S.; Newton, A.C. The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J. Biol. Chem. 2009, 284, 4921–4935. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef]
- Baffi, T.R.; Lordén, G.; Wozniak, J.M.; Feichtner, A.; Yeung, W.; Kornev, A.P.; King, C.C.; Del Rio, J.C.; Limaye, A.J.; Bogomolovas, J.; et al. mTORC2 controls the activity of PKC and Akt by phosphorylating a conserved TOR interaction motif. Sci. Signal 2021, 14, eabe4509. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C as a tumor suppressor. Semin. Cancer Biol. 2018, 48, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Balendran, A.; Hare, G.R.; Kieloch, A.; Williams, M.R.; Alessi, D.R. Further evidence that 3-phosphoinositide-dependent protein kinase-1 (PDK1) is required for the stability and phosphorylation of protein kinase C (PKC) isoforms. FEBS Lett. 2000, 484, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.J.M.; Escribano, C.; Saurin, A.T.; Kostelecky, B.; Parker, P.J. PKC maturation is promoted by nucleotide pocket occupation independently of intrinsic kinase activity. Nat. Struct. Mol. Biol. 2009, 16, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Regulation of the ABC kinases by phosphorylation: Protein kinase C as a paradigm. Biochem. J. 2003, 370 Pt 2, 361–371. [Google Scholar] [CrossRef]
- Newton, A. Regulation of Conventional and Novel Protein Kinase C Isozymes by Phosphorylation and Lipids. In Protein Kinase C in Cancer Signaling and Therapy; Kazanietz, M.G., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 9–23. [Google Scholar]
- Freeley, M.; Kelleher, D.; Long, A. Regulation of Protein Kinase C function by phosphorylation on conserved and non-conserved sites. Cell Signal 2011, 23, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Kumar, S.; Tomar, M.S.; Verma, P.K.; Kumar, A.; Kumar, S.; Kumar, N.; Singh, J.P.; Acharya, A. Putative role of natural products as Protein Kinase C modulator in different disease conditions. Daru 2021, 29, 397–414. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Zhang, H.; Gao, Y.; Huang, C.; Zhou, A.; Zhou, Y.; Li, Y. Sequential posttranslational modifications regulate PKC degradation. Mol. Biol. Cell 2016, 27, 410–420. [Google Scholar] [CrossRef]
- Drummond, M.L.; Prehoda, K.E. Molecular Control of Atypical Protein Kinase C: Tipping the Balance between Self-Renewal and Differentiation. J. Mol. Biol. 2016, 428, 1455–1464. [Google Scholar] [CrossRef]
- Violin, J.D.; Zhang, J.; Tsien, R.Y.; Newton, A.C. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J. Cell Biol. 2003, 161, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, P.J.; Young, K.W.; Nahorski, S.R.; Challiss, R.A.J. Single cell analysis and temporal profiling of agonist-mediated inositol 1, 4, 5-trisphosphate, Ca2+, diacylglycerol, and protein kinase C signaling using fluorescent biosensors. J. Biol. Chem. 2005, 280, 21837–21846. [Google Scholar] [CrossRef] [PubMed]
- Uchino, M.; Sakai, N.; Kashiwagi, K.; Shirai, Y.; Shinohara, Y.; Hirose, K.; Iino, M.; Yamamura, T.; Saito, N. Isoform-specific phosphorylation of metabotropic glutamate receptor 5 by protein kinase C (PKC) blocks Ca2+ oscillation and oscillatory translocation of Ca2+-dependent PKC. J. Biol. Chem. 2004, 279, 2254–2261. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, L.L.; Kunkel, M.T.; Newton, A.C. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J. Biol. Chem. 2006, 281, 30947–30956. [Google Scholar] [CrossRef] [PubMed]
- Marín-Vicente, C.; Nicolás, F.E.; Gómez-Fernández, J.C.; Corbalán-García, S. The PtdIns (4, 5) P2 ligand itself influences the localization of PKCα in the plasma membrane of intact living cells. J. Mol. Biol. 2008, 377, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, S.; Merida, I. Diacylglycerol-dependent binding recruits PKCθ and RasGRP1 C1 domains to specific subcellular localizations in living T lymphocytes. Mol. Biol. Cell 2004, 15, 2932–2942. [Google Scholar] [CrossRef]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar]
- Antal, C.E.; Newton, A.C. Tuning the signalling output of protein kinase C. Biochem. Soc. Trans. 2014, 42, 1477–1483. [Google Scholar] [CrossRef]
- Konishi, H.; Tanaka, M.; Takemura, Y.; Matsuzaki, H.; Ono, Y.; Kikkawa, U.; Nishizuka, Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc. Natl. Acad. Sci. USA 1997, 94, 11233–11237. [Google Scholar] [CrossRef]
- Konishi, H.; Yamauchi, E.; Taniguchi, H.; Yamamoto, T.; Matsuzaki, H.; Takemura, Y.; Ohmae, K.; Kikkawa, U.; Nishizuka, Y. Phosphorylation sites of protein kinase C delta in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 6587–6592. [Google Scholar] [CrossRef]
- Adwan, T.S.; Ohm, A.M.; Jones, D.N.M.; Humphries, M.J.; Reyland, M.E. Regulated binding of importin-alpha to protein kinase Cdelta in response to apoptotic signals facilitates nuclear import. J. Biol. Chem. 2011, 286, 35716–35724. [Google Scholar] [CrossRef]
- Kaul, S.; Anantharam, V.; Yang, Y.; Choi, C.J.; Kanthasamy, A.; Kanthasamy, A.G. Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cdelta in dopaminergic neuronal cells. J. Biol. Chem. 2005, 280, 28721–28730. [Google Scholar] [CrossRef] [PubMed]
- Langeberg, L.K.; Scott, J.D. Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 2015, 16, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Finger, E.C.; Castellini, L.; Rankin, E.B.; Vilalta, M.; Krieg, A.J.; Jiang, D.; Banh, A.; Zundel, W.; Powell, M.B.; Giaccia, A.J. Hypoxic induction of AKAP12 variant 2 shifts PKA-mediated protein phosphorylation to enhance migration and metastasis of melanoma cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4441–4446. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.H.; Glantz, S.B.; Li, Y.I.N.G.; You, Y.; Rubin, C.S. Cloning and expression of an intron-less gene for AKAP 75, an anchor protein for the regulatory subunit of cAMP-dependent protein kinase II beta. J. Biol. Chem. 1992, 267, 2131–2134. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Scott, J.D. AKAP signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef]
- Marin, W. A-kinase anchoring protein 1 (AKAP1) and its role in some cardiovascular diseases. J. Mol. Cell. Cardiol. 2020, 138, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kazanietz, M.G.; Lemmon, M.A. Protein Kinase C Regulation: C1 Meets C-tail. Structure 2011, 19, 144–146. [Google Scholar] [CrossRef]
- Scott, J.D.; Newton, A.C. Shedding light on local kinase activation. BMC Biol. 2012, 10, 61. [Google Scholar] [CrossRef]
- Mukherjee, A.; Roy, S.; Saha, B.; Mukherjee, D. Spatio-Temporal Regulation of PKC Isoforms Imparts Signaling Specificity. Front. Immunol. 2016, 7, 45. [Google Scholar] [CrossRef]
- Mochly-Rosen, D.; Khaner, H.; Lopez, J.; Smith, B. Intracellular receptors for activated protein kinase C. Identification of a binding site for the enzyme. J. Biol. Chem. 1991, 266, 14866–14868. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Smith, B.L.; Chen, C.H.; Disatnik, M.H.; Ron, D. Interaction of protein kinase C with RACK1, a receptor for activated C-kinase: A role in beta protein kinase C mediated signal transduction. Biochem. Soc. Trans. 1995, 23, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Gordon, A.S. Anchoring proteins for protein kinase C: A means for isozyme selectivity. Faseb J. 1998, 12, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.D.; Qvit, N.; Mochly-Rosen, D. Peptides and peptidomimetics as regulators of protein-protein interactions. Curr. Opin. Struct. Biol. 2017, 44, 59–66. [Google Scholar] [CrossRef]
- Obsilova, V.; Obsil, T. The 14-3-3 Proteins as Important Allosteric Regulators of Protein Kinases. Int. J. Mol. Sci. 2020, 21, 8824. [Google Scholar] [CrossRef]
- Hoque, M.; Rentero, C.; Cairns, R.; Tebar, F.; Enrich, C.; Grewal, T. Annexins—Scaffolds modulating PKC localization and signaling. Cell Signal 2014, 26, 1213–1225. [Google Scholar] [CrossRef]
- Li, K.; Sun, P.; Wang, Y.; Gao, T.; Zheng, D.; Liu, A.; Ni, Y. Hsp90 interacts with Cdc37, is phosphorylated by PKA/PKC, and regulates Src phosphorylation in human sperm capacitation. Andrology 2021, 9, 185–195. [Google Scholar] [CrossRef]
- Chen, J.; Wu, M.; Zhang, X.; Zhang, W.; Zhang, Z.; Chen, L.; He, J.; Zheng, Y.; Chen, C.; Wang, F.; et al. Hepatitis B virus polymerase impairs interferon-α–induced STA T activation through inhibition of importin-α5 and protein kinase C-δ. Hepatology 2013, 57, 470–482. [Google Scholar] [CrossRef]
- Pidoux, G.; Taskén, K. Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J. Mol. Endocrinol. 2010, 44, 271–284. [Google Scholar] [CrossRef]
- Limaye, A.J.; Bendzunas, G.N.; Kennedy, E.J. Targeted disruption of PKC from AKAP signaling complexes. RSC Chem. Biol. 2021, 2, 1227–1231. [Google Scholar] [CrossRef]
- Ron, D.; Chen, C.H.; Caldwell, J.; Jamieson, L.; Orr, E.; Mochly-Rosen, D. Cloning of an intracellular receptor for protein kinase C: A homolog of the beta subunit of G proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Harrison-Lavoie, K.; Lewis, V.; Hynes, G.; Collison, K.; Nutland, E.; Willison, K. A 102 kDa subunit of a Golgi-associated particle has homology to beta subunits of trimeric G proteins. Embo J. 1993, 12, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Stenbeck, G.; Harter, C.; Brecht, A.; Herrmann, D.; Lottspeich, F.; Orci, L.; Wieland, F. beta’-COP, a novel subunit of coatomer. Embo J. 1993, 12, 2841–2845. [Google Scholar] [CrossRef]
- Csukai, M.; Chen, C.H.; De Matteis, M.A.; Mochly-Rosen, D. The coatomer protein beta’-COP, a selective binding protein (RACK) for protein kinase Cepsilon. J. Biol. Chem. 1997, 272, 29200–29206. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Mochly-Rosen, D. An Autoregulatory Region in Protein-Kinase-C—The Pseudoanchoring Site. Proc. Natl. Acad. Sci. USA 1995, 92, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Luo, J.; Mochly-Rosen, D. C2 region-derived peptides inhibit translocation and function of beta protein kinase C in vivo. J. Biol. Chem. 1995, 270, 24180–24187. [Google Scholar] [CrossRef]
- Churchill, E.N.; Qvit, N.; Mochly-Rosen, D. Rationally designed peptide regulators of protein kinase C. Trends Endocrinol. Metab. 2009, 20, 25–33. [Google Scholar]
- Kim, J.; Mochly-Rosen, D. Regulation of PKC by Protein-Protein Interactions in Cancer, in Protein Kinase C in Cancer Signaling and Therapy; Humana Press: Totowa, NJ, USA, 2010; pp. 79–103. [Google Scholar]
- Palaniyandi, S.S.; Sun, L.; Ferreira, J.C.B.; Mochly-Rosen, D. Protein kinase C in heart failure: A therapeutic target? Cardiovasc. Res. 2009, 82, 229–239. [Google Scholar] [CrossRef]
- Qvit, N.; Mochly-Rosen, D. Highly specific modulators of protein kinase C localization: Applications to heart failure. Drug Discov. Today: Dis. Mech. 2010, 7, e87–e93. [Google Scholar] [CrossRef]
- Tsunoda, S.; Sierralta, J.; Sun, Y.; Bodner, R.; Suzuki, E.; Becker, A.; Socolich, M.; Zuker, C.S. A multivalent PDZ-domain protein assembles signalling complexes in a G-protein-coupled cascade. Nature 1997, 388, 243–249. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.A.; Riegel, K.; Pompaiah, M.; Juhl, H.; Rajalingam, K. Druggable genome and precision medicine in cancer: Current challenges. Febs J. 2021, 288, 6142–6158. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992, 258, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 1984, 308, 693–698. [Google Scholar] [CrossRef]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef]
- Berenblum, I.; Shubik, P. The Role of Croton Oil Applications, Associated with a Single Painting of a Carcinogen, in Tumour Induction of the Mouse’s Skin. Br. J. Cancer 1947, 1, 379–382. [Google Scholar] [CrossRef]
- Black, A.R.; Black, J.D. Protein kinase C signaling and cell cycle regulation. Front. Immunol. 2012, 3, 423. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Antal, C.E.; Hudson, A.M.; Kang, E.; Zanca, C.; Wirth, C.; Stephenson, N.L.; Trotter, E.W.; Gallegos, L.L.; Miller, C.J.; Furnari, F.B.; et al. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell 2015, 160, 489–502. [Google Scholar] [CrossRef]
- Parker, P.J.; Brown, S.J.; Calleja, V.; Chakravarty, P.; Cobbaut, M.; Linch, M.; Marshall, J.J.T.; Martini, S.; McDonald, N.Q.; Soliman, T.; et al. Equivocal, explicit and emergent actions of PKC isoforms in cancer. Nat. Rev. Cancer 2021, 21, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Baudot, A.D.; Jeandel, P.Y.; Mouska, X.; Maurer, U.; Tartare-Deckert, S.; Raynaud, S.D.; Cassuto, J.P.; Ticchioni, M.; Deckert, M. The tyrosine kinase Syk regulates the survival of chronic lymphocytic leukemia B cells through PKCδ and proteasome-dependent regulation of Mcl-1 expression. Oncogene 2009, 28, 3261–3273. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Qiu, Y.; Coombes, K.R.; Zhang, N.; Neeley, E.S.; Ruvolo, V.R.; Hail, N.; Borthakur, G.; Konopleva, M.; Andreeff, M.; et al. Phosphorylation of GSK3α/β correlates with activation of AKT and is prognostic for poor overall survival in acute myeloid leukemia patients. BBA Clin. 2015, 4, 59–68. [Google Scholar] [CrossRef]
- Black, A.R.; Black, J.D. The complexities of PKCα signaling in cancer. Adv. Biol. Regul. 2021, 80, 100769. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Lum, M.A.; Lewis, R.E.; Black, A.R.; Black, J.D. A novel antiproliferative PKCα-Ras-ERK signaling axis in intestinal epithelial cells. J. Biol. Chem. 2022, 298, 102121. [Google Scholar] [CrossRef] [PubMed]
