
APOBEC3G Is a p53-Dependent Restriction Factor in Respiratory Syncytial Virus Infection of Human Cells Included in the p53/Immune Axis
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
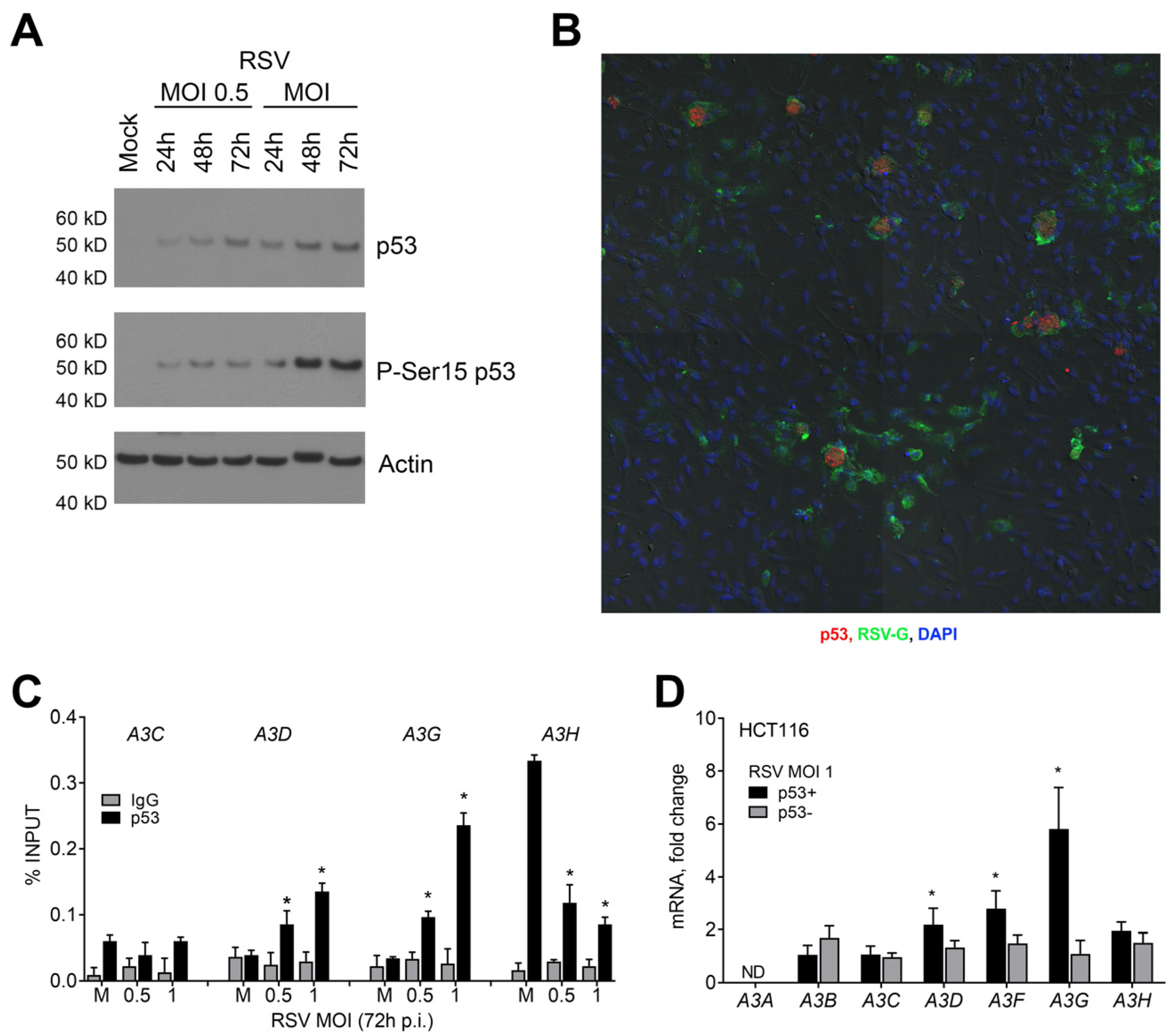
2.1. Influence of p53 on the Expression of APOBEC3 Genes during RSV Infection in Human Lung Cancer Cells
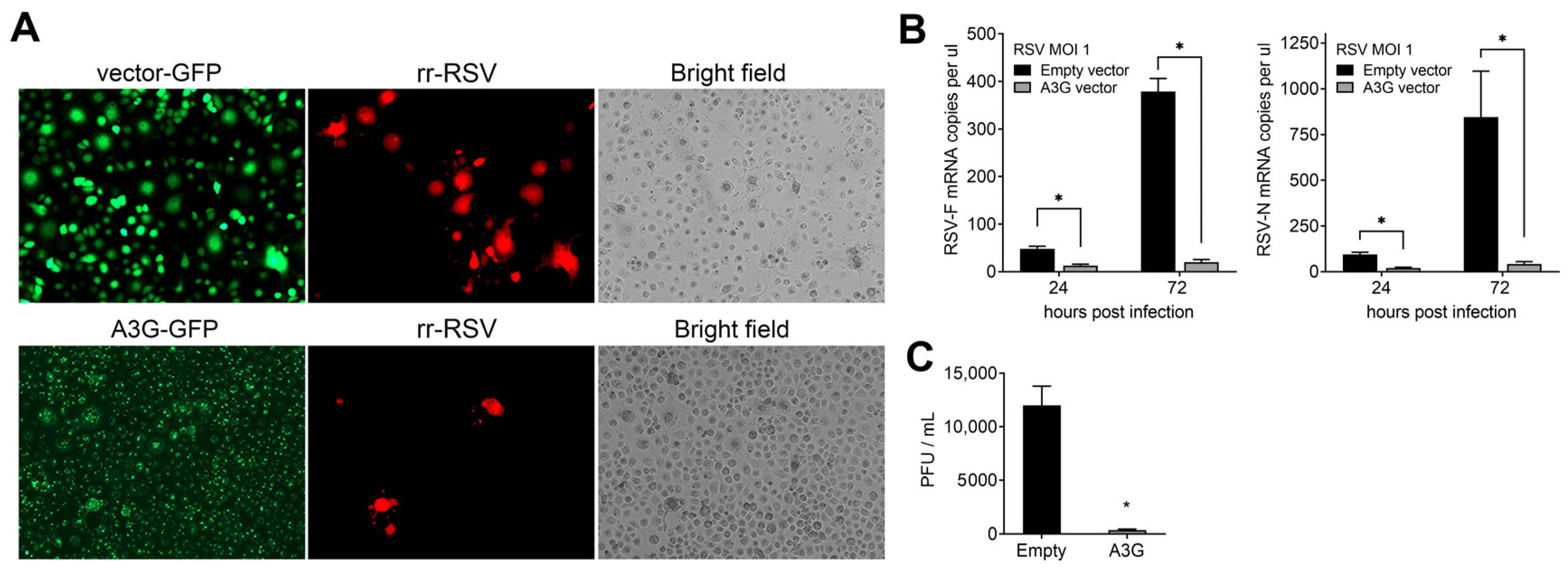
2.2. Effect of A3G on RSV Infectivity
2.3. p53 Functional Status Influences RSV-Induced Cytopathic Effect (CPE) and Cell Death