- Prévostel, C.; Alvaro, V.; De Boisvilliers, F.; Martin, A.; Jaffiol, C.; Joubert, D. The natural protein kinase C alpha mutant is present in human thyroid neoplasms. Oncogene 1995, 11, 669–674. [Google Scholar] [PubMed]
- Prevostel, C.; Alice, V.; Joubert, D.; Parker, P.J. Protein kinase C(alpha) actively downregulates through caveolae-dependent traffic to an endosomal compartment. J. Cell Sci. 2000, 113, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Lahn, M.M.; Sundell, K.L. The role of protein kinase C-alpha (PKC-alpha) in melanoma. Melanoma Res. 2004, 14, 85–89. [Google Scholar] [CrossRef]
- Palazzo, E.; Kellett, M.D.; Cataisson, C.; Bible, P.W.; Bhattacharya, S.; Sun, H.-W.; Gormley, A.C.; Yuspa, S.H.; Morasso, M.I. A novel DLX3–PKC integrated signaling network drives keratinocyte differentiation. Cell Death Differ. 2017, 24, 717–730. [Google Scholar] [CrossRef]
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Urtreger, A.J.; Kazanietz, M.G.; Bal de Kier Joffé, E.D. Contribution of individual PKC isoforms to breast cancer progression. IUBMB Life 2012, 64, 18–26. [Google Scholar] [CrossRef]
- Sadeghi, M.M.; Salama, M.F.; Hannun, Y.A. Protein Kinase C as a Therapeutic Target in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 5527. [Google Scholar] [CrossRef] [PubMed]
- Apostolatos, A.H.; Ratnayake, W.S.; Win-Piazza, H.; Apostolatos, C.A.; Smalley, T.; Kang, L.; Salup, R.; Hill, R.; Acevedo-Duncan, M. Inhibition of atypical protein kinase C-ι effectively reduces the malignancy of prostate cancer cells by downregulating the NF-κB signaling cascade. Int. J. Oncol. 2018, 53, 1836–1846. [Google Scholar] [CrossRef]
- Dowling, C.M.; Phelan, J.; Callender, J.A.; Cathcart, M.C.; Mehigan, B.; McCormick, P.; Dalton, T.; Coffey, J.C.; Newton, A.C.; O’sullivan, J.; et al. Protein kinase C beta II suppresses colorectal cancer by regulating IGF-1 mediated cell survival. Oncotarget 2016, 7, 20919–20933. [Google Scholar] [CrossRef] [PubMed]
- Engers, R.; Mrzyk, S.; Springer, E.; Fabbro, D.; Weissgerber, G.; Gerharz, C.D.; Gabbert, H.E. Protein kinase C in human renal cell carcinomas: Role in invasion and differential isoenzyme expression. Br. J. Cancer 2000, 82, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gobe, G. Protein kinase C activation and its role in kidney disease. Nephrology 2006, 11, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.-S.; Huang, C.-Y.; Chen, J.-Y.; Kang, W.-Y.; Lin, Y.-C.; Shiu, Y.-S.; Chuang, S.-J.; Yu, H.-J.; Lai, M.-K.; Tsai, Y.-C.; et al. Down-regulation of PKCζ in renal cell carcinoma and its clinicopathological implications. J. Biomed. Sci. 2012, 19, 39. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.M.A.; Patel, R.; Acevedo-Duncan, M. Protein Kinase C-ζ stimulates colorectal cancer cell carcinogenesis via PKC-ζ/Rac1/Pak1/β-Catenin signaling cascade. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Ahmedin, J.D.V.M. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmström, P.U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder cancer. Nat. Rev. Dis. Primers 2017, 3, 17022. [Google Scholar] [CrossRef]
- Jiang, Z.; Kong, C.; Zhang, Z.; Zhu, Y.; Zhang, Y.; Chen, X. Reduction of protein kinase C α (PKC-α) promote apoptosis via down-regulation of Dicer in bladder cancer. J. Cell. Mol. Med. 2015, 19, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Tachibana, Y.; Inokuchi, J.; Kang, J.-H.; Murata, M.; Eto, M. Identification of Activated Protein Kinase Cα (PKCα) in the Urine of Orthotopic Bladder Cancer Xenograft Model as a Potential Biomarker for the Diagnosis of Bladder Cancer. Int. J. Mol. Sci. 2021, 22, 9276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Zhang, H.; Liu, Y.; Yin, L.; Liu, X.; Li, X.; Yu, X.; Yao, J.; Zhang, Z.; et al. Exploring the five different genes associated with PKCα in bladder cancer based on gene expression microarray. J. Cell Mol. Med. 2021, 25, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Padala, S.A.; Barsouk, A.; Rawla, P. Epidemiology of Non-Hodgkin’s Lymphoma. Med. Sci. 2021, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Zeng, Q.; Zhang, X.; Ruan, W. Trends Analysis of Non-Hodgkin Lymphoma at the National, Regional, and Global Level, 1990–2019: Results from the Global Burden of Disease Study 2019. Front. Med. 2021, 8, 738693. [Google Scholar] [CrossRef] [PubMed]
- Su, T.T.; Guo, B.; Rawlings, D.J. Emerging roles for PKC isoforms in immune cell function. Mol. Interv. 2002, 2, 141–144. [Google Scholar] [CrossRef] [PubMed]
- McMahon, G. VEGF receptor signaling in tumor angiogenesis. Oncologist 2000, 5, 3–10. [Google Scholar] [CrossRef]
- Giles, F.J.; Vose, J.M.; Do, K.-A.; Johnson, M.M.; Manshouri, T.; Bociek, G.; Bierman, P.J.; O’Brien, S.M.; Kantarjian, H.M.; Armitage, J.O.; et al. Clinical relevance of circulating angiogenic factors in patients with non-Hodgkin’s lymphoma or Hodgkin’s lymphoma. Leuk. Res. 2004, 28, 595–604. [Google Scholar] [CrossRef]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Chan, W.C.; Aoun, P.; Cochran, G.T.; Pan, Z.; Smith, L.M.; Lynch, J.C.; Bociek, R.G.; et al. Expression of PKC-beta or cyclin D2 predicts for inferior survival in diffuse large B-cell lymphoma. Mod. Pathol. 2005, 18, 1377–1384. [Google Scholar] [CrossRef]
- Berditchevski, F.; Fennell, E.; Murray, P.G. Calcium-dependent signalling in B-cell lymphomas. Oncogene 2021, 40, 6321–6328. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.D.; Pantanowitz, L.; Hornick, J.L. A worldwide journey of thyroid cancer incidence centred on tumour histology. Lancet Diabetes Endocrinol. 2021, 9, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Prévostel, C.; Martin, A.; Alvaro, V.; Jaffiol, C.; Joubert, D. Protein kinase C alpha and tumorigenesis of the endocrine gland. Horm. Res. 1997, 47, 140–144. [Google Scholar] [PubMed]
- Alvaro, V.; Prévostel, C.; Joubert, D.; Slosberg, E.; Weinstein, B.I. Ectopic expression of a mutant form of PKCalpha originally found in human tumors: Aberrant subcellular translocation and effects on growth control. Oncogene 1997, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.N.; Knauf, J.A.; Gonsky, R.; Wang, M.; Lai, E.H.; Chissoe, S.; Fagin, J.A.; Korenberg, J.R. From amplification to gene in thyroid cancer: A high-resolution mapped bacterial-artificial-chromosome resource for cancer chromosome aberrations guides gene discovery after comparative genome hybridization. Am. J. Hum. Genet. 1998, 63, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Knauf, J.A.; Ward, L.S.; Nikiforov, Y.E.; Nikiforova, M.; Puxeddu, E.; Medvedovic, M.; Liron, T.; Mochly-Rosen, D.; Fagin, J.A. Isozyme-specific abnormalities of PKC in thyroid cancer: Evidence for post-transcriptional changes in PKC epsilon. J. Clin. Endocrinol. Metab. 2002, 87, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gong, T.T.; Liu, F.H.; Jiang, Y.T.; Sun, H.; Ma, X.X.; Zhao, Y.H.; Wu, Q.J. Global, Regional, and National Burden of Endometrial Cancer, 1990–2017: Results from the Global Burden of Disease Study, 2017. Front. Oncol. 2019, 9, 1440. [Google Scholar] [CrossRef]
- Haughian, J.M.; Reno, E.M.; Thorne, A.M.; Bradford, A.P. Protein kinase C alpha-dependent signaling mediates endometrial cancer cell growth and tumorigenesis. Int. J. Cancer 2009, 125, 2556–2564. [Google Scholar] [CrossRef]
- Reno, E.M.; Haughian, J.M.; Dimitrova, I.K.; Jackson, T.A.; Shroyer, K.R.; Bradford, A.P. Analysis of protein kinase C delta (PKC delta) expression in endometrial tumors. Human. Pathol. 2008, 39, 21–29. [Google Scholar] [CrossRef]
- Koo, K.-H.; Jeong, W.-J.; Cho, Y.-H.; Park, J.-C.; Min, D.S.; Choi, K.-Y. K-Ras stabilization by estrogen via PKCδ is involved in endometrial tumorigenesis. Oncotarget 2015, 6, 21328–21340. [Google Scholar] [CrossRef]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-X.; Zhao, C.-F.; Chen, W.-B.; Liu, Q.-C.; Li, Q.-W.; Lin, Y.-Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.; Cornford, P.A.; Dodson, A.; Neoptolemos, J.P.; Foster, C.S. Expression Patterns of Protein Kinase C Isoenzymes Are Characteristically Modulated in Chronic Pancreatitis and Pancreatic Cancer. Am. J. Clin. Pathol. 2003, 119, 392–402. [Google Scholar] [CrossRef]
- Storz, P. Targeting protein kinase C subtypes in pancreatic cancer. Expert. Rev. Anticancer. Ther. 2015, 15, 433–438. [Google Scholar] [CrossRef]
- Scotti, M.L.; Bamlet, W.R.; Smyrk, T.C.; Fields, A.P.; Murray, N.R. Protein kinase Ciota is required for pancreatic cancer cell transformed growth and tumorigenesis. Cancer Res. 2010, 70, 2064–2074. [Google Scholar] [CrossRef]
- Mauro, L.V.; Grossoni, V.C.; Urtreger, A.J.; Yang, C.; Colombo, L.L.; Morandi, A.; Pallotta, M.G.; Kazanietz, M.G.; de Kier Joffé, E.D.B.; Puricelli, L.L. PKCdelta promotes tumoral progression of human ductal pancreatic cancer. Pancreas 2010, 39, e31–e41. [Google Scholar] [CrossRef]
- Dowling, C.M.; Hayes, S.L.; Phelan, J.J.; Cathcart, M.C.; Finn, S.P.; Mehigan, B.; McCormick, P.; Coffey, J.C.; O’sullivan, J.; Kiely, P.A. Expression of protein kinase C gamma promotes cell migration in colon cancer. Oncotarget 2017, 8, 72096–72107. [Google Scholar] [CrossRef]
- Zhang, L.L.; Cao, F.F.; Wang, Y.; Meng, F.L.; Zhang, Y.; Zhong, D.S.; Zhou, Q.H. The protein kinase C (PKC) inhibitors combined with chemotherapy in the treatment of advanced non-small cell lung cancer: Meta-analysis of randomized controlled trials. Clin. Transl. Oncol. 2015, 17, 371–377. [Google Scholar] [CrossRef]
- Soares, A.C.; Fonseca, D.A. Cardiovascular diseases: A therapeutic perspective around the clock. Drug Discov. Today 2020, 25, 1086–1098. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics—2021 update: A report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Singh, V.P.; Baker, K.M. Kinase inhibitors for cardiovascular disease. J. Mol. Cell. Cardiol. 2007, 42, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fuller, S.J.; Osborne, S.A.; Leonard, S.J.; Hardyman, M.A.; Vaniotis, G.; Allen, B.G.; Sugden, P.H.; Clerk, A. Cardiac protein kinases: The cardiomyocyte kinome and differential kinase expression in human failing hearts. Cardiovasc. Res. 2015, 108, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Du, C.; Wei, T.; Shan, T.; Wang, L. Protein kinases in cardiovascular diseases. Chin. Med. J. 2022, 135, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Churchill, E.; Budas, G.; Vallentin, A.; Koyanagi, T.; Mochly-Rosen, D. PKC isozymes in chronic cardiac disease: Possible therapeutic targets? Annu. Rev. Pharmacol. Toxicol. 2008, 48, 569–599. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Cardiac actions of protein kinase C isoforms. Physiology 2012, 27, 130–139. [Google Scholar] [CrossRef]
- Pucéat, M.; Hilal-Dandan, R.; Strulovici, B.; Brunton, L.; Brown, J. Differential regulation of protein kinase C isoforms in isolated neonatal and adult rat cardiomyocytes. J. Biol. Chem. 1994, 269, 16938–16944. [Google Scholar] [CrossRef] [PubMed]
- Rybin, V.O.; Steinberg, S.F. Protein kinase C isoform expression and regulation in the developing rat heart. Circ. Res. 1994, 74, 299–309. [Google Scholar] [CrossRef]
- Bowling, N.; Walsh, R.A.; Song, G.; Estridge, T.; Sandusky, G.E.; Fouts, R.L.; Mintze, K.; Pickard, T.; Roden, R.; Bristow, M.R.; et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation 1999, 99, 384–391. [Google Scholar] [CrossRef]
- Simonis, G.; Briem, S.K.; Schoen, S.P.; Bock, M.; Marquetant, R.; Strasser, R.H. Protein kinase C in the human heart: Differential regulation of the isoforms in aortic stenosis or dilated cardiomyopathy. Mol. Cell. Biochem. 2007, 305, 103–111. [Google Scholar] [CrossRef]
- Singh, R.M.; Cummings, E.; Pantos, C.; Singh, J. Protein kinase C and cardiac dysfunction: A review. Heart Fail. Rev. 2017, 22, 843–859. [Google Scholar] [CrossRef]
- Weeks, K.L.; McMullen, J.R. Divergent Effects of PKC (Protein Kinase C) α in the Human and Animal Heart? Therapeutic Implications for PKC Inhibitors in Cardiac Patients. Circ. Genom. Precis. Med. 2018, 11, e002104. [Google Scholar] [CrossRef] [PubMed]
- Wetsel, W.; Khan, W.; Merchenthaler, I.; Rivera, H.; Halpern, A.; Phung, H.; Negro-Vilar, A.; Hannun, Y. Tissue and cellular distribution of the extended family of protein kinase C isoenzymes. J. Cell Biol. 1992, 117, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Parker, P.J.; Sugden, P.H. Characterization of protein kinase C isotype expression in adult rat heart. Protein kinase C-epsilon is a major isotype present, and it is activated by phorbol esters, epinephrine, and endothelin. Circ. Res. 1993, 72, 757–767. [Google Scholar] [CrossRef]
- Talosi, L.; Kranias, E.G. Effect of alpha-adrenergic stimulation on activation of protein kinase C and phosphorylation of proteins in intact rabbit hearts. Circ. Res. 1992, 70, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Ping, P.; Tang, X.L.; Manchikalapudi, S.; Rizvi, A.; Zhang, J.; Takano, H.; Wu, W.J.; Teschner, S.; Bolli, R. Direct evidence that protein kinase C plays an essential role in the development of late preconditioning against myocardial stunning in conscious rabbits and that epsilon is the isoform involved. J. Clin. Investig. 1998, 101, 2182–2198. [Google Scholar] [CrossRef] [PubMed]
- Karamchand, P.; Ball, N.A.; Dorn, G.W.; Walsh, R.A. Left ventricular stretch stimulates angiotensin II--mediated phosphatidylinositol hydrolysis and protein kinase C epsilon isoform translocation in adult guinea pig hearts. Circ. Res. 1997, 81, 643–650. [Google Scholar]
- Cai, J.J.; Lee, H.C. Protein kinase C isozyme-specific modulation of cyclic AMP-dependent phosphodiesterase in hypertrophic cardiomyopathic hamster hearts. Mol. Pharmacol. 1996, 49, 81–88. [Google Scholar]
- Domenech, R.J.; Macho, P.; Vélez, D.; Sánchez, G.; Liu, X.; Dhalla, N. Tachycardia preconditions infarct size in dogs: Role of adenosine and protein kinase C. Circulation 1998, 97, 786–794. [Google Scholar] [CrossRef]
- Goldberg, M.; Steinberg, S.F. Tissue-specific developmental regulation of protein kinase C isoforms. Biochem. Pharmacol. 1996, 51, 1089–1093. [Google Scholar] [CrossRef]
- Chen, C.-H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.-H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.B.; Mochly-Rosen, D.; Boutjdir, M. Regulation of cardiac excitability by protein kinase C isozymes. Front. Biosci. (Sch. Ed.) 2012, 4, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Churchill, E.N.; Mochly-Rosen, D. The roles of PKCdelta and epsilon isoenzymes in the regulation of myocardial ischaemia/reperfusion injury. Biochem. Soc. Trans. 2007, 35 Pt 5, 1040–1042. [Google Scholar] [CrossRef] [PubMed]
- Qvit, N.; Disatnik, M.-H.; Sho, E.; Mochly-Rosen, D. Selective phosphorylation inhibitor of delta protein kinase C-pyruvate dehydrogenase kinase protein-protein interactions: Application for myocardial injury in vivo. J. Am. Chem. Soc. 2016, 138, 7626–7635. [Google Scholar] [CrossRef]
- Miao, L.-N.; Pan, D.; Shi, J.; Du, J.-P.; Chen, P.-F.; Gao, J.; Yu, Y.; Shi, D.-Z.; Guo, M. Role and Mechanism of PKC-δ for Cardiovascular Disease: Current Status and Perspective. Front. Cardiovasc. Med. 2022, 9, 816369. [Google Scholar] [CrossRef] [PubMed]
- Paumelle, R.; Blanquart, C.; Briand, O.; Barbier, O.; Duhem, C.; Woerly, G.; Percevault, F.; Fruchart, J.C.; Dombrowicz, D.; Glineur, C.; et al. Acute antiinflammatory properties of statins involve peroxisome proliferator-activated receptor-alpha via inhibition of the protein kinase C signaling pathway. Circ. Res. 2006, 98, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Ruboxistaurin: LY 333531. Drugs R D 2007, 8, 193–199. [CrossRef]
- Wei, L.; Yin, Z.; Yuan, Y.; Hwang, A.; Lee, A.; Sun, D.; Li, F.; Di, C.; Zhang, R.; Cao, F.; et al. A PKC-β inhibitor treatment reverses cardiac microvascular barrier dysfunction in diabetic rats. Microvasc. Res. 2010, 80, 158–165. [Google Scholar] [CrossRef]
- Connelly, K.A.; Kelly, D.J.; Zhang, Y.; Prior, D.L.; Advani, A.; Cox, A.J.; Thai, K.; Krum, H.; Gilbert, R.E. Inhibition of Protein Kinase C–β by Ruboxistaurin Preserves Cardiac Function and Reduces Extracellular Matrix Production in Diabetic Cardiomyopathy. Circ. Heart Fail. 2009, 2, 129–137. [Google Scholar] [CrossRef]
- Ladage, D.; Tilemann, L.; Ishikawa, K.; Correll, R.N.; Kawase, Y.; Houser, S.R.; Molkentin, J.D.; Hajjar, R.J. Inhibition of PKCα/β with ruboxistaurin antagonizes heart failure in pigs after myocardial infarction injury. Circ. Res. 2011, 109, 1396–1400. [Google Scholar] [CrossRef]
- Zatta, A.J.; Kin, H.; Lee, G.; Wang, N.; Jiang, R.; Lust, R.; Reeves, J.G.; Mykytenko, J.; Guyton, R.A.; Zhao, Z.-Q.; et al. Infarct-sparing effect of myocardial postconditioning is dependent on protein kinase C signalling. Cardiovasc. Res. 2006, 70, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P. Rottlerin: An inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol. Sci. 2007, 28, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Fernández-Hernando, C.; Lai, J.H. Protein kinase C isoforms in atherosclerosis: Pro- or anti-inflammatory? Biochem. Pharmacol. 2014, 88, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Lien, C.-F.; Chen, S.-J.; Tsai, M.-C.; Lin, C.-S. Potential Role of Protein Kinase C in the Pathophysiology of Diabetes-Associated Atherosclerosis. Front. Pharmacol. 2021, 12, 716332. [Google Scholar] [CrossRef]
- Patel, J. Heart failure population health considerations. Am. J. Manag. Care 2021, 27 (Suppl. 9), S191–S195. [Google Scholar]
- Vlahos, C.J.; McDowell, S.A.; Clerk, A. Kinases as therapeutic targets for heart failure. Nat. Rev. Drug Discov. 2003, 2, 99–113. [Google Scholar] [CrossRef]
- Aslam, N. Increase in PKCα Activity during Heart Failure Despite the Stimulation of PKCα Braking Mechanism. Int. J. Mol. Sci. 2020, 21, 2561. [Google Scholar] [CrossRef]
- Liu, Q.; Molkentin, J.D. Protein kinase Cα as a heart failure therapeutic target. J. Mol. Cell Cardiol. 2011, 51, 474–478. [Google Scholar] [CrossRef]
- Sheng, J.; Chen, Y.; Chang, H.; Wang, Y.; Jiao, B.; Yu, Z. Multisite phosphorylation of Bcl-2 via protein kinase Cδ facilitates apoptosis of hypertrophic cardiomyocytes. Clin. Exp. Pharmacol. Physiol. 2014, 41, 891–901. [Google Scholar] [CrossRef]
- Fryer, L.; Holness, M.; Decock, J.; Sugden, M. Cardiac protein kinase C expression in two models of cardiac hypertrophy associated with an activated cardiac renin-angiotensin system: Effects of experimental hyperthyroidism and genetic hypertension (the mRen-2 rat). J. Endocrinol. 1998, 158, 27–33. [Google Scholar] [CrossRef]
- Cain, A.E.; Khalil, R.A. Pathophysiology of essential hypertension: Role of the pump, the vessel, and the kidney. Semin. Nephrol. 2002, 22, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Salamanca, D.A.; Khalil, R.A. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem. Pharmacol. 2005, 70, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Khalil, R.A. Role of Protein Kinase C and Related Pathways in Vascular Smooth Muscle Contraction and Hypertension. Neurovascular Med. Pursuing Cell. Longev. Healthy Aging 2008, 21. [Google Scholar] [CrossRef]
- Ringvold, H.C.; Khalil, R.A. Protein Kinase C as Regulator of Vascular Smooth Muscle Function and Potential Target in Vascular Disorders. Adv. Pharmacol. 2017, 78, 203–301. [Google Scholar]
- Khalil, R.A.; Lajoie, C.; Morgan, K.G. In situ determination of [Ca2+] i threshold for translocation of the alpha-protein kinase C isoform. Am. J. Physiol.-Cell Physiol. 1994, 266, C1544–C1551. [Google Scholar] [CrossRef]
- Liou, Y.M.; Morgan, K.G. Redistribution of protein kinase C isoforms in association with vascular hypertrophy of rat aorta. Am. J. Physiol. 1994, 267, C980–C989. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Shan, M.; Zhou, Y.; Huang, Y.; Shi, L. Aerobic exercise-induced inhibition of PKCα/CaV1.2 pathway enhances the vasodilation of mesenteric arteries in hypertension. Arch. Biochem. Biophys. 2019, 678, 108191. [Google Scholar] [CrossRef]
- Inagaki, K.; Iwanaga, Y.; Sarai, N.; Onozawa, Y.; Takenaka, H.; Mochly-Rosen, D.; Kihara, Y. Tissue angiotensin II during progression or ventricular hypertrophy to heart failure in hypertensive rats; differential effects on PKC epsilon and PKC beta. J. Mol. Cell. Cardiol. 2002, 34, 1377–1385. [Google Scholar] [CrossRef]
- Palaniyandi, S.S.; Ferreira, J.C.B.; Brum, P.C.; Mochly-Rosen, D. PKC beta II inhibition attenuates myocardial infarction induced heart failure and is associated with a reduction of fibrosis and pro-inflammatory responses. J. Cell. Mol. Med. 2011, 15, 1769–1777. [Google Scholar] [CrossRef]
- Palaniyandi, S.S.; Inagaki, K.; Mochly-Rosen, D. Mast cells and epsilonPKC: A role in cardiac remodeling in hypertension-induced heart failure. J. Mol. Cell. Cardiol. 2008, 45, 779–786. [Google Scholar] [CrossRef]
- Phipps, M.S.; Cronin, C.A. Management of acute ischemic stroke. BMJ 2020, 368, l6983. [Google Scholar] [CrossRef] [PubMed]
- Crumrine, R.C.; Dubyak, G.; LaManna, J.C. Decreased protein kinase C activity during cerebral ischemia and after reperfusion in the adult rat. J. Neurochem. 1990, 55, 2001–2007. [Google Scholar] [CrossRef] [PubMed]
- Domańska-Janik, K.; Zalewska, T. Effect of brain ischemia on protein kinase C. J. Neurochem. 1992, 58, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-H.; Messing, R.O. Protein Kinase C Isozymes in Stroke. Trends Cardiovasc. Med. 2005, 15, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.; Raval, A.P.; Dembner, J.M.; Pérez-Pinzón, M.A.; Steinberg, G.K.; Yenari, M.A.; Mochly-Rosen, D. Protein kinase C delta mediates cerebral reperfusion injury in vivo. J. Neurosci. 2004, 24, 6880–6888. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.; Mochly-Rosen, D. The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke 2005, 36, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Grune, J.; Yamazoe, M.; Nahrendorf, M. Electroimmunology and cardiac arrhythmia. Nat. Rev. Cardiol. 2021, 18, 547–564. [Google Scholar] [CrossRef]
- Kida, K.; Ichinose, F. Preventing ischemic brain injury after sudden cardiac arrest using NO inhalation. Crit. Care 2014, 18, 1–6. [Google Scholar] [CrossRef]
- Damluji, A.A.; Al-Damluji, M.S.; Pomenti, S.; Zhang, T.J.; Cohen, M.G.; Mitrani, R.D.; Moscucci, M.; Myerburg, R.J. Health Care Costs After Cardiac Arrest in the United States. Circ. Arrhythmia Electrophysiol. 2018, 11, e005689. [Google Scholar] [CrossRef]
- Perez-Pinzon, M.A.; Raval, A.P.; Dave, K.R. Protein kinase C and synaptic dysfunction after cardiac arrest. Pathophysiology 2005, 12, 29–34. [Google Scholar] [CrossRef]
- Lu, L.; Liu, M.; Sun, R.; Zheng, Y.; Zhang, P. Myocardial Infarction: Symptoms and Treatments. Cell Biochem. Biophys. 2015, 72, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hahn, H.; Wu, G.; Chen, C.H.; Liron, T.; Schechtman, D.; Cavallaro, G.; Banci, L.; Guo, Y.; Bolli, R.; et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc. Natl. Acad. Sci. USA 2001, 98, 11114–11119. [Google Scholar] [CrossRef] [PubMed]
- Churchill, E.N.; Ferreira, J.C.; Brum, P.C.; Szweda, L.I.; Mochly-Rosen, D. Ischaemic preconditioning improves proteasomal activity and increases the degradation of deltaPKC during reperfusion. Cardiovasc. Res. 2010, 85, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Jirousek, M.R.; Koya, D.; Takagi, C.; Xia, P.; Clermont, A.; Bursell, S.E.; Kern, T.S.; Ballas, L.M.; Heath, W.F.; et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science 1996, 272, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.; Zidovetzki, R. Amplification of diacylglycerol activation of protein kinase C by cholesterol. Biophys. J. 2008, 94, 4700–4710. [Google Scholar] [CrossRef]
- Gineste, R.; Sirvent, A.; Paumelle, R.; Helleboid, S.; Aquilina, A.; Darteil, R.; Hum, D.W.; Fruchart, J.-C.; Staels, B. Phosphorylation of farnesoid X receptor by protein kinase C promotes its transcriptional activity. Mol. Endocrinol. 2008, 22, 2433–2447. [Google Scholar] [CrossRef]
- Huang, W.; Bansode, R.; Mehta, M.; Mehta, K.D. Loss of protein kinase Cβ function protects mice against diet-induced obesity and development of hepatic steatosis and insulin resistance. Hepatology 2009, 49, 1525–1536. [Google Scholar] [CrossRef]
- Araki, T.; Hayashi, M.; Saruta, T. Cloning and characterization of a novel gene promoting ureteric bud branching in the metanephros. Kidney Int. 2003, 64, 1968–1977. [Google Scholar] [CrossRef]
- Lang, D.; Terstesse, M.; Dohle, F.; Bangen, P.; Banas, B.; Pauels, H.-G.; Heidenreich, S. Protein kinase C (PKC) dependent induction of tissue factor (TF) by mesangial cells in response to inflammatory mediators and release during apoptosis. Br. J. Pharmacol. 2002, 137, 1116–1124. [Google Scholar] [CrossRef]
- Noh, H.; King, G. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. 2007, 72, S49–S53. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R. Protein kinase C-beta inhibition for diabetic kidney disease. Diabetes Res. Clin. Pract. 2008, 82 (Suppl. 1), S70–S74. [Google Scholar] [CrossRef] [PubMed]
- Brenner, W.; Färber, G.; Herget, T.; Wiesner, C.; Hengstler, J.G.; Thüroff, J.W. Protein kinase C eta is associated with progression of renal cell carcinoma (RCC). Anticancer Res. 2003, 23, 4001–4006. [Google Scholar] [PubMed]
- Dempsey, E.C.; Newton, A.C.; Mochly-Rosen, D.; Fields, A.P.; Reyland, M.E.; Insel, P.A.; Messing, R.O.; Jackson, W.F.; Boerman, E.M.; Gottipati, K.R.; et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L429–L438. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, E.C.; Cool, C.D.; Littler, C.M. Lung disease and PKCs. Pharmacol. Res. 2007, 55, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, Y.; Fan, Z. Cellular Signal Transduction Pathways Involved in Acute Lung Injury Induced by Intestinal Ischemia-Reperfusion. Oxidative Med. Cell. Longev. 2021, 2021, 9985701. [Google Scholar] [CrossRef] [PubMed]
- Vachier, I.; Chanez, P.; Radeau, T.; Le Doucen, C.; Leger, C.; Godard, P. Cellular protein kinase C activity in asthma. Am. J. Respir. Crit. Care Med. 1997, 155, 1211–1216. [Google Scholar] [CrossRef]
- Do, J.-S.; Park, K.-S.; Seo, H.-J.; Kim, J.-H.; Hwang, J.-K.; Song, E.-J.; Nam, S.-Y. Therapeutic target validation of protein kinase C(PKC)-zeta for asthma using a mouse model. Int. J. Mol. Med. 2009, 23, 561–566. [Google Scholar]
- Mousavi, S.R.; Ahmadi, A.; Jamalkandi, S.A.; Salimian, J. Involvement of microRNAs in physiological and pathological processes in asthma. J. Cell Physiol. 2019, 234, 21547–21559. [Google Scholar] [CrossRef]
- Abdel-Halim, M.; Darwish, S.S.; ElHady, A.K.; Hoppstädter, J.; Abadi, A.H.; Hartmann, R.W.; Kiemer, A.K.; Engel, M. Pharmacological inhibition of protein kinase C (PKC)ζ downregulates the expression of cytokines involved in the pathogenesis of chronic obstructive pulmonary disease (COPD). Eur. J. Pharm. Sci. 2016, 93, 405–409. [Google Scholar] [CrossRef]
- Wang, J.; Sun, L.; Nie, Y.; Duan, S.; Zhang, T.; Wang, W.; Ye, R.D.; Hou, S.; Qian, F. Protein Kinase C δ (PKCδ) Attenuates Bleomycin Induced Pulmonary Fibrosis via Inhibiting NF-κB Signaling Pathway. Front. Physiol. 2020, 11, 367. [Google Scholar] [CrossRef] [PubMed]