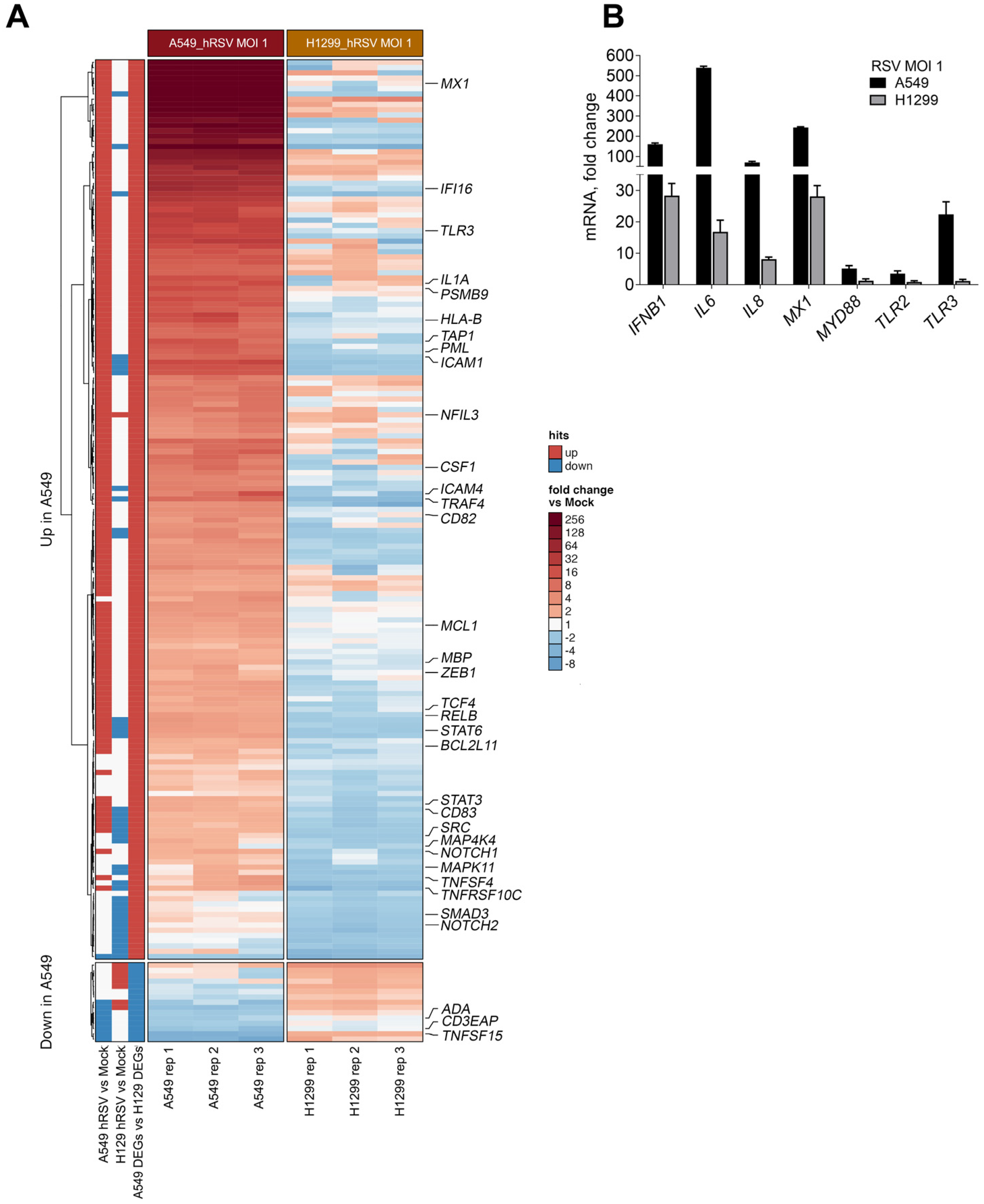
2.4. p53 Influences RSV-Induced Immune Responses at the Transcriptional Level
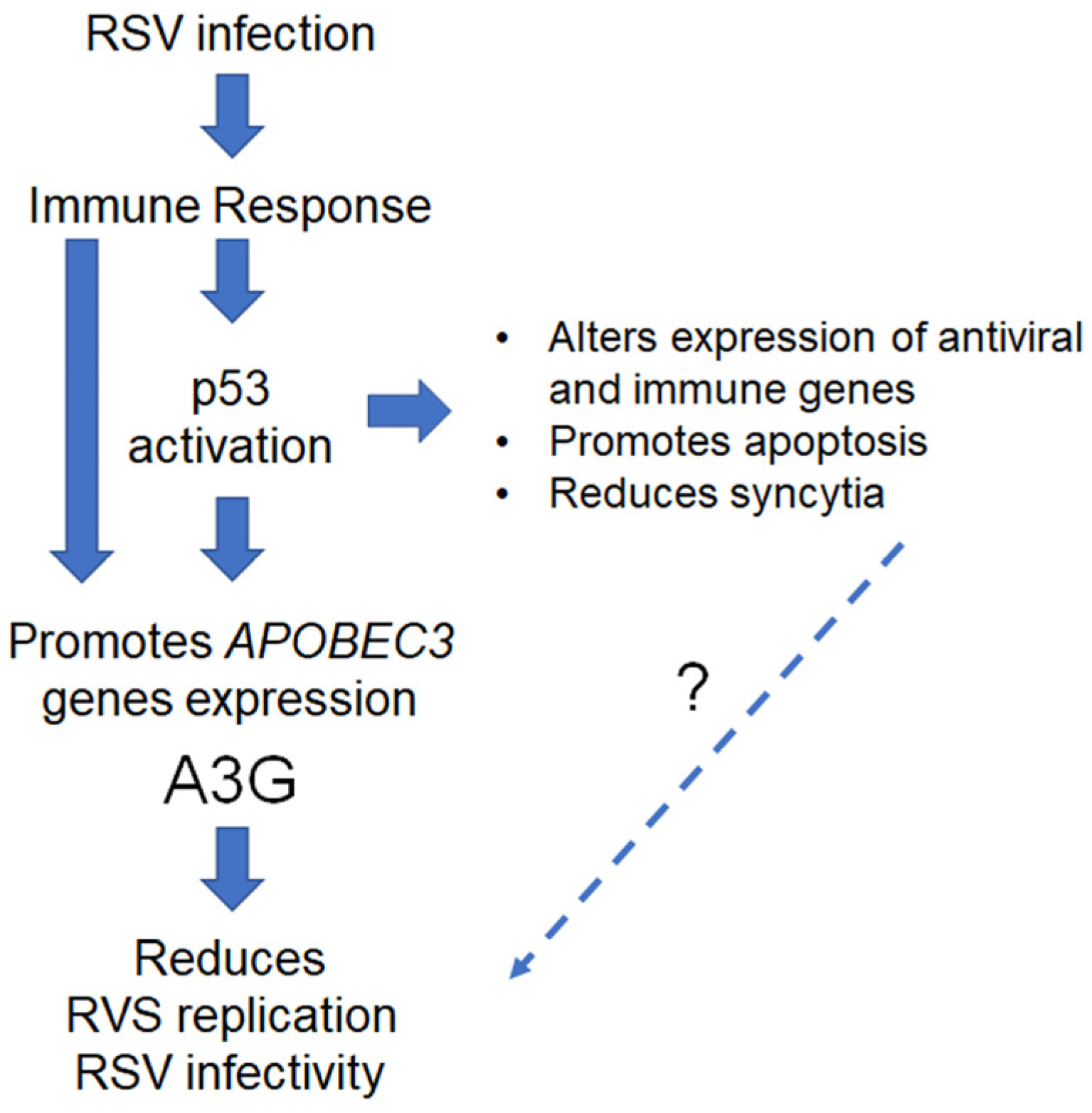
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Viral Infections and Plaque Assays
4.3. RNA Isolation and qRT-PCR
4.4. Apoptosis Evaluation
4.5. Droplet Digital PCR (ddPCR)
4.6. Confocal Microscopy
4.7. Western Blotting
4.8. Plasmid Transfection
4.9. Chromatin Immunoprecipitation (ChIP)
4.10. NanoString nCounter Gene Expression
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collins, P.L.; Graham, B.S. Viral and host factors in human respiratory syncytial virus pathogenesis. J. Virol. 2008, 82, 2040–2055. [Google Scholar] [CrossRef]
- Howard, T.S.; Hoffman, L.H.; Stang, P.E.; Simoes, E.A. Respiratory syncytial virus pneumonia in the hospital setting: Length of stay, charges, and mortality. J Pediatr 2000, 137, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Verwey, C.; Madhi, S.A. Review and Update of Active and Passive Immunization Against Respiratory Syncytial Virus. BioDrugs 2023, 37, 295–309. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA Approves First Respiratory Syncytial Virus (RSV) Vaccine. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-respiratory-syncytial-virus-rsv-vaccine (accessed on 2 November 2023).
- Agency, E.M. First RSV Vaccine to Protect Infants up to 6 Months of Age and Older Adults. Available online: https://www.ema.europa.eu/en/news/first-rsv-vaccine-protect-infants-6-months-age-older-adults (accessed on 2 November 2023).