- Mondrinos, M.J.; Kennedy, P.A.; Lyons, M.; Deutschman, C.S.; Kilpatrick, L.E. Protein kinase C and acute respiratory distress syndrome. Shock 2013, 39, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Hrenak, J.; Simko, F. Renin–Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8038. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.-C.; Fang, Y.-F.; Yamaguchi, H.; Wang, W.-J.; Chen, T.-C.; Hong, X.; Ke, B.; Xia, W.; Wei, Y.; Zha, Z.; et al. Targeting PKCδ as a Therapeutic Strategy against Heterogeneous Mechanisms of EGFR Inhibitor Resistance in EGFR-Mutant Lung Cancer. Cancer Cell 2018, 34, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Kishimoto, A.; Takai, Y.; Nishizuka, Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. II. Proenzyme and its activation by calcium-dependent protease from rat brain. J. Biol. Chem. 1977, 252, 7610–7616. [Google Scholar] [CrossRef] [PubMed]
- Arts, B.; Jabben, N.; Krabbendam, L.; van Os, J. Meta-analyses of cognitive functioning in euthymic bipolar patients and their first-degree relatives. Psychol. Med. 2008, 38, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Forbes, N.F.; Carrick, L.A.; McIntosh, A.M.; Lawrie, S.M. Working memory in schizophrenia: A meta-analysis. Psychol. Med. 2009, 39, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.N.; Rizavi, H.S.; Ren, X. Protein and mRNA expression of protein kinase C (PKC) in the postmortem brain of bipolar and schizophrenic subjects. J. Psychiatr. Res. 2020, 130, 362–371. [Google Scholar] [CrossRef]
- Young, K.W.; Garro, M.A.; Challiss, R.J.; Nahorski, S.R. NMDA-receptor regulation of muscarinic-receptor stimulated inositol 1, 4, 5-trisphosphate production and protein kinase C activation in single cerebellar granule neurons. J. Neurochem. 2004, 89, 1537–1546. [Google Scholar] [CrossRef]
- Choi, B.; Chae, H.; Park, T.; Oh, J.; Lim, J.; Kang, S.; Ha, H.; Kim, K. Protein kinase C regulates the activity and stability of serotonin N-acetyltransferase. J. Neurochem. 2004, 90, 442–454. [Google Scholar] [CrossRef]
- Poole, A.W.; Pula, G.; Hers, I.; Crosby, D.; Jones, M.L. PKC-interacting proteins: From function to pharmacology. Trends Pharmacol. Sci. 2004, 25, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.K.; Alkon, D.L. Protein kinase C activators as synaptogenic and memory therapeutics. Arch. Der Pharm. An. Int. J. Pharm. Med. Chem. 2009, 342, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.M.; Kornberg, M.D. Targeting PKC in microglia to promote remyelination and repair in the CNS. Curr. Opin. Pharmacol. 2022, 62, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Battaini, F.; Pascale, A. The different facets of protein kinases C: Old and new players in neuronal signal transduction pathways. Pharmacol. Res. 2006, 54, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Govoni, S.; Amadio, M.; Battaini, F.; Pascale, A. Senescence of the brain: Focus on cognitive kinases. Curr. Pharm. Des. 2010, 16, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Geribaldi-Doldán, N.; Gómez-Oliva, R.; Domínguez-García, S.; Nunez-Abades, P.; Castro, C. Protein Kinase C: Targets to Regenerate Brain Injuries? Front. Cell Dev. Biol. 2019, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Neuroprotective effect of protein kinase Cδ inhibitor rottlerin in cell culture and animal models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2007, 322, 913–922. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Deierborg, T.; Kavanagh, E.; Persson, A.; Hajji, N.; Garcia-Quintanilla, A.; Cano, J.; Brundin, P.; Englund, E.; Venero, J.L.; et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011, 472, 319–324. [Google Scholar] [CrossRef]
- Kaleli, H.N.; Ozer, E.; Kaya, V.O.; Kutlu, O. Protein kinase C isozymes and autophagy during neurodegenerative disease progression. Cells 2020, 9, 553. [Google Scholar] [CrossRef]
- Laperle, A.H.; Sances, S.; Yucer, N.; Dardov, V.J.; Garcia, V.J.; Ho, R.; Fulton, A.N.; Jones, M.R.; Roxas, K.M.; Avalos, P.; et al. iPSC modeling of young-onset Parkinson’s disease reveals a molecular signature of disease and novel therapeutic candidates. Nat. Med. 2020, 26, 289–299. [Google Scholar] [CrossRef]
- Giusto, E.; Yacoubian, T.A.; Greggio, E.; Civiero, L. Pathways to Parkinson’s disease: A spotlight on 14-3-3 proteins. NPJ Park. Dis. 2021, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-K.; Alkon, D.L. Activation of Protein Kinase C Isozymes for the Treatment of Dementias. In Advances in Pharmacology; Michaelis, E.K., Michaelis, M.L., Eds.; Academic Press: Cambridge, MA, USA, 2012; pp. 273–302. [Google Scholar]
- Sun, M.-K.; Alkon, D.L. Chapter Two—The “Memory Kinases”: Roles of PKC Isoforms in Signal Processing and Memory Formation. In Progress in Molecular Biology and Translational Science; Khan, Z.U., Muly, E.C., Eds.; Academic Press: Cambridge, MA, USA, 2014; pp. 31–59. [Google Scholar]
- Sweitzer, S.M.; Wong, S.M.; Peters, M.C.; Mochly-Rosen, D.; Yeomans, D.C.; Kendig, J.J. Protein kinase C epsilon and gamma: Involvement in formalin-induced nociception in neonatal rats. J. Pharmacol. Exp. Ther. 2004, 309, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Velzquez, K.T.; Mohammad, H.; Sweitzer, S.M. Protein kinase C in pain: Involvement of multiple isoforms. Pharmacol. Res. 2007, 55, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Leitges, M.; Gereau, R.W. Isozyme-specific effects of protein kinase C in pain modulation. Anesthesiology 2011, 115, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Giraud, F.; Pereira, E.; Anizon, F.; Moreau, P. Recent Advances in pain management: Relevant protein kinases and their inhibitors. Molecules 2021, 26, 2696. [Google Scholar] [CrossRef] [PubMed]
- Garrido, J.L.; Godoy, J.; Alvarez, A.; Bronfman, M.; Inestrosa, N.C. Protein kinase C inhibits amyloid beta peptide neurotoxicity by acting on members of the Wnt pathway. Faseb J. 2002, 16, 1982–1984. [Google Scholar] [CrossRef]
- Alkon, D.L.; Sun, M.K.; Nelson, T.J. PKC signaling deficits: A mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci. 2007, 28, 51–60. [Google Scholar] [CrossRef]
- Talman, V.; Pascale, A.; Jäntti, M.; Amadio, M.; Tuominen, R.K. Protein Kinase C Activation as a Potential Therapeutic Strategy in Alzheimer’s Disease: Is there a Role for Embryonic Lethal Abnormal Vision-like Proteins? Basic Clin. Pharmacol. Toxicol. 2016, 119, 149–160. [Google Scholar] [CrossRef]
- Callender, J.A.; Newton, A.C. Conventional protein kinase C in the brain: 40 years later. Neuronal Signal 2017, 1, Ns20160005. [Google Scholar] [CrossRef]
- Du, Y.; Zhao, Y.; Li, C.; Zheng, Q.; Tian, J.; Li, Z.; Huang, T.Y.; Zhang, W.; Xu, H. Inhibition of PKCδ reduces amyloid-β levels and reverses Alzheimer disease phenotypes. J. Exp. Med. 2018, 215, 1665–1677. [Google Scholar] [CrossRef]
- Chen, W.-H.; Chang, Y.-T.; Chen, Y.-C.; Cheng, S.-J.; Chen, C.-C. Spinal protein kinase C/extracellular signal–regulated kinase signal pathway mediates hyperalgesia priming. Pain 2018, 159, 907–918. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, Z.J. Spinal and afferent PKC signaling mechanisms that mediate chronic pain in sickle cell disease. Neurosci. Lett. 2019, 706, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Kopach, O.; Krotov, V.; Shysh, A.; Sotnic, A.; Viatchenko-Karpinski, V.; Dosenko, V.; Voitenko, N. Spinal PKCα inhibition and gene-silencing for pain relief: AMPAR trafficking at the synapses between primary afferents and sensory interneurons. Sci. Rep. 2018, 8, 10285. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.-H.; Sørgård, M.; Di Marzo, V.; Julius, D.; Högestätt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.R.; Urban, T.J.; Hsu, A.K.; Qvit, N.; Gross, G.J.; Mochly-Rosen, D. TRPV1 Mediates Remote and Direct Cardioprotection. Am. Heart Assoc. 2013. [Google Scholar] [CrossRef]
- Fang, J.; Wang, S.; Zhou, J.; Shao, X.; Sun, H.; Liang, Y.; He, X.; Jiang, Y.; Liu, B.; Jin, X.; et al. Electroacupuncture Regulates Pain Transition Through Inhibiting PKCε and TRPV1 Expression in Dorsal Root Ganglion. Front. Neurosci. 2021, 15, 685715. [Google Scholar] [CrossRef]
- Chuang, H.-H.; Prescott, E.D.; Kong, H.; Shields, S.; Jordt, S.-E.; Basbaum, A.I.; Chao, M.V.; Julius, D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 2001, 411, 957–962. [Google Scholar] [CrossRef] [PubMed]
- García-Bernal, F.; Geribaldi-Doldán, N.; Domínguez-García, S.; Carrasco, M.; Murillo-Carretero, M.; Delgado-Ariza, A.; Díez-Salguero, M.; Verástegui, C.; Castro, C. Protein Kinase C Inhibition Mediates Neuroblast Enrichment in Mechanical Brain Injuries. Front. Cell. Neurosci. 2018, 12, 462. [Google Scholar] [CrossRef]
- Brennan, H.A.; Vu, M.A.T.; Maciejewski, P.K.; van Dyck, C.H.; Gottron, M.; Arnsten, A.F. Inhibition of protein kinase C signaling protects prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Proc. Natl. Acad. Sci. USA 2009, 106, 17957–17962. [Google Scholar]
- Turner, R.S.; Raynor, R.L.; Mazzei, G.J.; Girard, P.R.; Kuo, J.F. Developmental studies of phospholipid-sensitive Ca2+-dependent protein kinase and its substrates and of phosphoprotein phosphatases in rat brain. Proc. Natl. Acad. Sci. USA 1984, 81, 3143–3147. [Google Scholar] [CrossRef]
- Corbit, K.C.; Soh, J.-W.; Yoshida, K.; Eves, E.M.; Weinstein, I.B.; Rosner, M.R. Different protein kinase C isoforms determine growth factor specificity in neuronal cells. Mol. Cell Biol. 2000, 20, 5392–5403. [Google Scholar] [CrossRef] [PubMed]
- Müller-Oerlinghausen, B.; Berghöfer, A.; Bauer, M. Bipolar disorder. Lancet 2002, 359, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.K.; Lenox, R.H. The nature of bipolar disorder. J. Clin. Psychiatry 2000, 61 (Suppl. 13), 42–57. [Google Scholar] [PubMed]
- Zarate, C.A.; Manji, H.K. Protein kinase C inhibitors: Rationale for use and potential in the treatment of bipolar disorder. CNS Drugs 2009, 23, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.J.; Currier, D.; Quiroz, J.A.; Manji, H.K. Chapter 60—Neurobiology of Severe Mood and Anxiety Disorders. In Basic Neurochemistry, 8th ed.; Brady, S.T., Siegel, G.J., Albers, R.W., Price, D.L., Eds.; Academic Press: New York, NY, USA, 2012; pp. 1021–1036. [Google Scholar] [CrossRef]
- Saxena, A.; Scaini, G.; Bavaresco, D.V.; Leite, C.; Valvassoria, S.S.; Carvalho, A.F.; Quevedo, J. Role of protein kinase C in bipolar disorder: A review of the current literature. Complex. Psychiatry 2017, 3, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.K.; Lenox, R.H. Protein kinase C signaling in the brain: Molecular transduction of mood stabilization in the treatment of manic-depressive illness. Biol. Psychiatry 1999, 46, 1328–1351. [Google Scholar] [CrossRef] [PubMed]
- Denning, M.F. Epidermal keratinocytes: Regulation of multiple cell phenotypes by multiple protein kinase C isoforms. Int. J. Biochem. Cell Biol. 2004, 36, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, A.M.; Robinson, J.K.; Maududi, T.; Chaturvedi, V.; Nickoloff, B.J.; Denning, M.F. The proapoptotic tumor suppressor protein kinase C-delta is lost in human squamous cell carcinomas. Oncogene 2006, 25, 378–386. [Google Scholar] [CrossRef]
- Jansen, A.P.; Verwiebe, E.G.; Dreckschmidt, N.E.; Wheeler, D.L.; Oberley, T.D.; Verma, A.K. Protein kinase C-epsilon transgenic mice: A unique model for metastatic squamous cell carcinoma. Cancer Res. 2001, 61, 808–812. [Google Scholar]
- Tibudan, S.S.; Wang, Y.; Denning, M.F. Activation of Protein Kinase C Triggers Irreversible Cell CycleWithdrawal In Human Keratinocytes. J. Investig. Dermatol. 2002, 119, 1282–1289. [Google Scholar] [CrossRef]
- Segrelles, C.; Moral, M.; Lara, M.F.; Ruiz, S.; Santos, M.; Leis, H.; García-Escudero, R.; Martínez-Cruz, A.B.; Martínez-Palacio, J.; Hernández, P.; et al. Molecular determinants of Akt-induced keratinocyte transformation. Oncogene 2006, 25, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Maioli, E.; Valacchi, G. Rottlerin: Bases for a possible usage in psoriasis. Curr. Drug Metab. 2010, 11, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Skvara, H.; Dawid, M.; Kleyn, E.; Wolff, B.; Meingassner, J.G.; Knight, H.; Dumortier, T.; Kopp, T.; Fallahi, N.; Stary, G.; et al. The PKC inhibitor AEB071 may be a therapeutic option for psoriasis. J. Clin. Investig. 2008, 118, 3151–3159. [Google Scholar] [CrossRef] [PubMed]
- Soloff, R.S.; Katayama, C.; Lin, M.Y.; Feramisco, J.R.; Hedrick, S.M. Targeted deletion of protein kinase C lambda reveals a distribution of functions between the two atypical protein kinase C isoforms. J. Immunol. 2004, 173, 3250–3260. [Google Scholar] [CrossRef] [PubMed]
- Varin, M.-M.; Guerrier, T.; Devauchelle-Pensec, V.; Jamin, C.; Youinou, P.; Pers, J.-O. In Sjögren’s syndrome, B lymphocytes induce epithelial cells of salivary glands into apoptosis through protein kinase C delta activation. Autoimmun. Rev. 2012, 11, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Mohanraj, M.; Sekar, P.; Liou, H.-H.; Chang, S.-F.; Lin, W.-W. The mycobacterial adjuvant analogue TDB attenuates neuroinflammation via Mincle-independent PLC-γ1/PKC/ERK signaling and microglial polarization. Mol. Neurobiol. 2019, 56, 1167–1187. [Google Scholar] [CrossRef]
- Birchall, A.M.; Bishop, J.; Bradshaw, D.; Cline, A.; Coffey, J.; Elliott, L.H.; Gibson, V.M.; Greenham, A.; Hallam, T.J.; Harris, W. Ro 32-0432, a selective and orally active inhibitor of protein kinase C prevents T-cell activation. J. Pharmacol. Exp. Ther. 1994, 268, 922–929. [Google Scholar]
- Zhong, C.; Wu, Y.; Chang, H.; Liu, C.; Zhou, L.; Zou, J.; Qi, Z. Effect of PKC inhibitor on experimental autoimmune myocarditis in Lewis rats. Oncotarget 2017, 8, 54187–54198. [Google Scholar] [CrossRef]
- DiMartino, M.; Wolff, C.; Patil, A.; Nambi, P. Effects of a protein kinase C inhibitor (PKCI) on the development of adjuvant-induced arthritis (AA) in rats. Inflamm. Res. 1995, 44 (Suppl. 2), S123–S124. [Google Scholar] [CrossRef]
- Leppänen, T.; Tuominen, R.K.; Moilanen, E. Protein Kinase C and its Inhibitors in the Regulation of Inflammation: Inducible Nitric Oxide Synthase as an Example. Basic. Clin. Pharmacol. Toxicol. 2014, 114, 37–43. [Google Scholar] [CrossRef]
- Jacobson, P.B.; Kuchera, S.L.; Metz, A.; Schächtele, C.; Imre, K.; Schrier, D.J. Anti-inflammatory properties of Gö 6850: A selective inhibitor of protein kinase C. J. Pharmacol. Exp. Ther. 1995, 275, 995–1002. [Google Scholar] [PubMed]
- Le, M.Q.; Kim, M.S.; Song, Y.S.; Ryu, H.W.; Oh, S.R.; Yoon, D.Y. 6-O-Veratroyl catalpol suppresses pro-inflammatory cytokines via regulation of extracellular signal-regulated kinase and nuclear factor-κB in human monocytic cells. Biochimie 2015, 119, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Anderson, R.G. Potocytosis. Robert Feulgen Lecture. Histochem. Cell Biol. 2001, 116, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Empig, C.J.; Goldsmith, M.A. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 2002, 76, 5266–5270. [Google Scholar] [CrossRef] [PubMed]
- Stuart, A.D.; Eustace, H.E.; McKee, T.A.; Brown, T.D.K. A novel cell entry pathway for a DAF-using human enterovirus is dependent on lipid rafts. J. Virol. 2002, 76, 9307–9322. [Google Scholar] [CrossRef] [PubMed]
- Marjomäki, V.; Pietiäinen, V.; Matilainen, H.; Upla, P.; Ivaska, J.; Nissinen, L.; Reunanen, H.; Huttunen, P.; Hyypiä, T.; Heino, J. Internalization of echovirus 1 in caveolae. J. Virol. 2002, 76, 1856–1865. [Google Scholar] [CrossRef]
- Mañes, S.; del Real, G.; Lacalle, R.A.; Lucas, P.; Gómez-Moutón, C.; Sánchez-Palomino, S.; Delgado, R.; Alcamí, J.; Mira, E.; Martínez-A, C. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 2000, 1, 190–196. [Google Scholar] [CrossRef]
- Guyader, M.; Kiyokawa, E.; Abrami, L.; Turelli, P.; Trono, D. Role for human immunodeficiency virus type 1 membrane cholesterol in viral internalization. J. Virol. 2002, 76, 10356–10364. [Google Scholar] [CrossRef]
- Warrilow, D.; Gardner, J.; Darnell, G.A.; Suhrbier, A.; Harrich, D. HIV Type 1 Inhibition by Protein Kinase C Modulatory Compounds. AIDS Res. Human. Retroviruses 2006, 22, 854–864. [Google Scholar] [CrossRef]
- Contreras, X.; Mzoughi, O.; Gaston, F.; Peterlin, M.B.; Bahraoui, E. Protein kinase C-delta regulates HIV-1 replication at an early post-entry step in macrophages. Retrovirology 2012, 9, 37. [Google Scholar] [CrossRef]
- Dang, Y.-F.; Qiu, T.-X.; Song, D.-W.; Liu, L. PMA-triggered PKCε activity enhances Nrf2-mediated antiviral response on fish rhabdovirus infection. Fish. Shellfish. Immunol. 2019, 87, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Souza-Souza, K.F.C.; Gonçalves-De-Albuquerque, C.F.; Cirne-Santos, C.; Paixão, I.C.N.P.; Burth, P. Alphavirus Replication: The Role of Cardiac Glycosides and Ion Concentration in Host Cells. Biomed. Res. Int. 2020, 2020, 2813253. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, M.A.; Lee, J.S.; Zhou, X.; Ramke, M.; Lee, J.Y.; Chodosh, J.; Rajaiya, J. Protein Kinase C Signaling in Adenoviral Infection. Biochemistry 2016, 55, 5938–5946. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, S.N.; Cernescu, C.D.; Popescu, L.M. Effects of protein kinase C inhibitors on viral entry and infectivity. FEBS Lett. 1991, 292, 31–33. [Google Scholar] [PubMed]
- Park, R.; Baines Joel, D. Herpes Simplex Virus Type 1 Infection Induces Activation and Recruitment of Protein Kinase C to the Nuclear Membrane and Increased Phosphorylation of Lamin B. J. Virol. 2006, 80, 494–504. [Google Scholar] [CrossRef]
- Sieczkarski, S.B.; Brown, H.A.; Whittaker, G.R. Role of protein kinase C βII in influenza virus entry via late endosomes. J. Virol. 2003, 77, 460–469. [Google Scholar] [CrossRef]
- Mondal, A.; Dawson, A.R.; Potts, G.K.; Freiberger, E.C.; Baker, S.F.; Moser, L.A.; Bernard, K.A.; Coon, J.J.; Mehle, A. Influenza virus recruits host protein kinase C to control assembly and activity of its replication machinery. eLife 2017, 6, e26910. [Google Scholar] [CrossRef]
- Noppakunmongkolchai, W.; Poyomtip, T.; Jittawuttipoka, T.; Luplertlop, N.; Sakuntabhai, A.; Chimnaronk, S.; Jirawatnotai, S.; Tohtong, R. Inhibition of protein kinase C promotes dengue virus replication. Virol. J. 2016, 13, 35. [Google Scholar] [CrossRef]
- Monick, M.M.; Staber, J.M.; Thomas, K.W.; Hunninghake, G.W. Respiratory Syncytial Virus Infection Results in Activation of Multiple Protein Kinase C Isoforms Leading to Activation of Mitogen-Activated Protein Kinase. J. Immunol. 2001, 166, 2681. [Google Scholar] [CrossRef]
- San-Juan-Vergara, H.; Peeples, M.E.; Lockey, R.F.; Mohapatra, S.S. Protein kinase C-α activity is required for respiratory syncytial virus fusion to human bronchial epithelial cells. J. Virol. 2004, 78, 13717–13726. [Google Scholar] [CrossRef]
- Yousuf, M.A.; Zhou, X.; Mukherjee, S.; Chintakuntlawar, A.V.; Lee, J.Y.; Ramke, M.; Chodosh, J.; Rajaiya, J. Caveolin-1 associated adenovirus entry into human corneal cells. PLoS ONE 2013, 8, e77462. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.K.; Vezina, C.M. Externalization of host cell protein kinase C during enteropathogenic Escherichia coli infection. Cell Death Differ. 2005, 12, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Gauron, M.C.; Newton, A.C.; Colombo, M.I. PKCα Is Recruited to Staphylococcus aureus-Containing Phagosomes and Impairs Bacterial Replication by Inhibition of Autophagy. Front. Immunol. 2021, 12, 662987. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, M.; Van Ngo, H.; Gyanwali, G.C.; Ireton, K. The Host Scaffolding Protein Filamin A and the Exocyst Complex Control Exocytosis during InlB-Mediated Entry of Listeria monocytogenes. Infect. Immun. 2019, 87, e00689-18. [Google Scholar] [CrossRef] [PubMed]
- Sah, P.; Nelson, N.H.; Shaw, J.H.; Lutter, E.I. Chlamydia trachomatis recruits protein kinase C during infection. Pathog. Dis. 2019, 77, ftz061. [Google Scholar] [CrossRef] [PubMed]
- LaFayette, S.L.; Collins, C.; Zaas, A.K.; Schell, W.A.; Betancourt-Quiroz, M.; Gunatilaka, A.A.L.; Perfect, J.R.; Cowen, L.E. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 2010, 6, e1001069. [Google Scholar] [CrossRef] [PubMed]
- Schwegmann, A.; Guler, R.; Cutler, A.J.; Arendse, B.; Horsnell, W.G.C.; Flemming, A.; Kottmann, A.H.; Ryan, G.; Hide, W.; Leitges, M.; et al. Protein kinase C δ is essential for optimal macrophage-mediated phagosomal containment of Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 2007, 104, 16251–16256. [Google Scholar] [CrossRef] [PubMed]
- Parihar, S.P.; Ozturk, M.; Marakalala, M.J.; Loots, D.T.; Hurdayal, R.; Maasdorp, D.B.; Van Reenen, M.; E Zak, D.; Darboe, F.; Penn-Nicholson, A.; et al. Protein kinase C-delta (PKCδ), a marker of inflammation and tuberculosis disease progression in humans, is important for optimal macrophage killing effector functions and survival in mice. Mucosal Immunol. 2018, 11, 496–511. [Google Scholar] [CrossRef]
- St-Denis, A.; Caouras, V.; Gervais, F.; Descoteaux, A. Role of protein kinase C-α in the control of infection by intracellular pathogens in macrophages. J. Immunol. 1999, 163, 5505–5511. [Google Scholar] [CrossRef]
- Guler, R.; Afshar, M.; Arendse, B.; Parihar, S.P.; Revaz-Breton, M.; Leitges, M.; Schwegmann, A.; Brombacher, F. PKCδ regulates IL-12p40/p70 production by macrophages and dendritic cells, driving a type 1 healer phenotype in cutaneous leishmaniasis. Eur. J. Immunol. 2011, 41, 706–715. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Ghosh, S.; Sen, P.; Roy, S.; Majumdar, S. Selective impairment of protein kinase C isotypes in murine macrophage by Leismania donovani. Mol. Cell. Biochem. 2001, 216, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.; Gratton, J.-P.; Rafikov, R.; Black, S.M.; Fulton, D.J.R. Calcium/calmodulin-dependent kinase II mediates the phosphorylation and activation of NADPH oxidase 5. Mol. Pharmacol. 2011, 80, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [PubMed]
- Rahimova, N.; Cooke, M.; Zhang, S.; Baker, M.J.; Kazanietz, M.G. The PKC universe keeps expanding: From cancer initiation to metastasis. Adv. Biol. Regul. 2020, 78, 100755. [Google Scholar] [CrossRef] [PubMed]
- Lordén, G.; Newton, A.C. Conventional protein kinase C in the brain: Repurposing cancer drugs for neurodegenerative treatment? Neuronal Signal 2021, 5, Ns20210036. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’shea, J.J. Selectivity and therapeutic inhibition of kinases: To be or not to be? Nat. Immunol. 2009, 10, 356–360. [Google Scholar] [CrossRef]
- Wu-Zhang, A.X.; Newton, A.C. Protein kinase C pharmacology: Refining the toolbox. Biochem. J. 2013, 452, 195–209. [Google Scholar] [CrossRef]
- Echols, N.; Harrison, P.; Balasubramanian, S.; Luscombe, N.M.; Bertone, P.; Zhang, Z.; Gerstein, M. Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes. Nucleic Acids Res. 2002, 30, 2515–2523. [Google Scholar] [CrossRef]
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Lee, K.-W.; Kim, S.G.; Kim, H.-P.; Kwon, E.; You, J.; Choi, H.-J.; Park, J.-H.; Kang, B.-C.; Im, S.-A.; Kim, T.-Y.; et al. Enzastaurin, a protein kinase C beta inhibitor, suppresses signaling through the ribosomal S6 kinase and bad pathways and induces apoptosis in human gastric cancer cells. Cancer Res. 2008, 68, 1916–1926. [Google Scholar] [CrossRef] [PubMed]
- Bourhill, T.; Narendran, A.; Johnston, R.N. Enzastaurin: A lesson in drug development. Crit. Rev. Oncol./Hematol. 2017, 112, 72–79. [Google Scholar] [CrossRef]
- Blumberg, P.M.; Jaken, S.; König, B.; Sharkey, N.A.; Leach, K.L.; Jeng, A.Y.; Yeh, E. Mechanism of action of the phorbol ester tumor promoters: Specific receptors for lipophilic ligands. Biochem. Pharmacol. 1984, 33, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Marquez, V.E.; Nacro, K.; Benzaria, S.; Lee, J.; Sharma, R.; Teng, K.; Milne, G.W.; Bienfait, B.; Wang, S.; Lewin, N.E.; et al. The transition from a pharmacophore-guided approach to a receptor-guided approach in the design of potent protein kinase C ligands. Pharmacol. Ther. 1999, 82, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-H.; Kim, S.Y.; Lee, J.; Marquez, V.E.; Lewin, N.E.; Pearce, L.V.; Blumberg, P.M. Macrocyclic diacylglycerol-bis-lactones as conformationally constrained analogues of diacylglycerol-lactones. Interactions with protein kinase C. J. Med. Chem. 2004, 47, 4000–4007. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.; Zhou, X.; Casado-Medrano, V.; Lopez-Haber, C.; Baker, M.J.; Garg, R.; Ann, J.; Lee, J.; Blumberg, P.M.; Kazanietz, M.G. Characterization of AJH-836, a diacylglycerol-lactone with selectivity for novel PKC isozymes. J. Biol. Chem. 2018, 293, 8330–8341. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bermejo, M.L.; Leskow, F.C.; Fujii, T.; Wang, Q.; Blumberg, P.M.; Ohba, M.; Kuroki, T.; Han, K.C.; Lee, J.; Marquez, V.E.; et al. Diacylglycerol (DAG)-lactones, a new class of protein kinase C (PKC) agonists, induce apoptosis in LNCaP prostate cancer cells by selective activation of PKCalpha. J. Biol. Chem. 2002, 277, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-K.; Alkon, D.L. Bryostatin-1: Pharmacology and therapeutic potential as a CNS drug. CNS Drug Rev. 2006, 12, 1–8. [Google Scholar] [CrossRef]
- Ruan, B.-F.; Zhu, H.-L. The chemistry and biology of the bryostatins: Potential PKC inhibitors in clinical development. Curr. Med. Chem. 2012, 19, 2652–2664. [Google Scholar] [CrossRef]
- Mackay, H.J.; Twelves, C.J. Targeting the protein kinase C family: Are we there yet? Nat. Rev. Cancer 2007, 7, 554–562. [Google Scholar] [CrossRef]
- Raghuvanshi, R.; Bharate, S.B. Preclinical and clinical studies on bryostatins, a class of marine-derived protein kinase C modulators: A mini-review. Curr. Top. Med. Chem. 2020, 20, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Müller, M.; Dean, N.M.; Fabbro, D. Antitumor activity of a PKC-alpha antisense oligonucleotide in combination with standard chemotherapeutic agents against various human tumors transplanted into nude mice. Anticancer Drug Des. 1998, 13, 35–45. [Google Scholar] [PubMed]
- Wang, C.; Wang, X.; Liang, H.; Wang, T.; Yan, X.; Cao, M.; Wang, N.; Zhang, S.; Zen, K.; Zhang, C.; et al. miR-203 Inhibits Cell Proliferation and Migration of Lung Cancer Cells by Targeting PKCα. PLoS ONE 2013, 8, e73985. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Ruetz, S.; Bodis, S.; Pruschy, M.; Csermak, K.; Man, A.; Campochiaro, P.; Wood, J.; O’Reilly, T.; Meyer, T. PKC412--a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000, 15, 17–28. [Google Scholar] [PubMed]
- Tremblay, G.; Dolph, M.; Patel, S.; Brandt, P.; Forsythe, A. Cost-effectiveness analysis for midostaurin versus standard of care in acute myeloid leukemia in the United Kingdom. Cost. Eff. Resour. Alloc. 2018, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Medicine, C.T.g.U.S.N.L.o. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=Midostaurin&cntry=&state=&city=&dist= (accessed on 15 July 2022).