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Aloni-Grinstein, R.; Charni-Natan, M.; Solomon, H.; Rotter, V. p53 and the Viral Connection: Back into the Future. Cancers 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Lasut-Szyszka, B.; Rusin, M. The Wheel of p53 Helps to Drive the Immune System. Int. J. Mol. Sci. 2023, 24, 7645. [Google Scholar] [CrossRef]
- Cardozo, C.M.; Hainaut, P. Viral strategies for circumventing p53: The case of severe acute respiratory syndrome coronavirus. Curr Opin Oncol 2021, 33, 149–158. [Google Scholar] [CrossRef]
- Levine, A.J. P53 and The Immune Response: 40 Years of Exploration-A Plan for the Future. Int. J. Mol. Sci. 2020, 21, 541. [Google Scholar] [CrossRef]
- Menendez, D.; Shatz, M.; Resnick, M.A. Interactions between the tumor suppressor p53 and immune responses. Curr. Opin. Oncol. 2013, 25, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Nguyen, T.A.; Freudenberg, J.M.; Mathew, V.J.; Anderson, C.W.; Jothi, R.; Resnick, M.A. Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013, 41, 7286–7301. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Nguyen, T.A.; Snipe, J.; Resnick, M.A. The Cytidine Deaminase APOBEC3 Family Is Subject to Transcriptional Regulation by p53. Mol. Cancer Res. 2017, 15, 735–743. [Google Scholar] [CrossRef]
- Menendez, D.; Shatz, M.; Azzam, K.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011, 7, e1001360. [Google Scholar] [CrossRef] [PubMed]
- Langlois, M.A.; Beale, R.C.; Conticello, S.G.; Neuberger, M.S. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 2005, 33, 1913–1923. [Google Scholar] [CrossRef]
- Taylor, B.J.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2013, 2, e00534. [Google Scholar] [CrossRef]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef]
- Milewska, A.; Kindler, E.; Vkovski, P.; Zeglen, S.; Ochman, M.; Thiel, V.; Rajfur, Z.; Pyrc, K. APOBEC3-mediated restriction of RNA virus replication. Sci. Rep. 2018, 8, 5960. [Google Scholar] [CrossRef]
- Vieira, V.C.; Soares, M.A. The role of cytidine deaminases on innate immune responses against human viral infections. Biomed Res. Int. 2013, 2013, 683095. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Iwatani, Y. APOBEC3G-Mediated G-to-A Hypermutation of the HIV-1 Genome: The Missing Link in Antiviral Molecular Mechanisms. Front. Microbiol. 2016, 7, 2027. [Google Scholar] [CrossRef]
- Bian, T.; Gibbs, J.D.; Örvell, C.; Imani, F. Respiratory syncytial virus matrix protein induces lung epithelial cell cycle arrest through a p53 dependent pathway. PLoS ONE 2012, 7, e38052. [Google Scholar] [CrossRef]
- Eckardt-Michel, J.; Lorek, M.; Baxmann, D.; Grunwald, T.; Keil, G.M.; Zimmer, G. The fusion protein of respiratory syncytial virus triggers p53-dependent apoptosis. J. Virol. 2008, 82, 3236–3249. [Google Scholar] [CrossRef]
- Groskreutz, D.J.; Monick, M.M.; Yarovinsky, T.O.; Powers, L.S.; Quelle, D.E.; Varga, S.M.; Look, D.C.; Hunninghake, G.W. Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J. Immunol. 2007, 179, 2741–2747. [Google Scholar] [CrossRef]
- Machado, D.; Pizzorno, A.; Hoffmann, J.; Traversier, A.; Endtz, H.; Lina, B.; Rosa-Calatrava, M.; Paranhos-Baccala, G.; Terrier, O. Role of p53/NF-κB functional balance in respiratory syncytial virus-induced inflammation response. J. Gen. Virol. 2018, 99, 489–500. [Google Scholar] [CrossRef]
- Menendez, D.; Snipe, J.; Marzec, J.; Innes, C.L.; Polack, F.P.; Caballero, M.T.; Schurman, S.H.; Kleeberger, S.R.; Resnick, M.A. p53-responsive TLR8 SNP enhances human innate immune response to respiratory syncytial virus. J. Clin. Invest. 2019, 129, 4875–4884. [Google Scholar] [CrossRef]
- Bisio, A.; Zamborszky, J.; Zaccara, S.; Lion, M.; Tebaldi, T.; Sharma, V.; Raimondi, I.; Alessandrini, F.; Ciribilli, Y.; Inga, A. Cooperative interactions between p53 and NFkappaB enhance cell plasticity. Oncotarget 2014, 5, 12111–12125. [Google Scholar] [CrossRef]
- Ghosh, M.; Saha, S.; Li, J.Y.; Montrose, D.C.; Martinez, L.A. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell 2023, 83, 266–280.e6. [Google Scholar] [CrossRef] [PubMed]
- Lokshin, M.; Tanaka, T.; Prives, C. Transcriptional regulation by p53 and p73. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Grimm, S.A.; Bushel, P.R.; Li, J.; Li, Y.; Bennett, B.D.; Lavender, C.A.; Ward, J.M.; Fargo, D.C.; Anderson, C.W.; et al. Revealing a human p53 universe. Nucleic Acids Res. 2018, 46, 8153–8167. [Google Scholar] [CrossRef]
- Lowe, J.M.; Menendez, D.; Bushel, P.R.; Shatz, M.; Kirk, E.L.; Troester, M.A.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. p53 and NF-κB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014, 74, 2182–2192. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database J. Biol. Databases Curation 2017, 2017, bax028. [Google Scholar] [CrossRef]
- Koning, F.A.; Newman, E.N.; Kim, E.Y.; Kunstman, K.J.; Wolinsky, S.M.; Malim, M.H. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar] [CrossRef]
- Bishop, K.N.; Holmes, R.K.; Sheehy, A.M.; Malim, M.H. APOBEC-mediated editing of viral RNA. Science 2004, 305, 645. [Google Scholar] [CrossRef]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun 2015, 6, 6881. [Google Scholar] [CrossRef]
- Salter, J.D.; Polevoda, B.; Bennett, R.P.; Smith, H.C. Regulation of Antiviral Innate Immunity Through APOBEC Ribonucleoprotein Complexes. Subcell. Biochem. 2019, 93, 193–219. [Google Scholar]
- Smith, H.C. RNA binding to APOBEC deaminases; Not simply a substrate for C to U editing. RNA Biol. 2017, 14, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Iwatani, Y.; Takeuchi, H.; Strebel, K.; Levin, J.G. Biochemical activities of highly purified, catalytically active human APOBEC3G: Correlation with antiviral effect. J. Virol. 2006, 80, 5992–6002. [Google Scholar] [CrossRef]
- McDougall, W.M.; Smith, H.C. Direct evidence that RNA inhibits APOBEC3G ssDNA cytidine deaminase activity. Biochem. Biophys. Res. Commun. 2011, 412, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Polevoda, B.; McDougall, W.M.; Tun, B.N.; Cheung, M.; Salter, J.D.; Friedman, A.E.; Smith, H.C. RNA binding to APOBEC3G induces the disassembly of functional deaminase complexes by displacing single-stranded DNA substrates. Nucleic Acids Res. 2015, 43, 9434–9445. [Google Scholar] [CrossRef] [PubMed]
- Fehrholz, M.; Kendl, S.; Prifert, C.; Weissbrich, B.; Lemon, K.; Rennick, L.; Duprex, P.W.; Rima, B.K.; Koning, F.A.; Holmes, R.K.; et al. The innate antiviral factor APOBEC3G targets replication of measles, mumps and respiratory syncytial viruses. J. Gen. Virol. 2012, 93, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.G.; Zhao, Z.Y.; Li, Y.P.; Wang, Y.P.; Hao, L.H.; Fan, B.; Li, Y.H.; Wang, Y.M.; Shan, Y.Q.; Han, Y.X.; et al. Host apolipoprotein B messenger RNA-editing enzyme catalytic polypeptide-like 3G is an innate defensive factor and drug target against hepatitis C virus. Hepatology 2011, 53, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Gallois-Montbrun, S.; Kramer, B.; Swanson, C.M.; Byers, H.; Lynham, S.; Ward, M.; Malim, M.H. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 2007, 81, 2165–2178. [Google Scholar] [CrossRef]
- Wichroski, M.J.; Robb, G.B.; Rana, T.M. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2006, 2, e41. [Google Scholar] [CrossRef]
- Tiwarekar, V.; Wohlfahrt, J.; Fehrholz, M.; Scholz, C.J.; Kneitz, S.; Schneider-Schaulies, J. APOBEC3G-Regulated Host Factors Interfere with Measles Virus Replication: Role of REDD1 and Mammalian TORC1 Inhibition. J. Virol. 2018, 92, e00835-18. [Google Scholar] [CrossRef]
- Li, Z.; Ning, S.; Su, X.; Liu, X.; Wang, H.; Liu, Y.; Zheng, W.; Zheng, B.; Yu, X.F.; Zhang, W. Enterovirus 71 antagonizes the inhibition of the host intrinsic antiviral factor A3G. Nucleic Acids Res. 2018, 46, 11514–11527. [Google Scholar] [CrossRef]