- Crump, M.; Leppä, S.; Fayad, L.; Lee, J.J.; Di Rocco, A.; Ogura, M.; Hagberg, H.; Schnell, F.; Rifkin, R.; Mackensen, A.; et al. Randomized, Double-Blind, Phase III Trial of Enzastaurin Versus Placebo in Patients Achieving Remission After First-Line Therapy for High-Risk Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2016, 34, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, E.; Leblond, V.; Maisonneuve, H.; Benhadji, K.A.; Hossain, A.M.; Nguyen, T.S.; Wooldridge, J.E.; Moreau, P. A multicenter phase II study of single-agent enzastaurin in previously treated multiple myeloma. Leuk. Lymphoma 2014, 55, 2013–2017. [Google Scholar] [CrossRef]
- Grønberg, B.H.; Ciuleanu, T.; Fløtten, Ø.; Knuuttila, A.; Abel, E.; Langer, S.W.; Krejcy, K.; Liepa, A.M.; Munoz, M.; Hahka-Kemppinen, M.; et al. A placebo-controlled, randomized phase II study of maintenance enzastaurin following whole brain radiation therapy in the treatment of brain metastases from lung cancer. Lung Cancer 2012, 78, 63–69. [Google Scholar] [CrossRef]
- Usha, L.; Sill, M.W.; Darcy, K.M.; Benbrook, D.M.; Hurteau, J.A.; Michelin, D.P.; Mannel, R.S.; Hanjani, P.; De Geest, K.; Godwin, A.K. A Gynecologic Oncology Group phase II trial of the protein kinase C-beta inhibitor, enzastaurin and evaluation of markers with potential predictive and prognostic value in persistent or recurrent epithelial ovarian and primary peritoneal malignancies. Gynecol. Oncol. 2011, 121, 455–461. [Google Scholar] [CrossRef]
- Morschhauser, F.; Seymour, J.F.; Kluin-Nelemans, H.C.; Grigg, A.; Wolf, M.; Pfreundschuh, M.; Tilly, H.; Raemaekers, J.; van’t Veer, M.B.; Milpied, N.; et al. A phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory mantle cell lymphoma. Ann. Oncol. 2008, 19, 247–253. [Google Scholar] [CrossRef]
- Querfeld, C.; Kuzel, T.M.; Kim, Y.H.; Porcu, P.; Duvic, M.; Musiek, A.; Rook, A.H.; Mark, L.A.; Pinter-Brown, L.; Hamid, O.; et al. Multicenter phase II trial of enzastaurin in patients with relapsed or refractory advanced cutaneous T-cell lymphoma. Leuk. Lymphoma 2011, 52, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Mina, L.; Krop, I.; Zon, R.T.; Isakoff, S.J.; Schneider, C.J.; Yu, M.; Johnson, C.; Vaughn, L.G.; Wang, Y.; Hristova-Kazmierski, M.; et al. A phase II study of oral enzastaurin in patients with metastatic breast cancer previously treated with an anthracycline and a taxane containing regimen. Investig. New Drugs 2009, 27, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Clément-Duchêne, C.; Natale, R.B.; Jahan, T.; Krupitskaya, Y.; Osarogiagbon, R.; Sanborn, R.E.; Bernstein, E.D.; Dudek, A.Z.; Latz, J.E.; Shi, P.; et al. A phase II study of enzastaurin in combination with erlotinib in patients with previously treated advanced non-small cell lung cancer. Lung Cancer 2012, 78, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.; Bepler, G.; Barlesi, F.; Soria, J.-C.; Reck, M.; Bearz, A.; Barata, F.; Scagliotti, G.; Park, K.; Wagle, A.; et al. Phase II, double-blinded, randomized study of enzastaurin plus pemetrexed as second-line therapy in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Butowski, N.; Chang, S.M.; Lamborn, K.R.; Polley, M.-Y.; Pieper, R.; Costello, J.F.; Vandenberg, S.; Parvataneni, R.; Nicole, A.; Sneed, P.K.; et al. Phase II and pharmacogenomics study of enzastaurin plus temozolomide during and following radiation therapy in patients with newly diagnosed glioblastoma multiforme and gliosarcoma. Neuro Oncol. 2011, 13, 1331–1338. [Google Scholar] [CrossRef]
- Dreicer, R.; Garcia, J.; Rini, B.; Vogelzang, N.; Srinivas, S.; Somer, B.; Shi, P.; Kania, M.; Raghavan, D. A randomized, double-blind, placebo-controlled, Phase II study with and without enzastaurin in combination with docetaxel-based chemotherapy in patients with castration-resistant metastatic prostate cancer. Investig. New Drugs 2013, 31, 1044–1050. [Google Scholar] [CrossRef]
- Vergote, I.B.; Chekerov, R.; Amant, F.; Harter, P.; Casado, A.; Emerich, J.; Bauknecht, T.; Mansouri, K.; Myrand, S.P.; Nguyen, T.S.; et al. Randomized, phase II, placebo-controlled, double-blind study with and without enzastaurin in combination with paclitaxel and carboplatin as first-line treatment followed by maintenance treatment in advanced ovarian cancer. J. Clin. Oncol. 2013, 31, 3127–3132. [Google Scholar] [CrossRef]
- Wolff, R.A.; Fuchs, M.; Di Bartolomeo, M.; Hossain, A.M.; Stoffregen, C.; Nicol, S.; Heinemann, V. A double-blind, randomized, placebo-controlled, phase 2 study of maintenance enzastaurin with 5-fluorouracil/leucovorin plus bevacizumab after first-line therapy for metastatic colorectal cancer. Cancer 2012, 118, 4132–4138. [Google Scholar] [CrossRef]
- Richards, D.A.; Kuefler, P.R.; Becerra, C.; Wilfong, L.S.; Gersh, R.H.; Boehm, K.A.; Zhan, F.; Asmar, L.; Myrand, S.P.; Hozak, R.R.; et al. Gemcitabine plus enzastaurin or single-agent gemcitabine in locally advanced or metastatic pancreatic cancer: Results of a phase II, randomized, noncomparative study. Investig. New Drugs 2011, 29, 144–153. [Google Scholar] [CrossRef]
- Kawano, T.; Inokuchi, J.; Eto, M.; Murata, M.; Kang, J.-H. Activators and Inhibitors of Protein Kinase C (PKC): Their Applications in Clinical Trials. Pharmaceutics 2021, 13, 1748. [Google Scholar] [CrossRef]
- Evenou, J.P.; Wagner, J.; Zenke, G.; Brinkmann, V.; Wagner, K.; Kovarik, J.; Welzenbach, K.A.; Weitz-Schmidt, G.; Guntermann, C.; Towbin, H.; et al. The potent protein kinase C-selective inhibitor AEB071 (sotrastaurin) represents a new class of immunosuppressive agents affecting early T-cell activation. J. Pharmacol. Exp. Ther. 2009, 330, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Narahara, K.A.; Elkayam, U.; Sullivan, J.M.; Pearle, D.L.; Massie, B.M.; Creager, M.A. Double-blind, placebo-controlled study of the efficacy of flosequinan in patients with chronic heart failure. J. Am. Coll. Cardiol. 1993, 22, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Alavi, J.B.; Supko, J.G.; Carson, K.A.; Priet, R.; Dorr, F.A.; Grundy, J.S.; Holmlund, J.T. Efficacy and toxicity of the antisense oligonucleotide aprinocarsen directed against protein kinase C-alpha delivered as a 21-day continuous intravenous infusion in patients with recurrent high-grade astrocytomas. Neuro Oncol. 2005, 7, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Douillard, J.-Y.; Koralewski, P.; Manegold, C.; Smit, E.F.; Reyes, J.M.; Chang, G.-C.; John, W.J.; Peterson, P.M.; Obasaju, C.K.; et al. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Ritch, P.; Rudin, C.M.; Bitran, J.D.; Edelman, M.J.; Makalinao, A.; Irwin, D.; Lilenbaum, R.; Peterson, P.; John, W.J. Phase II study of PKC-alpha antisense oligonucleotide aprinocarsen in combination with gemcitabine and carboplatin in patients with advanced non-small cell lung cancer. Lung Cancer 2006, 52, 173–180. [Google Scholar] [CrossRef]
- Advani, R.; Peethambaram, P.; Lum, B.L.; Fisher, G.A.; Hartmann, L.; Long, H.J.; Halsey, J.; Holmlund, J.T.; Dorr, A.; Sikic, B.I. A Phase II trial of aprinocarsen, an antisense oligonucleotide inhibitor of protein kinase C alpha, administered as a 21-day infusion to patients with advanced ovarian carcinoma. Cancer 2004, 100, 321–326. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Reyno, L.; Venner, P.M.; Ernst, S.D.; Moore, M.; Geary, R.S.; Chi, K.; Hall, S.; Walsh, W.; Dorr, A.; et al. A randomized phase II and pharmacokinetic study of the antisense oligonucleotides ISIS 3521 and ISIS 5132 in patients with hormone-refractory prostate cancer. Clin. Cancer Res 2002, 8, 2530–2535. [Google Scholar]
- Marshall, J.L.; Eisenberg, S.G.; Johnson, M.D.; Hanfelt, J.; Dorr, F.A.; El-Ashry, D.; Oberst, M.; Fuxman, Y.; Holmlund, J.; Malik, S. A phase II trial of ISIS 3521 in patients with metastatic colorectal cancer. Clin. Color. Cancer 2004, 4, 268–274. [Google Scholar] [CrossRef]
- Rao, S.; Watkins, D.; Cunningham, D.; Dunlop, D.; Johnson, P.; Selby, P.; Hancock, B.W.; Fegan, C.; Culligan, D.; Schey, S.; et al. Phase II study of ISIS 3521, an antisense oligodeoxynucleotide to protein kinase C alpha, in patients with previously treated low-grade non-Hodgkin’s lymphoma. Ann. Oncol. 2004, 15, 1413–1418. [Google Scholar] [CrossRef]
- Propper, D.; Macaulay, V.; O’Byrne, K.; Braybrooke, J.; Wilner, S.; Ganesan, T.; Talbot, D.; Harris, A. A phase II study of bryostatin 1 in metastatic malignant melanoma. Br. J. Cancer 1998, 78, 1337–1341. [Google Scholar] [CrossRef]
- Gonzalez, R.; Ebbinghaus, S.; Henthorn, T.K.; Miller, D.; Kraft, A.S. Treatment of patients with metastatic melanoma with bryostatin-1--a phase II study. Melanoma Res. 1999, 9, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Bedikian, A.Y.; Plager, C.; Stewart, J.R.; O’Brian, C.A.; Herdman, S.K.; Ross, M.; Papadopoulos, N.; Eton, O.; Ellerhorst, J.; Smith, T. Phase II evaluation of bryostatin-1 in metastatic melanoma. Melanoma Res. 2001, 11, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Brockstein, B.; Samuels, B.; Humerickhouse, R.; Arietta, R.; Fishkin, P.; Wade, J.; Sosman, J.; Vokes, E. Phase II studies of bryostatin-1 in patients with advanced sarcoma and advanced head and neck cancer. Investig. New Drugs 2001, 19, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Varterasian, M.L.; Pemberton, P.A.; Hulburd, K.; Rodriguez, D.H.; Murgo, A.; Al-Katib, A.M. Phase II study of bryostatin 1 in patients with relapsed multiple myeloma. Investig. New Drugs 2001, 19, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Zonder, J.A.; Shields, A.F.; Zalupski, M.; Chaplen, R.; Heilbrun, L.K.; Arlauskas, P.; Philip, P.A. A phase II trial of bryostatin 1 in the treatment of metastatic colorectal cancer. Clin. Cancer Res. 2001, 7, 38–42. [Google Scholar] [PubMed]
- Pfister, D.G.; McCaffrey, J.; Zahalsky, A.J.; Schwartz, G.K.; Lis, E.; Gerald, W.; Huvos, A.; Shah, J.; Kraus, D.; Shaha, A.; et al. A phase II trial of bryostatin-1 in patients with metastatic or recurrent squamous cell carcinoma of the head and neck. Investig. New Drugs 2002, 20, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Winegarden, J.D.; Mauer, A.M.; Gajewski, T.F.; Hoffman, P.C.; Krauss, S.A.; Rudin, C.M.; Vokes, E.E. A phase II study of bryostatin-1 and paclitaxel in patients with advanced non-small cell lung cancer. Lung Cancer 2003, 39, 191–196. [Google Scholar] [CrossRef]
- Nezhat, F.; Wadler, S.; Muggia, F.; Mandeli, J.; Goldberg, G.; Rahaman, J.; Runowicz, C.; Murgo, A.J.; Gardner, G.J. Phase II trial of the combination of bryostatin-1 and cisplatin in advanced or recurrent carcinoma of the cervix: A New York Gynecologic Oncology Group study. Gynecol. Oncol. 2004, 93, 144–148. [Google Scholar] [CrossRef]
- Lam, A.P.; Sparano, J.A.; Vinciguerra, V.; Ocean, A.J.; Christos, P.; Hochster, H.; Camacho, F.; Goel, S.; Mani, S.; Kaubisch, A. Phase II study of paclitaxel plus the protein kinase C inhibitor bryostatin-1 in advanced pancreatic carcinoma. Am. J. Clin. Oncol. 2010, 33, 121–124. [Google Scholar] [CrossRef]
- Haas, N.B.; Smith, M.; Lewis, N.; Littman, L.; Yeslow, G.; Joshi, I.D.; Murgo, A.; Bradley, J.; Gordon, R.; Wang, H.; et al. Weekly bryostatin-1 in metastatic renal cell carcinoma: A phase II study. Clin. Cancer Res. 2003, 9, 109–114. [Google Scholar]
- Madhusudan, S.; Protheroe, A.; Propper, D.; Han, C.; Corrie, P.; Earl, H.; Hancock, B.; Vasey, P.; Turner, A.; Balkwill, F.; et al. A multicentre phase II trial of bryostatin-1 in patients with advanced renal cancer. Br. J. Cancer 2003, 89, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Jiang, Y.; Faust, J.; Chang, B.B.; Ho, L.; Yao, J.C.; Rousey, S.; Dakhil, S.; Cherny, R.C.; Craig, C.; et al. A multi-center phase II study of sequential paclitaxel and bryostatin-1 (NSC 339555) in patients with untreated, advanced gastric or gastroesophageal junction adenocarcinoma. Investig. New Drugs 2006, 24, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Ku, G.Y.; Ilson, D.H.; Schwartz, L.H.; Capanu, M.; O’reilly, E.; Shah, M.A.; Kelsen, D.P.; Schwartz, G.K. Phase II trial of sequential paclitaxel and 1 h infusion of bryostatin-1 in patients with advanced esophageal cancer. Cancer Chemother. Pharmacol. 2008, 62, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Cripps, M.C.; Figueredo, A.T.; Oza, A.M.; Taylor, M.J.; Fields, A.L.; Holmlund, J.T.; McIntosh, L.W.; Geary, R.S.; Eisenhauer, E.A. Phase II randomized study of ISIS 3521 and ISIS 5132 in patients with locally advanced or metastatic colorectal cancer: A National Cancer Institute of Canada clinical trials group study. Clin. Cancer Res. 2002, 8, 2188–2192. [Google Scholar]
- Advani, R.; Lum, B.L.; Fisher, G.A.; Halsey, J.; Geary, R.S.; Holmlund, J.T.; Kwoh, T.J.; Dorr, F.A.; Sikic, B.I. A phase I trial of aprinocarsen (ISIS 3521/LY900003), an antisense inhibitor of protein kinase C-alpha administered as a 24-hour weekly infusion schedule in patients with advanced cancer. Investig. New Drugs 2005, 23, 467–477. [Google Scholar] [CrossRef]
- Robertson, M.J.; Kahl, B.S.; Vose, J.M.; De Vos, S.; Laughlin, M.; Flynn, P.J.; Rowland, K.; Cruz, J.C.; Goldberg, S.L.; Musib, L.; et al. Phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory diffuse large B-cell lymphoma. J. Clin. Oncol. 2007, 25, 1741–1746. [Google Scholar] [CrossRef]
- Oh, Y.; Herbst, R.S.; Burris, H.; Cleverly, A.; Musib, L.; Lahn, M.; Bepler, G. Enzastaurin, an oral serine/threonine kinase inhibitor, as second- or third-line therapy of non-small-cell lung cancer. J. Clin. Oncol. 2008, 26, 1135–1141. [Google Scholar] [CrossRef]
- Glimelius, B.; Lahn, M.; Gawande, S.; Cleverly, A.; Darstein, C.; Musib, L.; Liu, Y.; Spindler, K.L.; Frödin, J.E.; Berglund, Å.; et al. A window of opportunity phase II study of enzastaurin in chemonaive patients with asymptomatic metastatic colorectal cancer. Ann. Oncol. 2010, 21, 1020–1026. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Kotliarova, S.; Butman, J.A.; Albert, P.S.; Kim, L.; Musib, L.; Thornton, D.; Fine, H.A. A phase I/II trial of enzastaurin in patients with recurrent high-grade gliomas. Neuro Oncol. 2010, 12, 181–189. [Google Scholar] [CrossRef]
- Socinski, M.A.; Raju, R.N.; Stinchcombe, T.; Kocs, D.M.; Couch, L.S.; Barrera, D.; Rousey, S.R.; Choksi, J.K.; Jotte, R.; Patt, D.A.; et al. Randomized, phase II trial of pemetrexed and carboplatin with or without enzastaurin versus docetaxel and carboplatin as first-line treatment of patients with stage IIIB/IV non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 1963–1969. [Google Scholar] [CrossRef]
- Couldwell, W.T.; Hinton, D.R.; Surnock, A.A.; DeGiorgio, C.M.; Weiner, L.P.; Apuzzo, M.L.; Masri, L.; Law, R.E.; Weiss, M.H. Treatment of recurrent malignant gliomas with chronic oral high-dose tamoxifen. Clin. Cancer Res. 1996, 2, 619–622. [Google Scholar] [PubMed]
- Bergan, R.C.; Reed, E.; Myers, C.E.; Headlee, D.; Brawley, O.; Cho, H.K.; Figg, W.D.; Tompkins, A.; Linehan, W.M.; Kohler, D.; et al. A Phase II study of high-dose tamoxifen in patients with hormone-refractory prostate cancer. Clin. Cancer Res. 1999, 5, 2366–2373. [Google Scholar] [PubMed]
- Robins, H.I.; Won, M.; Seiferheld, W.F.; Schultz, C.J.; Choucair, A.K.; Brachman, D.G.; Demas, W.F.; Mehta, M.P. Phase 2 trial of radiation plus high-dose tamoxifen for glioblastoma multiforme: RTOG protocol BR-0021. Neuro Oncol. 2006, 8, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Weinberg, V.; Shaw, V.; Scott, J.; Bok, R.; Park, J.W.; Small, E.J. Time to disease progression to evaluate a novel protein kinase C inhibitor, UCN-01, in renal cell carcinoma. Cancer 2004, 101, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Welch, S.; Hirte, H.W.; Carey, M.S.; Hotte, S.J.; Tsao, M.-S.; Brown, S.; Pond, G.R.; Dancey, J.E.; Oza, A.M. UCN-01 in combination with topotecan in patients with advanced recurrent ovarian cancer: A study of the Princess Margaret Hospital Phase II consortium. Gynecol. Oncol. 2007, 106, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Bower, M.J.; McDevitt, P.J.; Zhao, H.; Davis, S.T.; Johanson, K.O.; Green, S.M.; Concha, N.O.; Zhou, B.-B.S. Structural basis for Chk1 inhibition by UCN-01. J. Biol. Chem. 2002, 277, 46609–46615. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Ward, D.; Saltz, L.; Casper, E.S.; Spiess, T.; Mullen, E.; Woodworth, J.; Venuti, R.; Zervos, P.; Storniolo, A.M.; et al. A pilot clinical/pharmacological study of the protein kinase C-specific inhibitor safingol alone and in combination with doxorubicin. Clin. Cancer Res. 1997, 3, 537–543. [Google Scholar]
- Ling, L.-U.; Tan, K.-B.; Lin, H.; Chiu, G.N.C. The role of reactive oxygen species and autophagy in safingol-induced cell death. Cell Death Dis. 2011, 2, e129. [Google Scholar] [CrossRef]
- Schaar, D.; Goodell, L.; Aisner, J.; Cui, X.X.; Han, Z.T.; Chang, R.; Martin, J.; Grospe, S.; Dudek, L.; Riley, J.; et al. A phase I clinical trial of 12- O-tetradecanoylphorbol-13-acetate for patients with relapsed/refractory malignancies. Cancer Chemother. Pharmacol. 2006, 57, 789–795. [Google Scholar] [CrossRef]
- Aiello, L.P.; Vignati, L.; Sheetz, M.J.; Zhi, X.; Girach, A.; Davis, M.D.; Wolka, A.M.; Shahri, N.; Milton, R.C. Oral protein kinase c β inhibition using ruboxistaurin: Efficacy, safety, and causes of vision loss among 813 patients (1,392 eyes) with diabetic retinopathy in the Protein Kinase C β Inhibitor-Diabetic Retinopathy Study and the Protein Kinase C β Inhibitor-Diabetic Retinopathy Study 2. Retina 2011, 31, 2084–2094. [Google Scholar] [PubMed]
- PKC-DRS Study Group. The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: Initial results of the Protein Kinase C beta Inhibitor Diabetic Retinopathy Study (PKC-DRS) multicenter randomized clinical trial. Diabetes 2005, 54, 2188–2197. [Google Scholar]
- Aiello, L.P.; Davis, M.D.; Girach, A.; Kles, K.A.; Milton, R.C.; Sheetz, M.J.; Vignati, L.; Zhi, X.E. Effect of ruboxistaurin on visual loss in patients with diabetic retinopathy. Ophthalmology 2006, 113, 2221–2230. [Google Scholar] [PubMed]
- Sheetz, M.J.; Aiello, L.P.; Shahri, N.; Davis, M.D.; Kles, K.A.; Danis, R.P.; Mbdv Study Group. Effect of ruboxistaurin (RBX) On visual acuity decline over a 6-year period with cessation and reinstitution of therapy: Results of an open-label extension of the Protein Kinase C Diabetic Retinopathy Study 2 (PKC-DRS2). Retina 2011, 31, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- PKC-DRS2 Group. Effect of ruboxistaurin in patients with diabetic macular edema: Thirty-month results of the randomized PKC-DMES clinical trial. Arch. Ophthalmol. 2007, 125, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Bakris, G.L.; Toto, R.D.; McGill, J.B.; Hu, K.; Anderson, P.W. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care 2005, 28, 2686–2690. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; McGill, J.B.; Haney, D.J.; Lin, T.E.; Anderson, P.W. Kidney outcomes in long-term studies of ruboxistaurin for diabetic eye disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A.I.; Bril, V.; Kempler, P.; Litchy, W.J.; Tesfaye, S.; Price, K.L.; Bastyr, E.J.; MBBQ Study Group. Treatment of symptomatic diabetic peripheral neuropathy with the protein kinase C beta-inhibitor ruboxistaurin mesylate during a 1-year, randomized, placebo-controlled, double-blind clinical trial. Clin. Ther. 2005, 27, 1164–1180. [Google Scholar] [CrossRef]
- Casellini, C.M.; Barlow, P.M.; Rice, A.L.; Casey, M.; Simmons, K.; Pittenger, G.; Bastyr, E.J.; Wolka, A.M.; Vinik, A.I. A 6-month, randomized, double-masked, placebo-controlled study evaluating the effects of the protein kinase C-beta inhibitor ruboxistaurin on skin microvascular blood flow and other measures of diabetic peripheral neuropathy. Diabetes Care 2007, 30, 896–902. [Google Scholar] [CrossRef]
- Bates, E.; Bode, C.; Costa, M.; Gibson, C.M.; Granger, C.; Green, C.; Grimes, K.; Harrington, R.; Huber, K.; Kleiman, N.; et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation 2008, 117, 886–896. [Google Scholar]
- Lincoff, M. Inhibition of delta-Protein Kinase C for Reduction of Infarct Size in Acute Myocardial Infarction—The PROTECTION AMI Trial, KAI. In Proceedings of the American College of Cardiology 60th Annual Scientific Sessions, New Orleans, LA, USA, 2–5 April 2011. [Google Scholar]
- Lincoff, A.M.; Roe, M.; Aylward, P.; Galla, J.; Rynkiewicz, A.; Guetta, V.; Zelizko, M.; Kleiman, N.; White, H.; McErlean, E.; et al. Inhibition of delta-protein kinase C by delcasertib as an adjunct to primary percutaneous coronary intervention for acute anterior ST-segment elevation myocardial infarction: Results of the PROTECTION AMI Randomized Controlled Trial. Eur. Heart J. 2014, 35, 2516–2523. [Google Scholar] [CrossRef] [PubMed]