- Martínez, I.; García-Carpizo, V.; Guijarro, T.; García-Gomez, A.; Navarro, D.; Aranda, A.; Zambrano, A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence 2016, 7, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Choudhary, S.; Tian, B.; Boldogh, I.; Yang, C.; Ivanciuc, T.; Ma, Y.; Garofalo, R.P.; Brasier, A.R. Ataxia telangiectasia mutated kinase mediates NF-kappaB serine 276 phosphorylation and interferon expression via the IRF7-RIG-I amplification loop in paramyxovirus infection. J. Virol. 2015, 89, 2628–2642. [Google Scholar] [CrossRef]
- Cervantes-Ortiz, S.L.; Zamorano Cuervo, N.; Grandvaux, N. Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology. Viruses 2016, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.P.; Kolli, D.; Casola, A. Respiratory syncytial virus infection: Mechanisms of redox control and novel therapeutic opportunities. Antioxid Redox Signal. 2013, 18, 186–217. [Google Scholar] [CrossRef]
- Cuddihy, A.R.; Li, S.; Tam, N.W.; Wong, A.H.; Taya, Y.; Abraham, N.; Bell, J.C.; Koromilas, A.E. Double-stranded-RNA-activated protein kinase PKR enhances transcriptional activation by tumor suppressor p53. Mol. Cell. Biol. 1999, 19, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Lee, J.F.; Lu, H.; Lee, M.H.; Yan, D.H. Interferon-inducible protein IFIXalpha1 functions as a negative regulator of HDM2. Mol. Cell. Biol. 2006, 26, 1979–1996. [Google Scholar] [CrossRef]
- Pampin, M.; Simonin, Y.; Blondel, B.; Percherancier, Y.; Chelbi-Alix, M.K. Cross talk between PML and p53 during poliovirus infection: Implications for antiviral defense. J. Virol. 2006, 80, 8582–8592. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, S.; Ross, S.R. APOBEC3 Proteins in Viral Immunity. J. Immunol. 2015, 195, 4565–4570. [Google Scholar] [CrossRef]
- Shatz, M.; Menendez, D.; Resnick, M.A. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012, 72, 3948–3957. [Google Scholar] [CrossRef]
- Menendez, D.; Lowe, J.M.; Snipe, J.; Resnick, M.A. Ligand dependent restoration of human TLR3 signaling and death in p53 mutant cells. Oncotarget 2016, 7, 61630–61642. [Google Scholar] [CrossRef]
- Taura, M.; Eguma, A.; Suico, M.A.; Shuto, T.; Koga, T.; Komatsu, K.; Komune, T.; Sato, T.; Saya, H.; Li, J.D.; et al. p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol. Cell. Biol. 2008, 28, 6557–6567. [Google Scholar] [CrossRef] [PubMed]
- Trapp, S.; Derby, N.R.; Singer, R.; Shaw, A.; Williams, V.G.; Turville, S.G.; Bess, J.W., Jr.; Lifson, J.D.; Robbiani, M. Double-stranded RNA analog poly(I:C) inhibits human immunodeficiency virus amplification in dendritic cells via type I interferon-mediated activation of APOBEC3G. J. Virol. 2009, 83, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Turpin, E.; Luke, K.; Jones, J.; Tumpey, T.; Konan, K.; Schultz-Cherry, S. Influenza virus infection increases p53 activity: Role of p53 in cell death and viral replication. J. Virol. 2005, 79, 8802–8811. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, W.J.; Planz, O.; Ehrhardt, C.; Giner, M.; Silberzahn, T.; Pleschka, S.; Ludwig, S. Caspase 3 activation is essential for efficient influenza virus propagation. EMBO J. 2003, 22, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Wei, J.; Deng, X.; Shi, Z.; Zhu, Z.; Shao, D.; Li, B.; Wang, S.; Tong, G.; Ma, Z. Transcriptional analysis of immune-related gene expression in p53-deficient mice with increased susceptibility to influenza A virus infection. BMC Med. Genom. 2015, 8, 52. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, Y.; Wei, J.; Shao, D.; Shi, Z.; Li, B.; Liu, K.; Qiu, Y.; Zheng, H.; Ma, Z. Type I interferon-mediated immune response against influenza A virus is attenuated in the absence of p53. Biochem. Biophys Res. Commun. 2014, 454, 189–195. [Google Scholar] [CrossRef]