- DeMets, D.L.; Califf, R.M. Lessons learned from recent cardiovascular clinical trials: Part II. Circulation 2002, 106, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Julier, K.; da Silva, R.; Garcia, C.; Bestmann, L.; Frascarolo, P.; Zollinger, A.; Chassot, P.G.; Schmid, E.R.; Turina, M.I.; von Segesser, L.K.; et al. Preconditioning by sevoflurane decreases biochemical markers for myocardial and renal dysfunction in coronary artery bypass graft surgery: A double-blinded, placebo-controlled, multicenter study. Anesthesiology 2003, 98, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Guarracino, F.; Landoni, G.; Tritapepe, L.; Pompei, F.; Leoni, A.; Aletti, G.; Scandroglio, A.M.; Maselli, D.; De Luca, M.; Marchetti, C.; et al. Myocardial damage prevented by volatile anesthetics: A multicenter randomized controlled study. J. Cardiothorac. Vasc. Anesth. 2006, 20, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Chen, C.H.; Kuo, M.C.; Kang, P.L.; Lo, A.; Liu, K. Isoflurane preconditioning-induced cardio-protection in patients undergoing coronary artery bypass grafting. Eur. J. Anaesthesiol. 2006, 23, 841–847. [Google Scholar] [CrossRef]
- Tritapepe, L.; Landoni, G.; Guarracino, F.; Pompei, F.; Crivellari, M.; Maselli, D.; De Luca, M.; Fochi, O.; D’Avolio, S.; Bignami, E.; et al. Cardiac protection by volatile anaesthetics: A multicentre randomized controlled study in patients undergoing coronary artery bypass grafting with cardiopulmonary bypass. Eur. J. Anaesthesiol. 2007, 24, 323–331. [Google Scholar] [CrossRef]
- De Hert, S.; Vlasselaers, D.; Barbé, R.; Ory, J.; Dekegel, D.; Donnadonni, R.; Demeere, J.; Mulier, J.; Wouters, P. A comparison of volatile and non volatile agents for cardioprotection during on-pump coronary surgery. Anaesthesia 2009, 64, 953–960. [Google Scholar] [CrossRef]
- Mentzer, R.M., Jr.; Birjiniuk, V.; Khuri, S.; Lowe, J.E.; Rahko, P.S.; Weisel, R.D.; Wellons, H.A.; Barker, M.L.; Lasley, R.D.; Adenosine Myocardial Protection Investigators. Adenosine myocardial protection: Preliminary results of a phase II clinical trial. Ann. Surg. 1999, 229, 643–650. [Google Scholar] [CrossRef]
- Belhomme, D.; Peynet, J.; Florens, E.; Tibourtine, O.; Kitakaze, M.; Menasché, P. Is adenosine preconditioning truly cardioprotective in coronary artery bypass surgery? Ann. Thorac. Surg. 2000, 70, 590–594. [Google Scholar] [CrossRef]
- Jin, Z.; Duan, W.; Chen, M.; Yu, S.; Zhang, H.; Feng, G.; Xiong, L.; Yi, D. The myocardial protective effects of adenosine pretreatment in children undergoing cardiac surgery: A randomized controlled clinical trial. Eur. J. Cardiothorac. Surg. 2011, 39, e90–e96. [Google Scholar] [CrossRef]
- Mangano, D.T.; Miao, Y.; Tudor, I.C.; Dietzel, C. Post-Reperfusion Myocardial Infarction: Long-Term Survival Improvement Using Adenosine Regulation with Acadesine. J. Am. Coll. Cardiol. 2006, 48, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.F.; Ferguson, T.B.; White, J.A.; Ambrosio, G.; Koglin, J.; Nussmeier, N.A.; Pearl, R.G.; Pitt, B.; Wechsler, A.S.; Weisel, R.D.; et al. Effect of adenosine-regulating agent acadesine on morbidity and mortality associated with coronary artery bypass grafting: The RED-CABG randomized controlled trial. JAMA 2012, 308, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Kingwell, B.A. Acadesine, an adenosine-regulating agent with the potential for widespread indications. Expert. Opin. Pharmacother. 2008, 9, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Sheikh, S.; Khan, M.A.; Chaturvedi, A.; Patel, P.; Patel, R.; Buch, B.C.; Anand, R.S.; Shah, T.C.; Vora, V.N.; et al. Endoxifen: A new, protein kinase C inhibitor to treat acute and mixed mania associated with bipolar I disorder. Bipolar Disord. 2021, 23, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Cousins, M.J.; Pickthorn, K.; Huang, S.; Critchley, L.; Bell, G. The safety and efficacy of KAI-1678- an inhibitor of epsilon protein kinase C (epsilonPKC)-versus lidocaine and placebo for the treatment of postherpetic neuralgia: A crossover study design. Pain Med. 2013, 14, 533–540. [Google Scholar] [CrossRef]
- Moodie, J.E.; Bisley, E.J.; Huang, S.; Pickthorn, K.; Bell, G. A single-center, randomized, double-blind, active, and placebo-controlled study of KAI-1678, a novel PKC-epsilon inhibitor, in the treatment of acute postoperative orthopedic pain. Pain. Med. 2013, 14, 916–924. [Google Scholar]
- Shoushtari, A.N.; Khan, S.; Komatsubara, K.; Feun, L.; Acquavella, N.; Singh-Kandah, S.; Negri, T.; Nesson, A.; Abbate, K.; Cremers, S.; et al. A Phase Ib Study of Sotrastaurin, a PKC Inhibitor, and Alpelisib, a PI3Kα Inhibitor, in Patients with Metastatic Uveal Melanoma. Cancers 2021, 13, 5504. [Google Scholar] [CrossRef]
- Scott, D.; Mazurkiewicz, M.; Leeman, P. The long-term monitoring of ventilation rhythms of the polychaetous annelid Nereis virens sars. Comp. Biochem. Physiol. A Comp. Physiol. 1976, 53, 65–68. [Google Scholar] [CrossRef]
- Jacob, J.S.; McDonald, H.S. Diving bradycardia in four species of North American aquatic snakes. Comp. Biochem. Physiol. A Comp. Physiol. 1976, 53, 69–72. [Google Scholar] [CrossRef]
- Pascher, A.; De, P.S.; Pratschke, J.; Salamé, E.; Pirenne, J.; Isoneimi, H.; Bijarnia, M.; Krishnan, I.; Klupp, J. Protein kinase C inhibitor sotrastaurin in de novo liver transplant recipients: A randomized phase II trial. Am. J. Transplant. 2015, 15, 1283–1292. [Google Scholar] [CrossRef]
- Farlow, M.R.; Thompson, R.E.; Wei, L.J.; Tuchman, A.J.; Grenier, E.; Crockford, D.; Wilke, S.; Benison, J.; Alkon, D.L. A Randomized, Double-Blind, Placebo-Controlled, Phase II Study Assessing Safety, Tolerability, and Efficacy of Bryostatin in the Treatment of Moderately Severe to Severe Alzheimer’s Disease. J. Alzheimers Dis. 2019, 67, 555–570. [Google Scholar] [CrossRef] [PubMed]

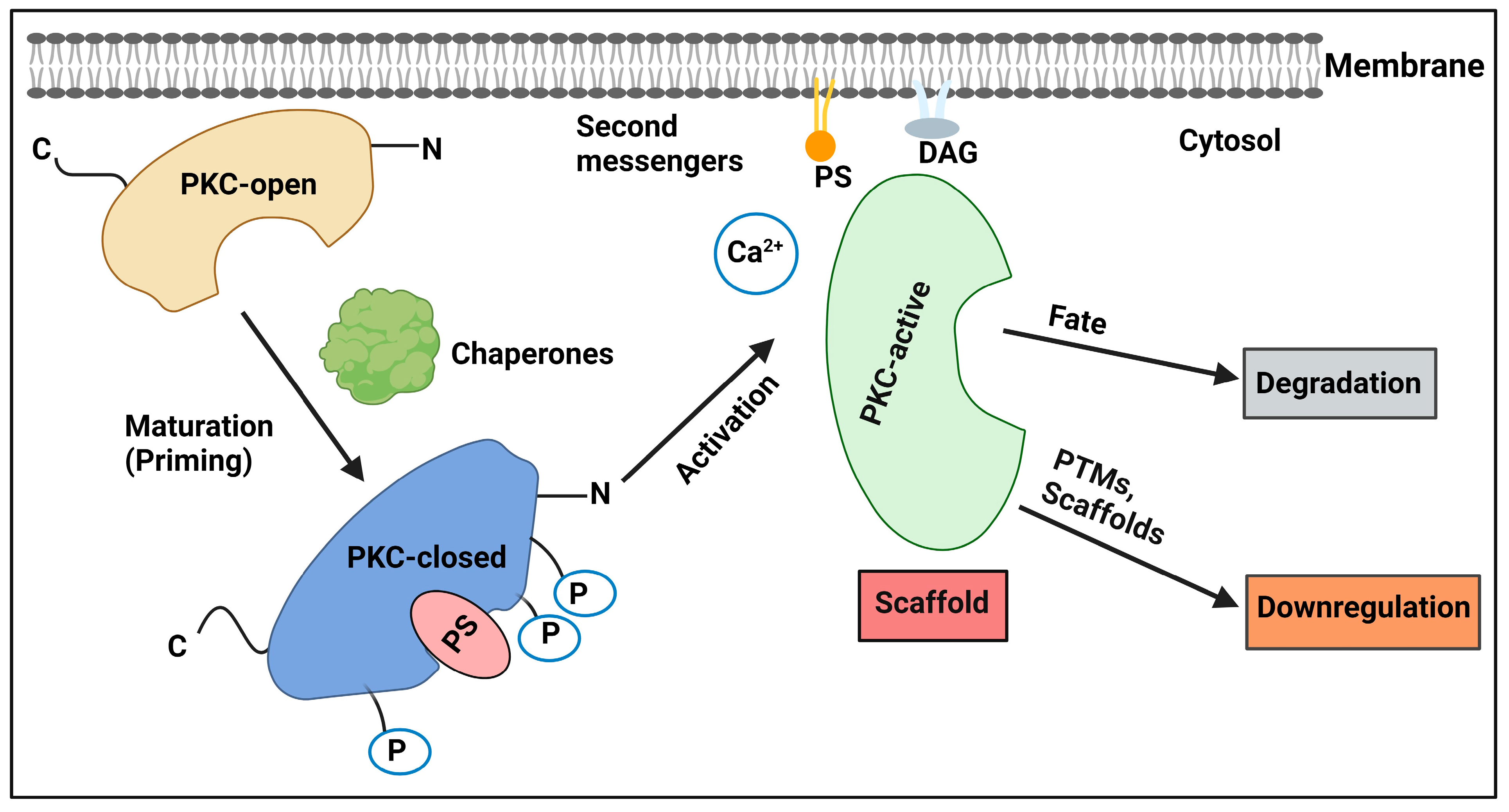
| Disease | PKC Isozyme | Associated Pathology |
|---|---|---|
| Cancer | PKCα | Proliferation; invasion; metastasis; RCC tumor progression; mutated in thyroid cancer; SCC tumorigenesis and progression; elevated in late-stage breast cancer; upregulated in lung cancer; activated in bladder cancer |
| PKCβ | Angiogenesis; invasion; elevated in early-stage breast cancer; downregulated in CRC; PKCβII tumor suppressor in CRC; promotes angiogenesis in NHLs and DLBCL | |
| PKCδ | Angiogenesis; apoptosis in endometrial cancer; elevated in early-stage breast cancer; downregulated in CRC | |
| PKCγ | Increased in CRC | |
| PKCε | Oncogene; proliferation; metastasis; chemotherapy resistance; mutated in thyroid cancer; SCC tumorigenesis, progression, and metastasis; keratinocyte cytoskeleton structure; upregulated in lung cancer; downregulated in CRC | |
| PKCη | Glioblastoma tumorigenesis; proliferation; radiation resistance; elevated in early-stage breast cancer; upregulated in lung cancer; downregulated in CRC | |
| PKCθ | Gastrointestinal stromal tumorigenesis | |
| PKCι/λ | Oncogene; upregulated in lung cancer; promotes prostate cancer invasion; conflicting roles in prostate cancer metastasis; elevation associated with poor outcomes in PDAC | |
| PKCζ | Elevated in late-stage breast cancer; upregulated in lung cancer; promotes RCC tumor progression | |
| Diabetes mellitus | PKCβ | Obesity; glucose transport; insulin resistance; cholesterol and fatty acid metabolism; PKCβII late-stage vascular complications |
| PKCδ | Islet cell function | |
| Ischemic heart disease | PKCβ | Pathologic postischemic cardiac remodeling |
| PKCδ | ROS production; apoptosis; necrosis; postischemic injury | |
| PKCε | Mitochondrial function; proteasome function; ALDH2 activation | |
| Heart failure | PKCα | Reduced contractility; β-adrenergic receptor uncoupling |
| PKCβII | Decreased proteasome function; calcium dysregulation; conflicting roles in hypertrophy; conflicting roles in contractility | |
| PKCδ | Abnormal activation in early HF | |
| PKCε | Fibrosis; inflammation; depleted in late-stage HF | |
| PKCζ | Generation of active NADPH oxidase; Reduced basal and TGF-βI induced fibroblast proliferation; increased MMP1,3,9 release | |
| Hypertension | PKCα | Elevated in hypertension |
| PKCβ | Elevated in hypertension | |
| PKCδ | Elevated in hypertension | |
| PKCε | Translocation and increased hypertrophic cellular growth | |
| Stroke | PKCδ | Mitochondrial fission; ROS production; blood–brain barrier dysfunction |
| PKCε | Cytoprotective; cerebral perfusion | |
| Neurodegeneration | PKCδ | Inflammation; neuronal cell death in Parkinson’s; β-amyloid formation in Alzheimer’s disease |
| PKCα | Associated with AD, promoting synaptic loss | |
| PKCγ | Associated with SCA, promoting neuronal death | |
| Bipolar disorder | PKCα | Abnormal gene expression |
| PKCε | Abnormal neuronal transmission | |
| Nociception | PKCα | Peripheral nociception signaling |
| PKCγ | Dorsal root ganglia signal transmission | |
| PKCε | Spinal cord signal transmission; peripheral nociception signaling | |
| Inflammation | PKCδ | B-cell development; keratinocyte proliferation and dysregulated angiogenesis in psoriasis; eosinophil activation in asthma |
| PKCθ | T-cell responses; airway inflammation; joint inflammation | |
| PKCζ | Lung inflammation |
| Compound Name | Structure | Indication(s) | Target Domain | Mechanism | Isozyme Selectivity |
|---|---|---|---|---|---|
| Midostaurin | 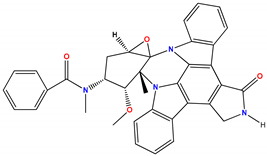 | Acute myeloid leukemia | Catalytic domain | Competitive inhibitor of ATP binding site | Multiple protein kinases |
| Riluzole |  | Amyotrophic lateral sclerosis | Catalytic domain | Competitive inhibitor of ATP binding site | PKCα and other kinases |
| Curcumin | 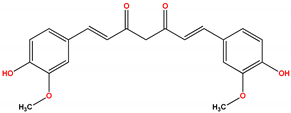 | Not approved; used as nutraceutical for ulcerative colitis and other inflammatory conditions | Regulatory domain | C1 inhibitor | PKCα |
| Resveratrol |  | Not approved; ongoing studies in cancer, cardiovascular disease, neurodegeneration, and aging | Regulatory domain | C1 inhibitor | PKCα; PKCβ; PKCε |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silnitsky, S.; Rubin, S.J.S.; Zerihun, M.; Qvit, N. An Update on Protein Kinases as Therapeutic Targets—Part I: Protein Kinase C Activation and Its Role in Cancer and Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 17600. https://doi.org/10.3390/ijms242417600
Silnitsky S, Rubin SJS, Zerihun M, Qvit N. An Update on Protein Kinases as Therapeutic Targets—Part I: Protein Kinase C Activation and Its Role in Cancer and Cardiovascular Diseases. International Journal of Molecular Sciences. 2023; 24(24):17600. https://doi.org/10.3390/ijms242417600
Chicago/Turabian StyleSilnitsky, Shmuel, Samuel J. S. Rubin, Mulate Zerihun, and Nir Qvit. 2023. "An Update on Protein Kinases as Therapeutic Targets—Part I: Protein Kinase C Activation and Its Role in Cancer and Cardiovascular Diseases" International Journal of Molecular Sciences 24, no. 24: 17600. https://doi.org/10.3390/ijms242417600