- Ghouzzi, V.E.; Bianchi, F.T.; Molineris, I.; Mounce, B.C.; Berto, G.E.; Rak, M.; Lebon, S.; Aubry, L.; Tocco, C.; Gai, M.; et al. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly and p53. Cell Death Dis. 2017, 8, e2567. [Google Scholar] [CrossRef]
- Castedo, M.; Roumier, T.; Blanco, J.; Ferri, K.F.; Barretina, J.; Tintignac, L.A.; Andreau, K.; Perfettini, J.L.; Amendola, A.; Nardacci, R. Sequential involvement of Cdk1, mTOR and p53 in apoptosis induced by the HIV-1 envelope. EMBO J. 2002, 21, 4070–4080. [Google Scholar] [CrossRef]
- Perfettini, J.L.; Roumier, T.; Castedo, M.; Larochette, N.; Boya, P.; Raynal, B.; Lazar, V.; Ciccosanti, F.; Nardacci, R.; Penninger, J.; et al. NF-kappaB and p53 are the dominant apoptosis-inducing transcription factors elicited by the HIV-1 envelope. J. Exp. Med. 2004, 199, 629–640. [Google Scholar] [CrossRef]
- Bando, S.Y.; Bertonha, F.B.; Vieira, S.E.; de Oliveira, D.B.L.; Chalup, V.N.; Durigon, E.L.; Palmeira, P.; Curi, A.C.P.; Faria, C.S.; Antonangelo, L. Blood leukocyte transcriptional modules and differentially expressed genes associated with disease severity and age in COVID-19 patients. Sci. Rep. 2023, 13, 898. [Google Scholar] [CrossRef]
- Lodi, G.; Gentili, V.; Casciano, F.; Romani, A.; Zauli, G.; Secchiero, P.; Zauli, E.; Simioni, C.; Beltrami, S.; Fernandez, M.; et al. Cell cycle block by p53 activation reduces SARS-CoV-2 release in infected alveolar basal epithelial A549-hACE2 cells. Front. Pharmacol 2022, 13, 1018761. [Google Scholar] [CrossRef] [PubMed]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gao, H.; Ji, Y.; Zhou, Q.; Du, Z.; Tian, L.; Jiang, Y.; Yao, K.; Zhou, Z. Targeting p53-MDM2 interaction by small-molecule inhibitors: Learning from MDM2 inhibitors in clinical trials. J. Hematol. Oncol. 2022, 15, 91. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; AlHilali, S.; Al-Swailem, S.; Secchiero, P.; Voltan, R. Therapeutic potential of the MDM2 inhibitor Nutlin-3 in counteracting SARS-CoV-2 infection of the eye through p53 activation. Front Med. 2022, 9, 902713. [Google Scholar] [CrossRef] [PubMed]
- Refsland, E.W.; Stenglein, M.D.; Shindo, K.; Albin, J.S.; Brown, W.L.; Harris, R.S. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nucleic Acids Res. 2010, 38, 4274–4284. [Google Scholar] [CrossRef]
- Tsang, H.F.; Xue, V.W.; Koh, S.P.; Chiu, Y.M.; Ng, L.P.; Wong, S.C. NanoString, a novel digital color-coded barcode technology: Current and future applications in molecular diagnostics. Expert Rev. Mol. Diagn. 2017, 17, 95–103. [Google Scholar] [CrossRef]
- Farris, S.; Ward, J.M.; Carstens, K.E.; Samadi, M.; Wang, Y.; Dudek, S.M. Hippocampal Subregions Express Distinct Dendritic Transcriptomes that Reveal Differences in Mitochondrial Function in CA2. Cell Rep. 2019, 29, 522–539.e6. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gladwell, W.; Yost, O.; Li, H.; Bell, W.J.; Chen, S.-H.; Ward, J.M.; Kleeberger, S.R.; Resnick, M.A.; Menendez, D. APOBEC3G Is a p53-Dependent Restriction Factor in Respiratory Syncytial Virus Infection of Human Cells Included in the p53/Immune Axis. Int. J. Mol. Sci. 2023, 24, 16793. https://doi.org/10.3390/ijms242316793
Gladwell W, Yost O, Li H, Bell WJ, Chen S-H, Ward JM, Kleeberger SR, Resnick MA, Menendez D. APOBEC3G Is a p53-Dependent Restriction Factor in Respiratory Syncytial Virus Infection of Human Cells Included in the p53/Immune Axis. International Journal of Molecular Sciences. 2023; 24(23):16793. https://doi.org/10.3390/ijms242316793
Chicago/Turabian StyleGladwell, Wesley, Oriana Yost, Heather Li, Whitney J. Bell, Shih-Heng Chen, James M. Ward, Steven R. Kleeberger, Michael A. Resnick, and Daniel Menendez. 2023. "APOBEC3G Is a p53-Dependent Restriction Factor in Respiratory Syncytial Virus Infection of Human Cells Included in the p53/Immune Axis" International Journal of Molecular Sciences 24, no. 23: 16793. https://doi.org/10.3390/ijms242316793